PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Anagrelide hydrochloride is a cyclic phosphodiesterase III inhibitor that was launched in 1997 in the U.S. by Shire Pharmaceuticals for the treatment of essential thrombocythemia and other myeloproliferative disorders
Anagrelide was assigned orphan drug status by the FDA in 1986, by the Japanese Ministry of Health in 2004 and by the European Commission in January 2001 for the treatment of essential thrombocythemia.
Anagrelide controlled release (GALE-401) is in phase III clinical trials by Galena Biopharma for the treatment of ET.[2]
Medical uses
Anagrelide is used to treat essential thrombocytosis, especially when the current treatment of the patient is insufficient.[3] Essential thrombocytosis patients who are suitable for anagrelide often meet one or more of the following factors:[4][5]
Common side effects are headache, diarrhea, unusual weakness/fatigue, hair loss, nausea and dizziness.
The same MRC trial mentioned above also analyzed the effects of anagrelide on bone marrow fibrosis, a common feature in patients with myelofibrosis. The use of anagrelide was associated with a rapid increase in the degree of reticulin deposition (the mechanism by which fibrosis occurs), when compared to those in whom hydroxyurea was used. Patients with myeloproliferative conditions are known to have a very slow and somewhat variable course of marrow fibrosis increase. This trend may be accelerated by anagrelide. Interestingly, this increase in fibrosis appeared to be linked to a drop in hemoglobin as it progressed. Fortunately, stopping the drug (and switching patients to hydroxyurea) appeared to reverse the degree of marrow fibrosis. Thus, patients on anagrelide may need to be monitored on a periodic basis for marrow reticulin scores, especially if anemia develops, or becomes more pronounced if present initially.[6]
Less common side effects include: congestive heart failure, myocardial infarction, cardiomyopathy, cardiomegaly, complete heart block, atrial fibrillation, cerebrovascular accident, pericarditis, pulmonary infiltrates, pulmonary fibrosis, pulmonary hypertension, pancreatitis, gastric/duodenal ulceration, renal impairment/failure and seizure.
Due to these issues, anagrelide should not generally be considered for first line therapy in ET.
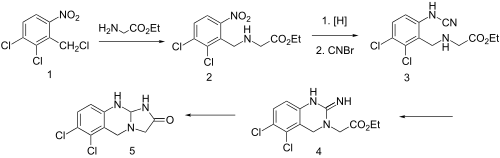
Condensation of benzyl chloride 1 with ethyl ester of glycine gives alkylated product 2. Reduction of the nitro group leads to the aniline and reaction of this with cyanogen bromidepossibly gives cyanamide 3 as the initial intermediate. Addition of the aliphatic would then lead to formation of the quinazoline ring (4). Amide formation between the newly formed imide and the ester would then serve to form the imidazolone ring, whatever the details of the sequence, there is obtained anagrelide (5).
The nitration of 1,2,3-trichlorobenzene (I) with concentrated HNO3 gives 2,3,4-trichloronitrobenzene (II), which by reaction with cuprous cyanide in hot pyridine is converted to 2,3-dichloro-6-nitrobenzonitrile (III). The reduction of (III) with borane in THF yields 2,3-dichloro-6-nitrobenzylamine (IV), which by reaction with ethyl bromoacetate (V) by means of triethylamine in refluxing dioxane affords ethyl N-(2,3-dichloro-6-nitrobenzyl)glycinate (VI). The reduction of (VI) with SnCl2 in concentrated HCl gives ethyl N-(6-amino-2,3-dichlorobenzyl)glycinate (VII), which is cyclized with cyanogen bromide (VIII) in toluene affording ethyl 5,6-dichloro-3,4-dihydro-2-(1H)-iminoquinazoline-3-acetate (IX). Finally, this compound is submitted to a new cyclization by means of triethylamine in refluxing ethanol.
The reaction of 3-chloroaniline (X) with choral hydrate (XI) and hydroxylamine gives isonitroso-3-chloroacetanilide (XII), which is cyclized by means of H2SO4 to 4-chloroisatin (XIII). Chlorination of (XIII) with SO2Cl2 affords 4,5-dichloroisatin (XIV), which is oxidized with H2O2 yielding 5,6-dichloroanthranilic acid (XV). The reduction of (XV) with borane in THF gives 6-amino-2,3-dichlorobenzyl alcohol (XVI), which by reaction with SOCl2 in benzene is converted to 6-amino-2,3-dichlorobenzyl chloride (XVII). This compound is condensed with ethyl glycinate (XVIII) by means of triethylamine in refluxing methylene chloride to give ethyl N-(6-amino-2,3-dichlorobenzyl)glycinate (VII), which is cyclized with cyanogen bromide (VIII) in toluene affording ethyl 5,6-dichloro-3,4-dihydro-2-(1H)-iminoquinazoline-3-acetate (IX). Finally, this compound is submitted to a new cyclization by means of triethylamine in refluxing ethanol.
SYN
WO 0208228
The nitration of 2,3-dichlorobenzaldehyde (I) with HNO3/H2SO4 gives 2,3-dichloro-6-nitrobenzaldehyde (II), which is reduced with NaBH4 in methanol, yielding 2,3-dichloro-6-nitrobenzyl alcohol (III). The reaction of (III) with SOCl2 and TEA affords the benzyl chloride (IV), which is condensed with glycine ethyl ester (V) by means of TEA to provide the adduct (VI). The reduction of the nitro group of (VI) with SnCl2 in aq. HCl or H2 over PtO2/C in ethanol gives the expected amino derivative (VII), which is cyclized with CN-Br in toluene to yield the iminoquinazoline (VIII). Finally, this compound is further cyclized by means of TEA in water to afford the target imidazoquinazolinone.
US 3932407
The condensation of 2-nitro-6-chlorobenzyl chloride (I) with ethyl glycinate (II) by means of triethylamine in refluxing ethanol gives ethyl N-(2-nitro-6-chlorobenzyl)glycinate (III), which is reduced with H2 over Pd/C in ethanol yielding ethyl N-(2-amino-6-chlorobenzyl)glycinate (IV). The cyclization of (IV) with cyanogen bromide (A) in refluxing ethanol affords 6-chloro-1,2,3,5-tetrahydroimidazo[2,1-b]quinazolin-2-one (V), which is finally chlorinated with Cl2 and FeCl3 in hot nitromethane.
Anagrelide, is a potent reducer of platelet count induced by a variety of aggregating agents and has the following structure
( Formula II)
TJS 4146718 disckre? the process for the preparation ->f ethyl-N-(2,3-dich’oro-6 n:tr^benzyl) glycine hydrochloride from 1,2,3-trichlorobenzene as depicted in Scheme I via 2,3-dichloro-6-nitrobenzonitrile, which involves the use of poisonous reagents, such as cuprous cyanide. Cyanation is carried out at a temperature of 1650C which is highly exothermic, uncontrollable and not scalable. 2, 3-dichloro-6-nitrobenzonitri]e has extreme toxic and skin-irritant properties. Diborane is a flammable gas, used for the reduction of 2, 3-dichloro-6-nitrobenzonitrile. The reduction reaction is exothermic, uncontrollable and not feasible industrially.
US 5801245 discloses process for the preparation of ethyl-N-(2,3-dichloro-6-nitrobenzyl)glycine hydrochloride from 2,3-dichloro toluene as depicted in Scheme II.
The reaction involves a radical halogenation of the toluene group. The material is purified by column chromatography at each stage which makes the process more tedious and it is not viable industrially. The use of a chromatographic solvent, such as chloroform (which is a known carcinogen), is disadvantageous with respect to industrial application.
US 2003/0060630 discloses a method for making ethyl-N-(2, 3-dichloro-6-nitro benzyl)glycine hydrochloride form 2,3-dichloro benzaldehyde as depicted in Scheme III.
In step (b), the reduction reaction is earned out in high boiling solvents like toluene. The reduction in step (b) and the chlorination in step (c) are sluggish. Also, the chlorination reaction is exothermic and uncontrollable, which leads to formation of more impurities and thereby resulting in low yield (page 4, column 2, and page 5, column 1 : 65 %) . Hence, this prior art process is not viable for industrial scale up.
Because of the difficulties encountered in the processes disclosed in the prior art, there is a need to develop more efficient and economical synthetic route for the preparation of ethyl-N- (2,3-dichloro-6-nitrobenzyl)glycine hydrochloride, which is suitable for industrial scale up. The present invention relates to a new process for the synthesis of Ethyl-N-(2, 3-dichloro-6-nitrobenzyl)glycine hydrochloride.
Scheme IV :
2,3-Dichloro-6-nitro 2, 3-Dichloro-6-nitro
benzaldehyde benzylalcohol
( III ) ( IV ) ( V )
Acetonitπle
H2NCH9COOEt
HCl(g) in DPA / Ethyl acetate
Ethyl-N-(2,3-dichloro-6-nitroberizyl)
glycine hydrochloride ( I )
EXAMPLES
Example 1
Preparation of 2, 3-dichloro-6-nitro benzyl methane sulphonate, a compound of formula
(V):
Methylene chloride (2000 ml) and sodium borohydride (120 g) were charged to a clean and dry flask and chilled to 0-50C. Methanol (100 ml) was added slowly over a period of 20 minutes followed by 2,3-dichloro-6-nitro benzaldehyde solution (500 g in 2000 ml of methylene chloride) over a period of 2 hours maintaining the temperature at 0-50C and the contents were stirred at 0-50C for 1 hour. After completion of reaction, water (3000 ml) was added and stirred for 10 minutes. The organic layer was separated, dried over sodium sulphate and was filtered to get a clear filtrate.
To the clear filtrate triethylamine (460 ml), was slowly added over a period of 1 hour at 10- 5 150C, then methane sulphonyl chloride (325 ml) was added drop wise over a period of 2 hours maintaining temperature of 10-150C and the reaction mass was allowed to attain room temperature. Further the reaction mass was stirred at room temperature for 5 hours and after completion of reaction, the organic layer was washed with water (1000 ml) twice, followed by IN HCl solution (1000 ml) twice, 5% Sodium bicarbonate solution (1000 ml) twice, water 0 (1000 ml) twice and was dried over sodium sulfate. The clear organic layer was concentrated under vacuum below 4O0C to give the title compound which was used in the next step.
Example 2
Preparation of ethyl N-(2,3-dichIoro-6-nitrobenzyl)gIycine hydrochloride, a compound of formula (I) :
2,3-dichloro-6-nitro benzyl methane sulphonate ( Examplel ) was dissolved in acetonitrile (2400 ml). To this reaction mass were charged anhydrous Potassium carbonate (480 g), dimethyl amino pyridine (480 mg) and glycine ethyl ester (240 g) at room temperature. The contents were stirred at 37-4O0C for 24 hours. After completion of reaction, the insolubles were filtered, washed with acetonitrile (120 ml). The clear filtrate was concentrated and stripped off usin” ethyl acetate (240 ml).
Further ethyl acetate (1200 ml) was added, chilled the contents to 5-100C, adjusted the pH to 2.0 using IP A-HCl at 5-1O0C. The contents were stirred at 5-100C for 1 hour. The solids were filtered, washed with chilled ethyl acetate (120 ml) and dried under vacuum at room temperature for 4 hours to give the title compound (595 g, 76 % yield, 98.5% HPLC purity).
Example 3
Preparation of Anagrelide , a compound of formula (II)
a) Preparation of Ethyl-5,6-dichloro-3,4-dihydro-2[lH]-imino quinazolin-3-acetate hydrobromide A solution of stannous chloride dihydrate (1850 gms) in concentrated HCl (6.7 liters ) was added slowly to a cooled solution of ethyl-N-(2,3-dichloro-6-nitrobenzyl)glycine hydrochloride (595gms) in concentrated HCl (5.15 liters) maintaining temperature 15-200C over a period of 2 hours. The contents were heated slowly to 40-450C and stirred for 1 hour at 40-450C. After completion of reaction, the contents were cooled to 15-2O0C, maintained for 15 minutes and filtered.
The solids thus obtained were suspended in water (2.9 liters), adjusted the pH of the reaction mass to 8.0-9.0 using potassium carbonate solution (prepared by dissolving 376 gms of potassium carbonate in 4.25 liters of water) at 0-50C, extracted into toluene (3.0 liters><3), dried over sodium sulphate and clarified.
To the clear toluene layer, added Cyanogen bromide solution (prepared by dissolving 222 gms of cyanogen bromide in 655 ml of toluene) in 30 minutes maintaining temperature 15-200C and stirred at 25-300C for 2 hours. The contents were heated slowly to 105-1100C and maintained for 16 hours at 105-1100C. After completion of reaction, the mass was cooled to 15-2O0C and stirred for 45 minutes. Filtered the material, washed with chilled toluene (1.3 liters). The material was slurried in toluene (470 ml) at 15-200C for 1 hour, filtered, washed with cold toluene (160 ml) and dried under vacuum at 50-600C for 8 hours to give the title compound (445 gms ).
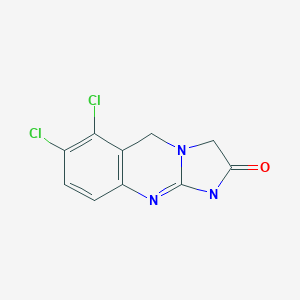
b) Preparation of 6,7-Dichloro-l,5-dihydroimidazo[2,l-b]quinazolin-2(3H)-one [Anagrelide]
A mixture of ethyl-5,6-dichloro-3,4-dihydro-2(lH)-iminoquinazolin-3-acetate hydrobromide (445 gms), isopropyl alcohol (4.45 liters) and triethylamine (246 ml) was refluxed for 2 hours. After completion of reaction, the mixture was cooled to 20-250C, filtered, washed with chilled isopropyl alcohol (1.0 liters) and dried under vacuum at 50-550C for 6 hours to give the title compound (285 gms).
US4208521A *1978-07-311980-06-17Bristol-Myers CompanyProcess for the preparation of imidazo[2,1-b]quinazolinones
EP0514917A1 *1991-05-221992-11-25Egis GyogyszergyarProcess for and 2-(cyanoimino)-quinazoline derivatives useful as intermediates in the preparation of 6,7-di-(chloro)-1,5-di(hydro)-imidazo-[2,1-b]quinazolin-2[3H]-one and process for preparing the 2-(cyanoimino)-quinazoline derivatives
US20030060630A1 *2000-07-262003-03-27Shire Us Inc.Method for the manufacture of Anagrelide
WO2008096145A12007-02-062008-08-14Cipla LimitedProcess for the preparation of ethyl-n-(2, 3-dichloro-6-nitrobenzyl) glycine hydrochloride
REF
Jump up^Voglová J, Maisnar V, Beránek M, Chrobák L (2006). “[Combination of imatinib and anagrelide in treatment of chronic myeloid leukemia in blastic phase]”. Vnitr̆ní lékar̆ství (in Czech). 52 (9): 819–22. PMID17091608.
^ Jump up to:abHarrison CN, Campbell PJ, Buck G, et al. (July 2005). “Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia”. N. Engl. J. Med. 353 (1): 33–45. doi:10.1056/NEJMoa043800. PMID16000354.
Jump up^Reilly, John T. (1 February 2009). “Anagrelide for the treatment of essential thrombocythemia: a survey among European hematologists/oncologists”. Hematology. 14(1): 1–10. doi:10.1179/102453309X385115. PMID19154658.
Jump up^Jones GH, Venuti MC, Alvarez R, Bruno JJ, Berks AH, Prince A (February 1987). “Inhibitors of cyclic AMP phosphodiesterase. 1. Analogues of cilostamide and anagrelide”. J. Med. Chem. 30 (2): 295–303. doi:10.1021/jm00385a011. PMID3027338.
Jump up^Harrison CN, Bareford D, Butt N, et al. (May 2010). “Guideline for investigation and management of adults and children presenting with a thrombocytosis”. Br. J. Haematol. 149(3): 352–75. doi:10.1111/j.1365-2141.2010.08122.x. PMID20331456.
Jump up^Yamaguchi, Hitoshi; Ishikawa, Fumiyoshi (1981). “Synthesis and reactions of 2-chloro-3,4-dihydrothienopyrimidines and -quinazolines”. Journal of Heterocyclic Chemistry. 18: 67. doi:10.1002/jhet.5570180114.
Percent Composition: C 46.90%, H 2.76%, Cl 27.69%, N 16.41%, O 6.25%
Literature References: Phosphodiesterase inhibitor with antiplatelet activity. Prepn: W. N. Beverung, A. Partyka, US3932407; USRE 31617; T. A. Jenks et al.,US4146718 (1976, 1984, 1979 all to Bristol-Myers); H. Yamaguchi, F. Ishikawa, J. Heterocycl. Chem.18, 67 (1981). Antithrombotic and platelet aggregation inhibiting properties: J. S. Fleming, J. P. Buyniski, Thromb. Res.15, 373 (1979). Mode of action studies: S. S. Tang, M. M. Frojmovic, J. Lab. Clin. Med.95, 241 (1980); S. Seiler et al.,J. Pharmacol. Exp. Ther.243, 767 (1987). GC-MS determn in human plasma: E. H. Kerns et al.,J. Chromatogr.416, 357 (1987). Clinical reduction of platelet counts: W. A. Andes et al.,Thromb. Haemostasis52, 325 (1984). Clinical trials to control thrombocytosis in chronic myeloproliferative diseases: M. N. Silverstein et al.,N. Engl. J. Med.318, 1292 (1988); Anagrelide Study Group, Am. J. Med.92,69 (1992). Review of pharmacology and clinical experience: P. E. Petrides, Expert Opin. Pharmacother.5, 1781-1798 (2004).
Percent Composition: C 38.67%, H 3.25%, Cl 34.25%, N 13.53%, O 10.30%
Properties: Off-white powder. Very slightly sol in water; sparingly sol in DMSO, DMF. Also prepd as the hemihydrate; crystals from ethanolic HCl, mp >280°.
Prucalopride, a first in class dihydro-benzofuran-carboxamide, is a selective, high affinity serotonin (5-HT4) receptor agonist with enterokinetic activities.[13] Prucalopride alters colonic motility patterns via serotonin 5-HT4 receptor stimulation: it stimulates colonic mass movements, which provide the main propulsive force for defecation.
The observed effects are exerted via highly selective action on 5-HT4 receptors:[13] prucalopride has >150-fold higher affinity for 5-HT4 receptors than for other receptors.[1][14] Prucalopride differs from other 5-HT4 agonists such as tegaserod and cisapride, which at therapeutic concentrations also interact with other receptors (5-HT1B/D and the cardiac human ether-a-go-go K+ or hERG channelrespectively) and this may account for the adverse cardiovascular events that have resulted in the restricted availability of these drugs.[14]Clinical trials evaluating the effect of prucalopride on QT interval and related adverse events have not demonstrated significant differences compared with placebo.[13]
Pharmacokinetics
Prucalopride is rapidly absorbed (Cmax attained 2–3 hours after single 2 mg oral dose) and is extensively distributed. Metabolism is not the major route of elimination. In vitro, human liver metabolism is very slow and only minor amounts of metabolites are found. A large fraction of the active substance is excreted unchanged (about 60% of the administered dose in urine and at least 6% in feces).Renal excretion of unchanged prucalopride involves both passive filtration and active secretion. Plasma clearance averages 317 ml/min, terminal half-life is 24–30 hours,[15] and steady-state is reached within 3–4 days. On once daily treatment with 2 mg prucalopride, steady-state plasma concentrations fluctuate between trough and peak values of 2.5 and 7 ng/ml, respectively.[13]
In vitro data indicate that prucalopride has a low interaction potential, and therapeutic concentrations of prucalopride are not expected to affect the CYP-mediated metabolism of co-medicated medicinal products.[13]
Efficacy
The primary measure of efficacy in the clinical trials is three or more spontaneous complete bowel movements per week; a secondary measure is an increase of at least one complete spontaneous bowel movement per week.[7][16][17] Further measures are improvements in PAC-QOL[18] (a quality of life measure) and PAC-SYM[19] (a range of stool,abdominal, and rectal symptoms associated with chronic constipation). Infrequent bowel movements, bloating, straining, abdominal pain, and defecation urge with inability to evacuate can be severe symptoms, significantly affecting quality of life.[20][21][22][23][24]
In three large clinical trials, 12 weeks of treatment with prucalopride 2 and 4 mg/day resulted in a significantly higher proportion of patients reaching the primary efficacy endpoint of an average of ≥3 spontaneous complete bowel movements than with placebo.[7][16][17] There was also significantly improved bowel habit and associated symptoms, patient satisfaction with bowel habit and treatment, and HR-QOL in patients with severe chronic constipation, including those who did not experience adequate relief with prior therapies (>80% of the trial participants).[7][16][17] The improvement in patient satisfaction with bowel habit and treatment was maintained during treatment for up to 24 months; prucalopride therapy was generally well tolerated.[25][26]
Side effects
Prucalopride has been given orally to ~2700 patients with chronic constipation in controlled clinical trials. The most frequently reported side effects are headache andgastrointestinal symptoms (abdominal pain, nausea or diarrhea). Such reactions occur predominantly at the start of therapy and usually disappear within a few days with continued treatment.[13]
Approval
In the European Economic Area, prucalopride was originally approved for the symptomatic treatment of chronic constipation in women in whom laxatives fail to provide adequate relief.[13] Subsequently, it has been approved by the European Commission for use in adults – that is, including male patients – for the same indication.[27]
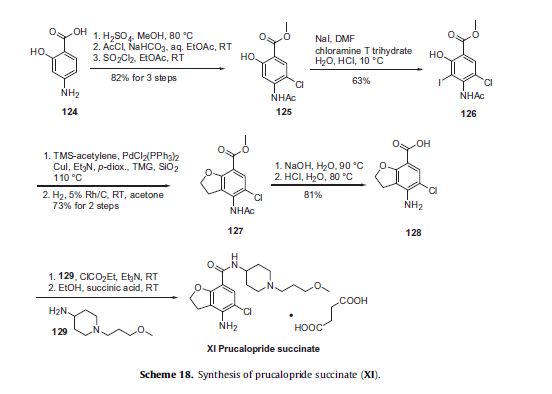
Prucalopride succinate, a first-in-class dihydrobenzofurancarboxamide, is a selective serotonin (5-HT4) receptor agonist.86–94 The drug, marketed under the brand name Resolor, possesses enterokinetic activity and was developed by the Belgian-based pharmaceutical firm Movetis. Prucalopride alters colonic motility patterns via serotonin 5-HT4 receptor stimulation, triggering the central propulsive force for defecation.95–97 The preparation of prucalopride succinate begins with the commercially available salicylic aniline 124 (Scheme 18). Acidic esterification, acetylation of the aniline nitrogen atom, and ambient-temperature chlorination via sulfuryl chloride (SO2Cl2) converted aminophenol 124 to acetamidoester 125 in 83% yield over the course of three steps.98–102 An unique set of conditions involving sodium tosylchloramide (chloramine T) trihydrate and sodium iodide were then employed to convert 125 to o-phenolic iodide 126, which then underwent sequential Sonogashira/cyclization reaction utilizing TMS-acetylene with tetramethylguanidine (TMG) in the presence of silica gel to furnish the benzofuran progenitor of 127.103 Hydrogenation of this intermediate benzofuranyl Sonagashira product saturated the 2,3-benzofuranyl bond while leaving the chlorine atom intact, ultimately delivering dihydrobenzofuran 127 in excellent yield for the two step sequence. Base-induced saponification and acetamide removal gave rise to acid 128. This acid was activated as the corresponding mixed anhydride and treated with commercial piperidine 129 to construct prucalopride which was stirred at room temperature for 24 h in ethanolic succinic acid to provide prucalopride succinate (XI). The yield for the formation of the salt was not provided.
86. Briejer, M. R.; Bosmans, J. P.; Van Daele, P.; Jurzak, M.; Heylen, L.; Leysen, J. E.;Prins, N. H.; Schuurkes, J. A. J. Eur. J. Pharmacol. 2001, 423, 71.
87. Briejer, M. R.; Prins, N. H.; Schuurkes, J. A. J. Neurogastroenterol. Motil. 2001, 13,465.
88. Coggrave, M.; Wiesel, P. H.; Norton, C. Cochrane Database Syst. Rev. 2006.CD002115.
89. Coremans, G.; Kerstens, R.; De Pauw, M.; Stevens, M. Digestion 2003, 67, 82.
90. De Winter, B. Y.; Boeckxstaens, G. E.; De Man, J. G.; Moreels, T. G.; Schuurkes, J.A. J.; Peeters, T. L.; Herman, A. G.; Pelckmans, P. A. Gut 1999, 45, 713.
91. Emmanuel, A. V.; Roy, A. J.; Nicholls, T. J.; Kamm, M. A. Aliment. Pharmacol.Ther. 2002, 16, 1347.
92. Frampton, J. E. Drugs 2009, 69, 2463.
93. Krogh, K.; Bach Jensen, M.; Gandrup, P.; Laurberg, S.; Nilsson, J.; Kerstens, R.;De Pauw, M. Scand. J. Gastroenterol. 2002, 37, 431.
94. Pau, D.; Workman, A. J.; Kane, K. A.; Rankin, A. C. J. Pharmacol. Exp. Ther. 2005,313, 146.
95. De Maeyer, J. H.; Schuurkes, J. A. J.; Lefebvre, R. A. Br. J. Pharmacol. 2009, 156,362.
96. Irving, H. R.; Tochon-Danguy, N.; Chinkwo, K. A.; Li, J. G.; Grabbe, C.; Shapiro,M.; Pouton, C. W.; Coupar, I. M. Pharmacology 2010, 85, 224.
97. Ray, A. M.; Kelsell, R. E.; Houp, J. A.; Kelly, F. M.; Medhurst, A. D.; Cox, H. M.;Calver, A. R. Eur. J. Pharmacol. 2009, 604, 1.
98. Baba, Y.; Usui, T.; Iwata, N. EP 640602 A1, 1995.
99. Fancelli, D.; Caccia, C.; Severino, D.; Vaghi, F.; Varasi, M. WO 9633186 A1,1996.
100. Hirokawa, Y.; Fujiwara, I.; Suzuki, K.; Harada, H.; Yoshikawa, T.; Yoshida, N.;Kato, S. J. Med. Chem. 2003, 46, 702.
101. Kakigami, T.; Usui, T.; Tsukamoto, K.; Kataoka, T. Chem. Pharm. Bull. 1998, 46,42.
102. Van Daele, G. H. P.; Bosmans, J.-P. R. M. A.; Schuurkes, J. A. J. WO 9616060 A1,1996.
103. Candiani, I.; DeBernadinis, S.; Cabri, W.; Marchi, M.; Bedeschi, A.; Penco, S.Synlett 1993, 269.
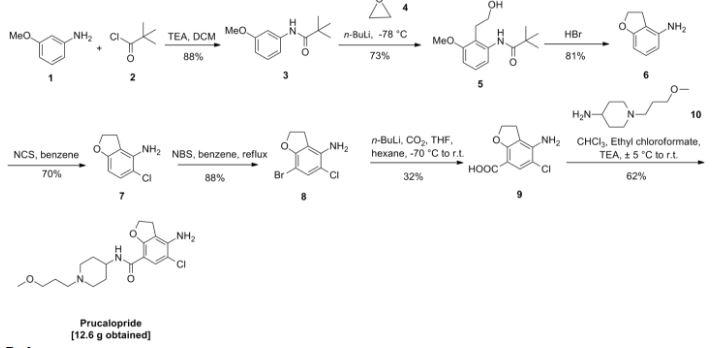
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ±5° C. N,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10° C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10° C. The resulting mixture was added dropwise over a 20-min period to a solution of 1-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCl3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml)+a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50° C.), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-N- 1-(3-methoxypropyl)-4-piperidinyl!-7-benzofurancarboxamide monohydrochloride (comp. 1).
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ±5° C. N,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10° C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10° C. The resulting mixture was added dropwise over a 20-min period to a solution of 1-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCl3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml)+ a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50° C.), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-N- 1-(3-methoxypropyl)-4-piperidinyl!-7-benzofurancarboxamide monohydrochloride (comp. 1).
EP-0,389,037-A, published on September 26, 1990, N-(3-hydroxy-4-piperidin- yl) (dihydrobenzofuran or dihydro-2H-benzopyran)carboxamide derivatives are disclosed as having gastrointestinal motility stimulating properties. In our EP-0,445,862-A, published on September 11, 1991, N-(4-piperidinyl) (dihydrobenzo¬ furan or dihydro-2H-benzopyran)carboxamide derivatives are disclosed also having gastrointestinal motility stimulating properties.
The compound subject to the present application differs therefrom by showing superior enterokinetic properties.
The present invention concerns a compound of formula
and the pharmaceutically acceptable acid addition salts thereof.
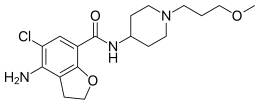
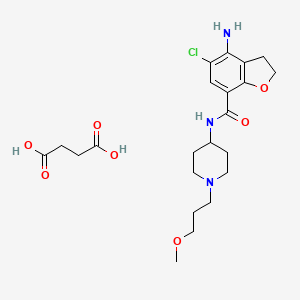
The chemical name of the compound of formula (I) is 4-amino-5-chloro-2,3-dihydro-N- [l-(3-methoxypropyl)-4-piperidinyl]-7-benzofurancarboxamide.
Example 1
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ± 5 °C. H,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10 °C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10°C. The resulting mixture was added dropwise over a 20-min period to a solution of l-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCI3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml) + a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50 °C), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-M-[ 1 -(3-methoxypropyl)-4-piperidinyl]-7- benzofurancarboxamide monohydrochloride (comp. 1).
Example 2
A mixture of 4-amino-5-chloro-2,3-dihydro-N-(4-piperidinyl)-7-benzofuran- carboxamide(O.Olmol), l-chloro-3-methoxypropane (0.012mol), M,M-diethyl- ethanamine (2Jml) and KI (catalytic amount) in N,M-dimethylformamide (75ml) was stirred overnight at 50°C. The reaction mixture was cooled. The solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent: CHCl3/(CH3OH/NH3) 97/3). The pure fractions were collected and the solvent was evaporated. The residue was dissolved in 2-propanol and converted into the hydrochloric acid salt (1:1) with HCl/2-propanol. The precipitate was filtered off and dried (vacuum; 80°C), yielding 1.40g (35%) of 4-amino-5-chloro-2,3-dihydro-N-[l-(3-methoxypropyl)- 4-piperidinyl]-7-benzofurancarboxamide monohydrochloride (comp. 1).
PAPER
Chinese Journal of Pharmaceuticals2012, 43, 5-8.
CLIP
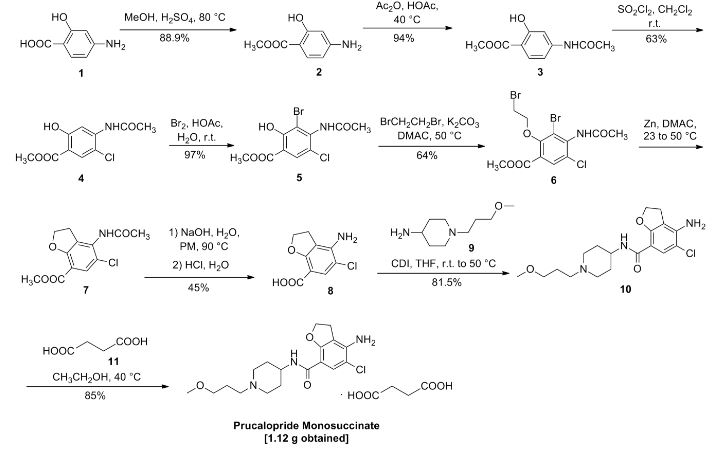
Chinese Patent CN 103012337 A report is as follows:
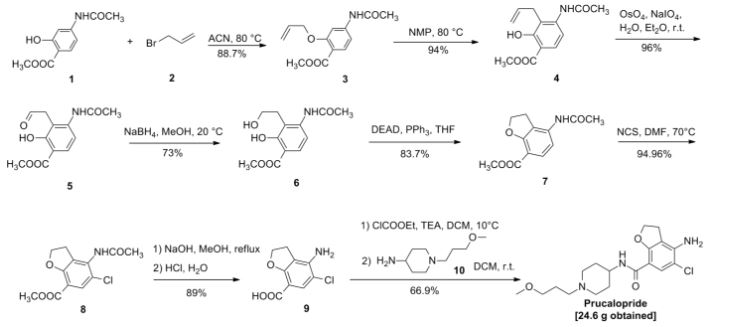
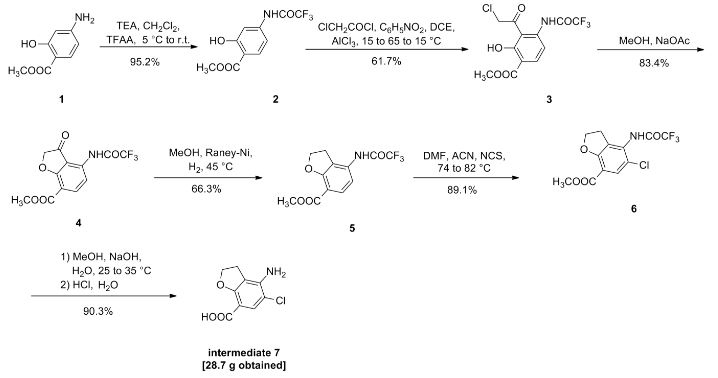
Compound I (167. lg, Imol), triethylamine (111. lg, I. Imol) and methylene chloride (KMOg) added to the reaction flask, nitrogen cooled to 5 ° C, was slowly added dropwise trifluoroacetic anhydride (220. 5g, 1.05mol) / methylene chloride (150g) solution, maintaining the temperature throughout 5~15 ° C, dropping was completed, the reaction after 3 hours at room temperature, TLC (DCM = MeOH = 25: 1) The reaction was monitored to complete the reaction; the reaction mixture was slowly poured into ice water (560g) and stirred for 20 minutes, standing layer, the aqueous phase was separated, the organic phase was washed with saturated aqueous sodium bicarbonate (IOOg) wash sash; IM hydrochloric acid (IlOg) wash sash, then with saturated brine (200g) washed sash, magnesium sulfate (40g) dried, filtered and concentrated to give compound II (250. Ig), yield: 952%.
[0066] 2. Preparation of Compound III
[0067] Chloroacetyl chloride (101. 7g, 0. 9mol), nitrobenzene (20g) and dichloroethane (580 g) added to the reaction flask, nitrogen cooled to 5 ° C, was slowly added anhydrous trichloro aluminum powder (359. 2g, 2. 7mol), to keep the whole temperature 5~20 ° C, plus complete, insulation 15~25 ° C for 30 minutes to obtain a mixture A.
[0068] Compound II (. 236. 7g, 0 9mol) and dichloroethane (500g) added to the reaction flask, nitrogen cooled to 15 ° C; the mixture was added Compound II A quick solution, plus complete, rapid heating 65~75 ° C, 1 hours later once every 15 minutes in the control, monitoring TLC (DCM = MeOH = 50: 1) to complete the reaction; the reaction mixture was immediately poured into ice water (800g) and stirred for 30 minutes, controlling the temperature between 15~25 ° C, the organic phase was separated, the organic phase washed with water (180g) was washed with saturated brine (240g), dried over magnesium sulfate (45g) was dried, filtered and concentrated to give crude compound III (303 . 2g).
[0069] Take the crude compound III (291. 3g) / ethanol 1 dichloromethane: 1 solution (1500ml) was dissolved, and then adding activated carbon (14. 5g) was refluxed for one hour, cooled to room temperature filtered and the filtrate concentrated at room temperature to 600~ 650g, stop and concentrated down to 5~10 ° C, filtered to give a yellow solid (204. 7g); the resulting yellow solid (207. 6g) in tetrahydrofuran (510g) was purified, reduced to 10~15 ° C, filtered, The filter cake was washed with tetrahydrofuran (90g) dip, dried under vacuum to give compound III (181. 3g), yield: 61.7% billion
[0070] 3. Preparation of Compound IV
[0071] Compound 111 (! 169.68,0.5 11〇1), methanol (5,801,111) and sodium acetate (123.38,1.5111〇1) was added to the reaction flask. After 6 hours of reaction, began TLC (DCM: MeOH = 30: 1 ) the reaction was monitored to completion of the reaction; the reaction mixture was cooled to room temperature, concentrated, and the residue with ethyl acetate (500g) and water (200g) was dissolved, the organic phase was separated, the organic phase was washed with 2M sodium hydrogen carbonate (120g) was washed, then with saturated brine (IOOg), dried over magnesium sulfate (50g) was dried, filtered and concentrated to 250~280g, cooled to room temperature with stirring was added cyclohexane (200 g of), after stirring for 1 hour and then filtered and dried to obtain compound IV (126. 7g), yield: 83.4% billion
[0072] 4. Preparation of Compound V
[0073] Compound IV (12L 2g, 0. 4mol), methanol (380g) and Raney-Ni (12. 5g) added to the autoclave, purged with nitrogen, hydrogen is introduced (3. Ompa), the reaction was heated to 45 ° C after 8 hours, TLC (DCM = MeOH = 30: 1) to monitor the reaction, to complete the reaction, cooled to room temperature and pressure, and then purged with nitrogen, the reaction solution was filtered and concentrated to give crude compound V (103. 7g), taking compound V crude product (103g) was refluxed with ethyl acetate (420g) (1 hour) was purified, cooled to room temperature and stirred for 30 minutes and filtered to give a yellow solid was dried in vacuo to give compound V (76 8g.), yield: 663 %.
[0074] 5. Preparation of Compound VI
[0075] Compound ¥ (57.88,0.2111〇1), 1 ^ dimethylformamide (4.58) and acetonitrile (30 (^) was added to the reaction flask and heated 74~76 ° C; solution of N- chlorosuccinimide imide (. 26. 7g, 0 2mol) and acetonitrile (45g) was added dropwise over 30 minutes and maintaining the temperature finished 76~82 ° C, dropping was completed, the reaction was kept, after one hour the reaction started TLC (DCM: MeOH = 30: 1) to monitor the reaction, the reaction is complete the reaction solution cooled to 5~8 ° C, the filter cake was washed with water (210g) washed stirred, filtered, and dried in vacuo to give compound VI (57. 6g), yield. rate of 89.1%.
6. Preparation of Compound VII
Compound VI (48. 5g, 0. 15mol) and methanol (80g) added to the reaction flask, stirring at room temperature was added dropwise 4M aqueous sodium hydroxide (HOg), dropwise complete, for the reaction, 25 ° C~35 after 4 hours of reaction ° C, samples of about 7:00 adjust PH TLC (DCM = MeOH = 30: 1) to monitor the reaction, until the reaction was complete, down to 5~10 ° C, with 6M hydrochloric acid solution PH ~ 7. 5, half the solution was concentrated, then 2M hydrochloric acid solution PH ~ 7, reduced to 15~20 ° C was stirred for 30 minutes, filtered, the filter cake with methyl tert-butyl ether (70g) beating, filtration, and dried in vacuo to give compound VII (28. 7g), yield: 903%.
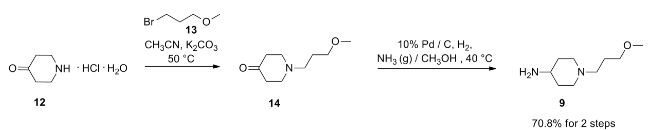
PAPER
Chem Pharm Bull 46 (1), 42-52 (1998) and Pharmaceutical and clinical study based on 2011 (4) 306-307 reported synthetic route is as follows:
Biological Activity
Description
Prucalopride is a selective, high affinity 5-HT4 receptor agonist, inhibiting human 5-HT(4a) and 5-HT(4b) receptor with Ki value of 2.5 nM and 8 nM, respectively.
Prucalopride induces contractions in a concentration-dependent manner with pEC50 of 7.5. Prucalopride (1 mM) significantly amplifies the rebound contraction of the guinea-pig proximal colon after electrical field stimulation. Prucalopride induces relaxation of the rat oesophagus preparation of rat oesophagus tunica muscularis mucosae with pEC50 of 7.8, yielding a monophasic concentration–response curve. [1] Prucalopride (0.1 μM) concentration-dependently increases the amplitude of submaximal cholinergic contractions and of acetylcholine release induced by electrical field stimulation in pig gastric circular muscle, and the effect is induced and enhanced IBMX (10 μM). [2] Prucalopride (1 μM) significantly enhances the electrically induced cholinergic contractions in pig descending colon, and the facilitating effect is significantly enhanced by Rolipram. [3]
In vivo
Prucalopride alters colonic contractile motility patterns in a dose-dependent fashion by stimulating high-amplitude clustered contractions in the proximal colon and by inhibiting contractile activity in the distal colon of fasted dogs. Prucalopride also causes a dose-dependent decrease in the time to the first giant migrating contraction (GMC); at higher doses of prucalopride, the first GMC generally occurres within the first half-hour after treatment. [4]
Features
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
Species
Mouse
Rat
Rabbit
Guinea pig
Hamster
Dog
Weight (kg)
0.02
0.15
1.8
0.4
0.08
10
Body Surface Area (m2)
0.007
0.025
0.15
0.05
0.02
0.5
Km factor
3
6
12
8
5
20
Animal A (mg/kg) = Animal B (mg/kg) multiplied by
Animal B Km
Animal A Km
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
Briejer, M. R.; Bosmans, J. P.; Van Daele, P.; Jurzak, M.; Heylen, L.; Leysen, J. E.; Prins, N. H.; Schuurkes, J. A. (2001). “The in vitro pharmacological profile of prucalopride, a novel enterokinetic compound”. European Journal of Pharmacology423 (1): 71–83.doi:10.1016/S0014-2999(01)01087-1. PMID11438309.
Smart, C. J.; Ramesh, A. N. (2011). “The successful treatment of acute refractory pseudo-obstruction with Prucalopride”. Colorectal Disease: no. doi:10.1111/j.1463-1318.2011.02929.x.
Jump up^Bouras, E. P.; Camilleri, M.; Burton, D. D.; Thomforde, G.; McKinzie, S.; Zinsmeister, A. R. (2001). “Prucalopride accelerates gastrointestinal and colonic transit in patients with constipation without a rectal evacuation disorder”. Gastroenterology120 (2): 354–360.doi:10.1053/gast.2001.21166. PMID11159875.
^ Jump up to:abcdTack, J.; Van Outryve, M.; Beyens, G.; Kerstens, R.; Vandeplassche, L. (2008). “Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives”. Gut58 (3): 357–365. doi:10.1136/gut.2008.162404.PMID18987031.
Briejer, M. R.; Prins, N. H.; Schuurkes, J. A. (2001). “Effects of the enterokinetic prucalopride (R093877) on colonic motility in fasted dogs”. Neurogastroenterology and motility : the official journal of the European Gastrointestinal Motility Society13 (5): 465–472. doi:10.1046/j.1365-2982.2001.00280.x. PMID11696108.
Oustamanolakis, P.; Tack, J. (2012). “Prucalopride for chronic intestinal pseudo-obstruction”. Alimentary Pharmacology & Therapeutics35 (3): 398–9. doi:10.1111/j.1365-2036.2011.04947.x. PMID22221087.
SmPC. Summary of product characteristics Resolor (prucalopride) October, 2009: 1-9.
De Maeyer, JH; Lefebvre, RA; Schuurkes, JA (Feb 2008). “5-HT(4) receptor agonists: similar but not the same”. Neurogastroenterol Motil20 (2): 99–112. doi:10.1111/j.1365-2982.2007.01059.x. PMID18199093.
Camilleri, M.; Kerstens, R.; Rykx, A.; Vandeplassche, L. (2008). “A Placebo-Controlled Trial of Prucalopride for Severe Chronic Constipation”. New England Journal of Medicine358 (22): 2344–2354. doi:10.1056/NEJMoa0800670. PMID18509121.
^ Jump up to:abcQuigley, E. M. M.; Vandeplassche, L.; Kerstens, R.; Ausma, J. (2009). “Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation – a 12-week, randomized, double-blind, placebo-controlled study”.Alimentary Pharmacology & Therapeutics29 (3): 315–328. doi:10.1111/j.1365-2036.2008.03884.x. PMID19035970.
Marquis, P.; De La Loge, C.; Dubois, D.; McDermott, A.; Chassany, O. (2005). “Development and validation of the Patient Assessment of Constipation Quality of Life questionnaire”. Scandinavian Journal of Gastroenterology40 (5): 540–551.doi:10.1080/00365520510012208. PMID16036506.
Frank, L.; Kleinman, L.; Farup, C.; Taylor, L.; Miner Jr, P. (1999). “Psychometric validation of a constipation symptom assessment questionnaire”. Scandinavian journal of gastroenterology34 (9): 870–877. doi:10.1080/003655299750025327.PMID10522604.
Johanson, JF; Kralstein, J (2007). “Chronic constipation: a survey of the patient perspective.”. Alimentary pharmacology & therapeutics25 (5): 599–608. doi:10.1111/j.1365-2036.2006.03238.x. PMID17305761.
Koch, A.; Voderholzer, W. A.; Klauser, A. G.; Müller-Lissner, S. (1997). “Symptoms in chronic constipation”. Diseases of the colon and rectum40 (8): 902–906.doi:10.1007/BF02051196. PMID9269805.
McCrea, G. L.; Miaskowski, C.; Stotts, N. A.; MacEra, L.; Paul, S. M.; Varma, M. G. (2009). “Gender differences in self-reported constipation characteristics, symptoms, and bowel and dietary habits among patients attending a specialty clinic for constipation”.Gender Medicine6 (1): 259–271. doi:10.1016/j.genm.2009.04.007. PMID19467522.
Pare, P.; Ferrazzi, S.; Thompson, W. G.; Irvine, E. J.; Rance, L. (2001). “An epidemiological survey of constipation in Canada: definitions, rates, demographics, and predictors of health care seeking”. The American Journal of Gastroenterology96 (11): 3130–3137. doi:10.1111/j.1572-0241.2001.05259.x. PMID11721760.
Wald, A.; Scarpignato, C.; Kamm, M. A.; Mueller-Lissner, S.; Helfrich, I.; Schuijt, C.; Bubeck, J.; Limoni, C.; Petrini, O. (2007). “The burden of constipation on quality of life: results of a multinational survey”. Alimentary Pharmacology & Therapeutics26 (2): 227–236. doi:10.1111/j.1365-2036.2007.03376.x. PMID17593068.
Camilleri, M; Beyens, G; Kerstens, R; Vandeplassche, L (2009). “Long-term follow-up of safety and satisfaction with bowel function in response to oral prucalopride in patients with chronic constipation [Abstract]”. Gastroenterology136 (Suppl 1): 160. doi:10.1016/s0016-5085(09)60143-8.
Van Outryve, MJ; Beyens, G; Kerstens, R; Vandeplassche, L (2008). “Long-term follow-up study of oral prucalopride (Resolor) administered to patients with chronic constipation [Abstract T1400]”. Gastroenterology134 (4 (suppl 1)): A547. doi:10.1016/s0016-5085(08)62554-8.
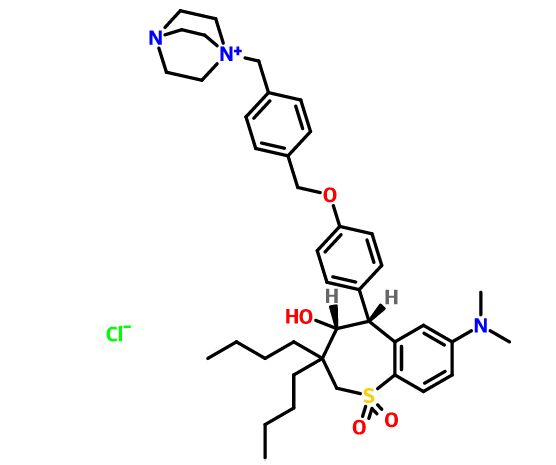
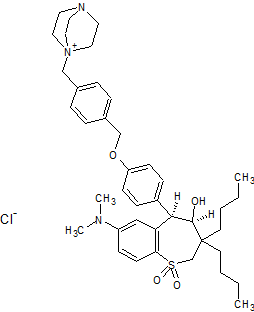
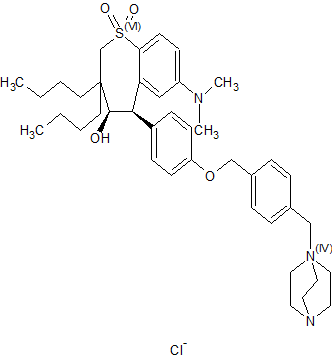
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt
It is well established that agents which inhibit the 20 transport of bile acids across the ileum can also cause a decrease in the level of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255- 25287, discusses biochemistry, physiology, and known active agents affecting bile acids and cholesterol.
A class of ileal bile acid transport-inhibiting compounds which was recently discovered to be useful for influencing the level of blood serum cholesterol is 30 tetrahydrobenzothiepine-l,l-dioxides (THBDO compounds). (U.S. Patent Application No. 08/816,065)
Some classes of compounds show enhanced potency as pharmaceutical therapeutics after they have been enantiomerically-enriched (see, for example, Richard B. Silverman, The Organic Chemistry of Drug Design and Drug Action, Academic Press, 1992, pp. 76-82) . Therefore, THBDO compounds that have been enantiomerically-enriched are of particular interest.
A class of chemistry useful as intermediates in the preparation of racemic THBDO compounds is tetrahydrobenzothiepine-1-oxides (THBO compounds) . THBDO compounds and THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring. A method of preparing enantiomerically-enriched samples of another phenyl/seven-member fused ring system, the benzothiazepines, is described by Higashikawa (JP 59144777) , where racemic benzothiazepine derivatives are optically resolved on a chromatographic column containing chiral crown ethers as a stationary phase. Although optical resolution is achieved, the Higashikawa method is limited to producing only small quantities of the enantiomerically-enriched benzothiazepine derivatives. Giordano (CA 2068231) reports the cyclization of (2S, 3S) -aminophenylthiopropionates in the presence of a phosphonic acid to produce (2S, 3S) -benzothiazepin-4-ones . However, that preparation is constrained by the need to use enantiomerically-enriched starting materials rather than racemic starting materials. In addition, the Giordano method controls the stereochemistry of the seven-member ring of the benzothiazepin-4-one only at the 2- and 3 -positions. The 4- and 5-positions of the seven-member ring of the benzothiazepin-4-one are not asymmetric centers, and the stereochemistry at these sites therefore cannot be controlled by the Giordano method. A method by which enantiomerically-enriched 1,5- benzothiazepin-3-hydroxy-4 (5H) -one compounds have been produced is through the asymmetric reduction of 1,5- benzothiazepin-3,4 (2H, 5H) -dione compounds, reported by Yamada, et al . (J“. Org. Chem. 1996, 61 (24), 8586-8590). The product is obtained by treating the racemic 1,5- benzothiazepin-3,4 (2H, 5H) -dione with the reaction product of an optically active alpha-amino acid and a reducing agent, for example sodium borohydride. Although a product with high optical purity was achieved, the method is limited by the use of a relatively expensive chemical reduction step.
The microbial reduction of racemic 1, 5-benzothiazepin- 3 , 4 (2H, 5H) -dione compounds to produce enantiomerically- enriched 1, 5-benzothiazepin-3-hydroxy-4 (5H) -one compounds is reported by Patel et al . , U.S. Patent 5,559,017. This method is limited by the inherent problems of maintaining a viable and pure bacterial culture of the appropriate species and variety. In addition, that method is limited in scale, producing only microgram quantities of the desired product. Until now, there have been no reported processes for preparing enantiomerically-enriched THBDO compounds or enantiomerically-enriched THBO compounds. Furthermore, there have been no reported processes for controlling the stereochemistry at the 4- and 5-positions of the seven- member rings of THBDO compounds or THBO compounds
FDA Grants Breakthrough Designation to Shire’s Rare GI Therapies
Tue, 06/14/2016
Shire announced that the U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy Designation for two investigational products for rare diseases: SHP621 (budesonide oral suspension, or BOS) for eosinophilic esophagitis (EoE), and SHP625 (maralixibat) for progressive familial intrahepatic cholestasis type 2 (PFIC2).
“Receiving Breakthrough Therapy Designation on two pipeline products this past week reflects the potential of our strong and innovative pipeline of more than 60 programs,” said Flemming Ornskov, M.D., MPH, and CEO, Shire. “Shire is committed to bringing innovation to the rare and specialty areas we focus on. We persevere to see compounds through the many stages of development through their challenges and successes, and always keep patients with unmet needs top of mind.”
EoE is a serious, chronic and rare disease that stems from an elevated number of eosinophils, a type of white blood cell, that infiltrate the walls of the esophagus. EoE is characterized by an inflammation of the esophagus that may lead to difficulty swallowing (dysphagia). The diagnosed prevalence of EoE ranges from approximately 15-55 cases per 100,000 persons, with high-end estimates reported by studies in Western regions.
PFIC refers to a group of autosomal-recessive liver disorders of childhood that disrupt bile formation and present with cholestasis. The symptoms of PFIC include severe itching of the skin (pruritus), and jaundice. PFIC is estimated to affect 1 in 50,000 to 1 in 100,000 births. PFIC2 is the most common type of PFIC, accounting for around half of cases.
According to the FDA, Breakthrough Therapy Designation is granted to a therapy that is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on one or more clinically significant endpoints over current standard of care. Under the designation, the FDA provides intensive guidance, organizational commitment involving senior managers, and eligibility for rolling and priority review of the application; this process helps ensure patients have access to therapies as soon as possible, pending approval. Breakthrough Therapy Designation does not guarantee that FDA will ultimately approve BOS for EoE or maralixibat for PFIC2, and the timing of any such approval is uncertain.
“On behalf of patients in the United States with EoE and PFIC2, we are so pleased that the FDA has granted Breakthrough Therapy Designation to BOS and maralixibat,” said Philip J. Vickers, Ph.D., Head of R&D, Shire. “We look forward to working with the agency to continue their development and, pending FDA approval, deliver these therapeutic options to the patients who need them most.”
It is well established that agents which inhibit the transport of bile acids across the tissue of the ileum can also cause a decrease in the levels of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255-287 discusses biochemistry, physiology, and known active agents surrounding bile acids and cholesterol. Bile acids are actively transported across the tissue of the ileum by an apical sodium co-dependent bile acid transporter (ASBT), alternatively known as an ileal bile acid transporter (IBAT). A class of ASBT-inhibiting compounds that was recently discovered to be useful for influencing the level of blood serum cholesterol comprises tetrahydrobenzothiepine oxides (THBO compounds, PCT Patent Application No. WO 96/08484). Further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 97/33882. Additional THBO compounds useful as ASBT inhibitors are described in U.S. Patent No. 5,994,391. Still further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 99/64409. Included in the THBO class are tetrahydrobenzo-thiepine-l -oxides and tetrahydrobenzothiepine- 1,1 -dioxides. THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring.
Published methods for the preparation of THBO compounds include the synthesis through an aromatic sulfone aldehyde intermediate. For example l-(2,2-dibutyl-3-oxopropylsulfonyl)-2-((4-methoxyphenyl)methyl)benzene (29) was cyclized with potassium t-butoxide to form tetrahydrobenzothiepine- 1,1 -dioxide (svn-24) as shown in Eq. 1.
Compound 29 was prepared by reacting 2-chloro-5-nitrobenzoic acid chloride with anisole in the presence of aluminum trichloride to produce a chlorobenzophenone compound; the chlorobenzophenone compound was reduced in the presence of trifluoromethanesulfonic acid and triethylsilane to produce a chlorodiphenylmethane compound; the chlorodiphenylmethane compound was treated with lithium sulfide and 2,2-dibutyl-3-(methanesulfonato)propanal to produce l-(2,2-dibutyl-3-oxopropylthio)-2-((4-methoxyphenyl)methyl)-4-dimethylaminobenzene (40); and 40 was oxidized with m-chloroperbenzoic acid to produce 29. The first step of that method of preparing compound 29 requires the use of a corrosive and reactive carboxylic acid chloride that was prepared by the reaction of the corresponding carboxylic acid with phosphorus pentachloride. Phosphorus pentachloride readily hydrolyzes to produce volatile and hazardous hydrogen chloride. The reaction of 2,2-dibutyl-3-(methanesulfonato)propanal with the lithium sulfide and the chlorodiphenylmethane compound required the intermediacy of a cyclic tin compound to make the of 2,2-dibutyl-3-(methanesulfonato)propanal. The tin compound is expensive and creates a toxic waste stream. In WO 97/33882 compound syn-24 was dealkylated using boron tribromide to produce the phenol compound 28. Boron tribromide is a corrosive and hazardous material that generates hydrogen bromide gas and requires special handling. Upon hydrolysis, boron tribromide also produces borate salts that are costly and time-consuming to separate and dispose of.
An alternative method of preparing THBO compounds was described in WO 97/33882, wherein a 1,3-propanediol was reacted with thionyl chloride to form a cyclic sulfite compound. The cyclic sulfite compound was oxidized to produce a cyclic sulfate compound. The cyclic sulfate was condensed with a 2-methylthiophenol that had been deprotonated with sodium hydride. The product of the condensation was a (2-methylphenyl) (3′-hydroxypropyl)thioether compound. The thioether compound was oxidized to form an thioether aldehyde compound. The thioether aldehyde compound was further oxidized to form an aldehyde sulfone compound which in turn was cyclized in the presence of potassium t-butoxide to form a 4-hydroxytetrahydrobenzothiepine 1,1 -dioxide compound. This cyclic sulfate route to THBO compounds requires an expensive catalyst. Additionally it requires the use of SOCI2, which in turn requires special equipment to handle. PCT Patent Application No. WO 97/33882 describes a method by which the phenol compound 28 was reacted at its phenol hydroxyl group to attach a variety of functional groups to the molecule, such as a quaternary ammonium group. For example, (4R,5R)-28 was reacted with l,4-bis(chloromethyl)benzene (?,??’-dichloro-p-xylene) to produce the chloromethyl benzyl- ether (4R,5R)-27. Compound (4R,5R)-27 was treated with diazabicyclo[2.2.2]octane (DABCO) to produce (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro-4-hydroxy-l , 1 -dioxido-1 -benzothiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza-l-azomabicyclo[2.2.2]octane chloride (41). This method suffers from low yields because of a propensity for two molecules of compound (4R,5R)-28 to react with one molecule of l,4-bis(chloromethyl)benzene to form a bis(benzothiepine) adduct. Once the bis-adduct forms, the reactive chloromethyl group of compound (4R,5R)-27 is not available to react with an amine to form the quaternary ammonium product.
A method of preparing enantiomerically enriched tetrahydrobenzothiepine oxides is described in PCT Patent Application No. WO 99/32478. In that method, an aryl-3- hydroxypropylsulfide compound was oxidized with an asymmetric oxidizing agent, for example (lR (->(8,9-dichloro-10-camphorsulfonyl)oxaziridine, to yield a chiral aryl-3-hydroxypropylsulfoxide. Reaction of the aryl-3-hydroxypropylsulfoxide with an oxidizing agent such as sulfur trioxide pyridine complex yielded an aryl-3-propanalsulfoxide. The aryl- 3-propanalsulfoxide was cyclized with a base such as potassium t-butoxide to enantioselectively produce a tetrahydrobenzothiepine- 1 -oxide. The tetrahydrobenzothiepine- 1 -oxide was further oxidized to produce a tetrahydrobenzothiepine- 1 , 1 -dioxide. Although this method could produce tetrahydrobenzothiepine- 1,1 -dioxide compounds of high enantiomeric purity, it requires the use of an expensive asymmetric oxidizing agent. Some 5-amidobenzothiepine compounds and methods to make them are described in
PCT Patent Application Number WO 92/18462. In Svnlett. 9, 943-944(1995) 2-bromophenyl 3-benzoyloxy-l-buten-4-yl sulfone was treated with tributyl tin hydride and AIBN to produce 3-benzoyloxytetrahydrobenzothiepine-1,1 -dioxide. In addition to forming the desired ASBT inhibitors, it is also desirable to form such
ASBT inhibitors of higher purity and having lower levels of residual solvent impurities. This is especially so with respect to ASBT inhibitors having a positively charged substituent, for example, the compounds designated as 41 (supra) and 60 (infra). It is further desirable to provide methods for making such high purity ASBT inhibitors.
( 4R, 5R) -26 A 1000 mL 4 neck jacketed Ace reactor flask was fitted with a mechanical stirrer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask was purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). To this was added 4.2 grams of 50% sodium hydroxide. The mixture was heated to 50°C and stiπed for 15 minutes. To the flask was added 8.3 grams of 55 dissolved in 10 mL of DMAC, all at once. The temperature was held at 50°C for 24 hrs. To the flask was added 250 mL of toluene followed by 125 mL of dilution water. The mixture was stiπed for 15 minutes and the layers were then allowed to separate at 50°C. The flask was then charged with 125 mL of saturated sodium chloride solution and stiπed 15 minutes. Layers separated cleanly in 30 seconds at 50°C. Approximately half of the solvent was distilled off under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
( 4R, 5R) -27 Toluene was charged back to the reaction mixture of Step 1 and the mixture was cooled to 35°C. To the mixture was then added 7.0 grams of thionyl chloride over 5 minutes. The reaction was exothermic and reached 39°C. The reaction turned cloudy on first addition of thionyl chloride, partially cleared then finally remained cloudy. The mixture was stirred for 0.5 hr and was then washed with 0.25N NaOH. The mixture appeared to form a small amount of solids that diminished on stirring, and the layers cleanly separated. The solvent was distilled to a minimum stir volume under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 was charged with 350 mL of methyl ethyl ketone (MEK) followed by 10.5 mL water and 6.4 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 10 mL of MEK. The mixture was heated to reflux, and HPLC showed <0.5% of (4R,5R)-27. The reaction remained homogenous initially then crystallized at the completion of the reaction. An additional 5.3 mL of water was charged to the flask to redissolve product. Approximately 160 mL of solvent was then distilled off at atmospheric pressure. The mixture started to form crystals after 70 mL of solvent was distilled. Water separated out of distillate indicating a ternary azeotrope between toluene, water and methyl ethyl ketone (MEK). The mixture was then cooled to 25°C. The solids were filtered and washed with 150 mL MEK, and let dry under vacuum at 60°C. Isolated 29.8.0 g of off-white crystalline 4 Example 11a. Alternate Preparation of (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 100 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 4.02 grams of 50% sodium hydroxide. The mixture is stiπed for 30 minutes. To the flask is added 8.7 grams of 55 dissolved in 12.5 mL of DMAC, all at once. The charge vessel is washed with 12.5 mL DMAC and the wash is added to the reactor. The reactor is stiπed for 3 hours. To the reactor is added 0.19 mL of 49.4% aq. NaOH and the mixture is stirred for 2 hours. To the mixture is added 0.9 g DABCO dissolved in 12.5 mL DMAC. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of dilution water. The mixture is stiπed for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stiπed 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (225 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 30°C. To the mixture is then added 6.7 grams of thionyl chloride over 30 to 45 minutes. The temperature is maintained below 35°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is stiπed for 0.5 hr and is then charged with 156.6 mL of 4% NaOH wash over a 30 minute period. The addition of the wash is stopped when the pH of the mixture reaches’ 8.0 to 10.0. The bottom aqueous layer is removed at 30°C and any rag layer is retained with the organic layer. To the mixture is charged 175 mL of saturated NaCl wash with agitation. The layers are separated at 30°C and the bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 325 mL of methyl ethyl ketone (MEK) and 13 mL water. Next, the reactor is charged 6.2 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 25 mL of MEK. The mixture is heated to reflux and held for 30 minutes. Approximately 10% of solvent volume is then distilled off. The mixture starts to form crystals during distillation. The mixture is then cooled to 20°C for 1 hour. The off-white crystalline 41 (Form U) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example lib. Alternate Preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 7.11 grams of 30% sodium hydroxide over a period of 15 to 30 minutes with agitation. The mixture is stiπed for 30 minutes. To the flask is added 9.5 grams of solid 55. The reactor is stiπed for 3 hours. To the mixture is added 1.2 g of solid DABCO. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of water. The mixture is stirred for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained with the organic layer. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stirred 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. The flask is then charged with 225 mL of water and stirred 15 minutes. The layers are allowed to separate at 50°C. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (112.5 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 25°C. To the mixture is then added 7.3 grams of thionyl chloride over 15 to 45 minutes. The temperature of the mixture is maintained above 20°C and below 40°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is then charged with 179.5 mL of 4% NaOH wash over a 30 minute period. The mixture is maintained above 20°C and below 40°C during this time. The addition of the wash is stopped when the pH of the mixture reaches 8.0 to 10.0. The mixture is then allowed to separate at 40°C for at least one hour.
The bottom aqueous layer is removed and any rag layer is retained with the organic layer. To the mixture is charged 200 mL of dilution water. The mixture is stiπed for 15 minutes and then allowed to separate at 40°C for at least one hour. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-2 .
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 350 mL of methyl ethyl ketone (MEK) and 7 mL water. The mixture is stiπed for 15 minutes and the temperature of the mixture is adjusted to 25°C. Next, the reactor is charged with 6.7 grams of solid diazabicyclo[2.2.2]octane (DABCO). The mixture is maintained at 25°C for three to four hours. It is then heated to 65°C and maintained at that temperature for 30 minutes. The mixture is then cooled to 25°C for 1 hour. The off-white crystalline 41 (Form II) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example 12. Alternate preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form I of 41
(4R,5R)-27 (2.82 kg dry basis, 4.7 mol) was dissolved in MTBE (9.4 L). The solution of (4R,5R)-22 was passed through a 0.2 mm filter cartridge into the feeding vessel. The flask and was rinsed with MTBE (2 x 2.5 L). The obtained solution as passed through the cartridge filter and added to the solution of (4R,5R)-2 in the feeding vessel. DABCO (diazabicyclo[2.2.2]octane, 0.784 kg, 7.0 mol) was dissolved in MeOH (14.2 L). The DABCO solution was passed through the filter cartridge into the 100 L nitrogen-flushed reactor. The Pyrex bottle and the cartridge filter were rinsed with MeOH (7.5 L) and the solution was added to the reactor. The (4R,5R)-22 solution was added from the feeding vessel into the reactor at 37°C over a period of 10 min, while stirring. Methanol (6.5 L) was added to the Pyrex bottle and via the cartridge filter added to the feeding vessel to rinse the remaining (4R,5R)-2 into the reactor. The reaction mixture was brought to 50-60°C over 10-20 min and stiπed at that temperature for about 1 h. The mixture was cooled to 20-25°C over a period of 1 h. To the reaction mixture, methyl t-butyl ether (MTBE) (42 L) was added over a period of 1 h and stiπed for a minimum of 1 h at 20 – 25°C. The suspension was filtered through a Buchner funnel. The reactor and the filter cake were washed with MTBE (2 x 14 L). The solids were dried on a rotary evaporator in a 20 L flask at 400 – 12 mbar, 40°C, for 22 h. A white crystalline solid was obtained. The yield of 4 . (Form I) was 3.08 kg (2.97 kg dry, 93.8 %) and the purity 99.7 area % (HPLC; Kromasil C 4, 250 x 4.6 mm column; 0.05% TFA in H2O/0.05% TFA in ACN gradient, UV detection at 215 nm).
Example 12a. Conversion of Form I of Compound 41 into Form II of Compound 41.
To 10.0 grams of Form I of 4 . in a 400 mL jacketed reactor is added 140 mL of MEK. The reactor is stirred (358 φm) for 10 minutes at 23 °C for 10 minutes and the stirring rate is then changed to 178 φm. The suspension is heated to reflux over 1 hour using a programmed temperature ramp (0.95°C/minute) using batch temperature control (cascade mode). The delta Tmaχ is set to 5°C. The mixture is held at reflux for 1 hour. The mixture is cooled to
25°C. After 3 hours at 25°C, a sample of the mixture is collected by filtration. Filtration is rapid (seconds) and the filtrate is clear and colorless. The white solid is dried in a vacuum oven (80°C, 25 in. Hg) to give a white solid. The remainder of the suspension is stirred at 25°C for 18 hours. The mixture is filtered and the cake starts to shrink as the mother liquor reaches the top of the cake. The filtration is stopped and the reactor is rinsed with 14 mL of MEK. The reactor stirrer speed is increased from 100 to 300 φm to rinse the reactor. The rinse is added to the filter and the solid is dried with a rapid air flow for 5 minutes. The solid is dried in a vacuum oven at 25 in. Hg for 84 hours to give Form II of 4
Department of Discovery Chemistry and Department of Cardiovascular Disease, Pharmacia, 700 Chesterfield Parkway W, Chesterfield, Missouri 63017, Office of Science and Technology, Chemical Science Division, Pharmacia, 800 Lindbergh Boulevard, Creve Coeur, Missouri 63167, Department of Pharmaceutical Sciences, Pharmacia, Skokie, Illinois, and Department of Chemistry, University of Missouri, St. Louis, Missouri
In the preceding paper several compounds were reported as potent apical sodium-codependent bile acid transporter (ASBT) inhibitors. Since the primary site for active bile acid reabsorption is via ASBT, which is localized on the luminal surface of the distal ileum, we reasoned that a nonsystemic inhibitor would be desirable to minimize or eliminate potential systemic side effects of an absorbed drug. To ensure bioequivalency and product stability, it was also essential that we identify a nonhygroscopic inhibitor in its most stable crystalline form. A series of benzothiepines were prepared to refine the structure−activity relationship of the substituted phenyl ring at the 5-position of benzothiepine ring and to identify potent, crystalline, nonhygroscopic, and efficacious ASBT inhibitors with low systemic exposure.
compd
R
IC50 (nM)b
hygroscp I wt gain (%)c
hygroscp II % wt gain (%)d
crystallinitye
74
OCH2C6H4(p)CH2(N+)DB
0.28
1.59
2.1
yes
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt(74). Following a similar procedure as in General Method B, the title compound 74 was prepared from the corresponding chloromethyl benzyl ether and DABCO as a white solid, mp 223−230 °C (dec); 1H NMR (CDCl3) δ 0.89 (m, 6H), 1.27−1.52 (br m, 10H), 1.63 (m, 1H), 2.20 (m, 1H), 2.81 (s, 6H), 3.06 (ABq, JAB = 15.1 Hz, J = 43.3 Hz, 2H), 3.16 (s, 6H), 3.76 (s, 6H), 4.11 (d, J = 7.7 Hz, 1H), 5.09 (s, 2H), 5.14 (s, 2H), 5.48 (s, 1H), 5.96 (s, 1H), 6.49 (d, J = 8.9 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 7.26 (m, 1H), 7.44 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 7.4 Hz, 2H), 7.68 (d, J = 7.4 Hz, 2H), 7.87 (d, J = 8.9 Hz, 1H). HRMS calcd for C40H56N3O4S: 674.3992; found, 674.4005. Anal. Calcd for C40H56N3O4S: ‘ C, 67.62; H, 7.95; N, 5.92; S, 4.51. Found: C, 67.48; H, 8.32; N, 5.85; S, 4.60.
a All compounds were prepared using method B in Scheme 3.b Taurocholate is transported across the baby hamster kidney cell membrane.c % weight gain in a 25 °C, 57% humidity chamber for 2 weeks.d % weight gain in a 40 °C, 80% humidity chamber for 2 weeks.e Crystallinity as determined by X-ray powder diffraction analysis.f (N+)DB is a DABCO terminal group with the quaternary ammonium attached to the linke
Example 10. Preparation of enantiomerically-enriched (4R.5R)-1- r.4- r _4- .3.3 -Dibutyl-7- (dimethylamino) -2.3 ,4.5- tetrahydro-4-hydroxy-1, l-dioxido-l-benzothiepin-5- yl] henoxy] ethyl] phenyl1methyl] -4-aza-l- azoniabicyclo [2.2.2] octane chloride ( (4R,5R) -XXVII) ♦
( (4R,5R) -XXVII) * = chiral center
Step 1. Preparation of 4-flUoro-2- ( (4- methoxyphenyl) methyl) -phenol To a stirred solution of 23.66 g of 95% sodium hydride (0.94 mol) in 600 mL of dry toluene was added 100.0 g of 4- fluorophenol (0.89 mol) at 0°C. The mixture was stirred at 90°C for 1 hour until gas evolution stopped. The mixture was cooled down to room temperature and a solution of 139.71 g of 3 -methoxybenzyl chloride (0.89 mol) in 400 mL of dry toluene was added. After refluxing for 24 hours, the mixture was cooled to room temperature and quenched with 500 mL of water. The organic layer was separated, dried over MgS04, and concentrated under high vacuum. The remaining starting materials were removed by distillation. The crude dark red oil was filtered through a layer of 1 L of silica gel with neat hexane to yield 53.00 g (25.6%) of the product as a pink solid: *H NMR (CDC13) d 3.79 (s, 3H) , 3.90 (s, 2H) , 4.58 (s, IH) , 6.70-6.74 (m, IH) , 6.79-6.88 (m, 4H) , 7.11-7.16 (m, 2H) .
Step 2. Preparation of 4-fluoro-2- ( (4- methoxyphenyl) methyl) -thiophenol
Step 2a. Preparation of thiocarbamate To a stirred solution of 50.00 g (215.30 mmol) of 4- fluoro-2- ( ( -methoxyphenyl) methyl) -phenol in 500 mL of dry DMF was added 11.20 g of 60% sodium hydride dispersion in mineral oil (279.90 mmol) at 2°C. The mixture was allowed to warm to room temperature and 26.61 g of dimethylthiocarbamoyl chloride (215.30 mmol) was added. The reaction mixture was stirred at room temperature overnight. The mixture was quenched with 100 mL of water in an ice bath. The solution was extracted with 500 mL of diethyl ether. The ether solution was washed with 500 mL of water and 500 mL of brine. The ether solution was dried over MgS04 and stripped to dryness. The crude product was filtered through a plug of 500 mL silica gel using 5% ethyl acetate/hexane to yield 48.00 g (69.8%) of the product as a pale white solid: XH NMR (CDC13) d 3.21 (s, 3H) , 3.46 (s, 3H) , 3.80 (s, 3H) , 3.82 (s, 2H) , 6.78-6.86 (m, 3H) , 6.90- 7.00 (m, 2H) , 7.09 (d, J = 8.7 Hz, 2H) .
Step 2b. Rearrangement and hydrolysis of thiocarbamate to 4-fluoro-2- ( (4 -methoxyphenyl) methyl) -thiophenol A stirred solution of 48.00 g (150.29 mmol) of thiocarbamate (obtained from Step 2a) in 200 mL of diphenyl ether was refluxed at 270°C overnight. The solution was cooled down to room temperature and filtered through 1 L of silica gel with 2 L of hexane to remove phenyl ether. The rearrangement product was washed with 5% ethyl acetate/hexane to give 46.00 g (95.8%) of the product as a pale yellow solid: XH NMR (CDC13) d 3.02 (s, 3H) , 3.10 (s, 3H) , 3.80 (s, 3H) , 4.07 (s, 2H) , 6.82-6.86 (m, 3H) , 6.93 (dt, J = 8.4 Hz, 2.7 Hz, IH) , 7.08 (d, J = 8.7 Hz, 2H) , 7.49 (dd, J = 6.0 Hz, 8.7 Hz, IH) . To a solution of 46.00 g (144.02 mmol) of the rearrangement product (above) in 200 mL of methanol and 200 mL of THF was added 17.28 g of NaOH (432.06 mmol) . The mixture was refluxed under nitrogen overnight . The solvents were evaporated off and 200 mL of water was added. The aqueous solution was washed with 200 mL of diethyl ether twice and placed in an ice bath. The aqueous mixture was acidified to pH 6 with concentrated HCl solution. The solution was extracted with 300 mL of diethyl ether twice. The ether layers were combined, dried over MgS04 and stripped to dryness to afford 27.00 g (75.5%) of the product as a brown oil: XH NMR (CDC13) d 3.24 (s, IH) , 3.80 (s, 3H) , 3.99 (s, 2H) , 6.81-6.87 (m, 4H) , 7.09 (d, J = 8.7 Hz, 2H) , 7.27- 7.33 (m, IH) .
Step 3. Preparation of dibutyl cyclic sulfate
Step 3a. Preparation of 2 , 2-dibutyl-l, 3-propanediol . To a stirred solution of di-butyl-diethylmalonate (Aldrich) (150g, 0.55 mol in dry THF (700ml) in an acetone/dry ice bath was added LAH (1 M THF) 662 ml (1.2 eq. , 0.66 mol) dropwise maintaining the temperature between -20 to 0°C. The reaction was stirred at RT overnight. The reaction was cooled to -20°C and 40 ml of water, and 80 mL of 10% NaOH and 80 ml of water were added dropwise. The resulting suspension was filtered. The filtrate was dried over sodium sulphate and concentrated in vacuo to give diol 598.4 g (yield 95%) as an oil. MS spectra and proton and carbon NMR spectra were consistent with the product.
Step 3b. Preparation of dibutyl cyclic sulfite
A solution of 2 , 2-dibutyl-l, 3-propanediol (103 g, 0.548 0 mol, obtained from Step 3a) and triethylamine (221 g, 2.19 mol) in anhydrous methylene chloride (500 ml) was stirred at 0°C under nitrogen. To the mixture, thionyl chloride (97.8* g, 0.‘82 mol) was added dropwise and within 5 min the solution turned yellow and then black when the addition was 5 completed within half an hour. The reaction mixture was stirred for 3 hrs. at 0°C. GC showed that there was no starting material left. The mixture was washed with ice water twice then with brine twice . The organic phase was dried over magnesium sulfate and concentrated under vacuum 0 to give 128 g (100%) of the dibutyl cyclic sulfite as a black oil. Mass spectrum (MS) was consistent with the product .
Step 3c. Oxidation of dibutyl cyclic sulfite to 5 dibutyl cyclic sulfate
To a solution of the dibutyl cyclic sulfite (127.5 g , 0.54 mol, obtained from Step 3b) in 600 ml acetonitrile and 500 ml of water cooled in an ice bath under nitrogen was added ruthenium (III) chloride (1 g) and sodium periodate 0 (233 g, 1.08 mol) . The reaction was stirred overnight and the color of the solution turned black. GC showed that there was no starting material left. The mixture was extracted with 300 ml of ether and the ether extract was washed three times with brine. The organic phase was dried over magnesium sulfate and passed through celite. The filtrate was 5 concentrated under vacuum and to give 133 g (97.8%) of the dibutyl cyclic sulfate as an oil. Proton and carbon NMR and MS were consistent with the product.
Step 4. Preparation of aryl-3-hydroxypropylsulfide
10 To a stirred solution of 27.00 g (108.73 mmol) of 4- fluoro-2- ( (4-methoxyphenyl) methyl) thiophenol (obtained from Step 2) in 270 mL of diglyme was added 4.35 g of 60% sodium-, hydride dispersion in mineral oil (108.73 mmol) at 0°C. After gas evolution ceased, 29.94 g (119.60 mmol) of the
15 dibutyl cyclic sulfate (obtained from Step 3c) was added at 0°C and stirred for 10 minutes. The mixture was allowed to warm up to room temperature and stirred overnight. The solvent was evaporated and 200 mL of water was added. The solution was washed with 200 mL of diethyl ether and added
2025 mL of concentrated sulfuric acid to make a 2.0 M solution that was refluxed overnight. The solution was extracted with ethyl acetate and the organic solution was dried over MgS04 and concentrated in vacuo. The crude aryl-3 – hydroxypropylsulfide was purified by silica gel
25 chromatography (Waters Prep 500) using 8% ethyl acetate/hexane to yield 33.00 g (72.5%) of the product as a light brown oil: E NMR (CDC13) d 0.90 (t, J = 7.1 Hz, 6H) , 1.14-1.34 (m, 12H) , 2.82 (s, 2H) , 3.48 (s, 2H) , 3.79 (s, 3H) , 4.10 (s, 2H) , 6.77-6.92 (m, 4H) , 7.09 (d, J = 8.7 Hz,
To a stirred solution of 20.00 g (47.78 mmol) of aryl- 53 -hydroxypropylsulfide (obtained from Step 4) in 1 L of methylene chloride was added 31.50 g of 96% (12?) – ( -) – (8 , 8- dichloro-10-camphor-sulfonyl) oxaziridine (100.34 mmol, Aldrich) at 2°C. After all the oxaziridine dissolved the mixture was placed into a -30 °C freezer for 72 hours. The
10 solvent was evaporated and the crude solid was washed with 1 L of hexane. The white solid was filtered off and the hexane solution was concentrated in vacuo. The crude oil was purified on a silica gel column (Waters Prep 500) using 15% ethyl acetate/hexane to afford 19.00 g (95%) of the
207.00 (d, J = 8.1 Hz, 2H) , 7.18-7.23 (m, IH) , 7.99-8.04 (m, IH) . Enantiomeric excess was determined by chiral HPLC on a (2?,2?) -Whelk-0 column using 5% ethanol/hexane as the eluent. It showed to be 78% e.e. with the first eluting peak as the major product.
25
Step 6. Preparation of enantiomerically-enriched aryl-3- propanalsulfoxide
To a stirred solution of 13.27 g of triethylamine (131.16 mmol, Aldrich) in 200 mL dimethyl sulfoxide were
30 added 19.00 g (43.72 mmol) of enantiomerically-enriched aryl-3 -hydroxypropylsulfoxide (obtained from Step 5) and 20.96 g of sulfur trioxide-pyridine (131.16 mmol, Aldrich) at room temperature. After the mixture was stirred at room temperature for 48 hours, 500 mL of water was added to the mixture and stirred vigorously. The mixture was then 5 extracted with 500 mL of ethyl acetate twice. The ethyl acetate layer was separated, dried over MgS04, and concentrated in vacuo. The crude oil was filtered through 500 mL of silica gel using 15% ethyl acetate/hexane to give 17.30 g (91%) of the enantiomerically-enriched aryl-3-
Step 7. Preparation of the enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R, 5R)
20 To a stirred solution of 17.30 g (39.99 mmol) of enantiomerically-enriched aryl-3 -propanalsulfoxide (obtained from Step 6) in 300 mL of dry THF at -15°C was added 48 mL of 1.0 M potassium t-butoxide in THF (1.2 equivalents) under nitrogen. The solution was stirred at -15°C for 4 hours.
25 The solution was then quenched with 100 mL of water and neutralized with 4 mL of concentrated HCl solution at 0°C. The THF layer was separated, dried over MgS04, and concentrated in vacuo. The enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R,5R) was purified by
To a stirred solution of 13.44 g (31.07 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (obtained from Step 7) in 150 mL of methylene chloride was added 9.46 g of 68% m-chloroperoxybenzoic acid (37.28 mmol,
15 Sigma) at 0 °C. After stirring at 0 °C for 2 hours, the mixture was allowed to warm up to room temperature and stirred for 4 hours. 50 mL of saturated Na2S03 was added into the mixture and stirred for 30 minutes. The solution was then neutralized with 50 mL of saturated NaHC03 solution.
20 The methylene chloride layer was separated, dried over MgS04, and concentrated in vacuo to give 13.00 g (97.5%) of the enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (4R,5R) as a light yellow solid: ‘H NMR (CDC13) d 0.89-0.95 (m, 6H) , 1.09-1.42 (m, 12H) , 2.16-2.26 (m, IH) ,
30 Step 9. Preparation of enantiomerically-enriched 7-
(dimethylamino) tetrahydrobenzothiepine-1 , 1-dioxide (4R.5R) – To a solution of 13.00 g (28.98 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (obtained from Step 8) in 73 mL of dimethylamine (2.0 M in THF, 146 mmol) in a Parr Reactor was added ca . 20 5 mL of neat dimethylamine . The mixture was sealed and stirred at 110 °C overnight, and cooled to ambient temperature. The excess dimethylamine was evaporated. The crude oil was dissolved in 200 mL of ethyl acetate and washed with 100 mL of water, dried over MgS04 and
10 concentrated in vacuo. Purification on a silica gel column (Waters Prep 500) using 20% ethyl acetate/hexane gave 12.43 g (90.5%) of the enantiomerically- enriched 7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (4R, 5R) as a colorless solid: *H NMR (CDC13) d 0.87-0.93 (m, 6H) ,
20 IH) . The product was determined to have 78% e.e. by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. Recrystallization of this solid from ethyl acetate/hexane gave 1.70 g of the racemic product. The remaining solution was concentrated and recrystallized to
25 give 9.8 g of colorless solid. Enantiomeric excess of this solid was determined by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. It showed to have 96% e.e with the first eluting peak as the major product.
30 Step 10: Demethylation of 5- (4 ‘ -methoxyphenyl) -7-
(dimethylamino) tetrahydrobenzothiepine-1.1-dioxide (4R, 5R) To a solution of 47 g (99 mmol) of enantiomeric- enriched (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (obtained from Step 9) in 500 mL of methylene chloride at -10 °C was added dropwise a solution of boron tribromide (297 mL, 1M in methylene chloride, 297 mmol), and the resulting solution was stirred cold (-5 °C to 0 °C) for 1 hour or until the reaction was complete. The reaction was cooled in an acetone-dry ice bath at -10 °C, and slowly quenched with 300 mL of water. The mixture was warmed to 10 °C, and further diluted with 300 mL of saturated sodium bicarbonate solution to neutralize the mixture. The aqueous layer was separated and extracted with 300 mL of methylene chloride, and the combined extracts were washed with 200 mL of water, brine, dried over MgS04 and concentrated in vacuo. The residue was dissolved in 500 mL of ethyl acetate and stirred with 50 mL of glacial acetic acid for 30 minutes at ambient temperature. The mixture was washed twice with 200 mL of water, 200 mL of brine, dried over MgS04 and concentrated in vacuo to give the crude 4-hydroxyphenyl intermediate. The solid residue was recrystallized from methylene chloride to give 37.5 g (82%) of the desired (4R, 5R) -5- (4′ – hydoxyphenyl) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1- dioxide as a white solid: *H NMR (CDC13) d 0.84-0.97 (m, 6H) , 1.1-1.5 (m, 10H) , 1.57-1.72 (m, IH) , 2.14-2.28 (m, IH) , 2.83 (s, 6H) , 3.00 (d, J = 15.3 Hz, IH) , 3.16 (d, J – 15.3 Hz, IH) , 4.11 (s, 2H) , 5.48 (s, IH) , 6.02 (d, J – 2.4 Hz, IH) , 6.55 (dd, J“ = 9, 2.4 Hz, IH) , 6.88 (d, 8 , 7 Hz , 2H) , 7.38 (d, J – 8.7 Hz, 2H) , 7.91 (d, J“ = 9 Hz, 2H) .
Step 11: Preparation of enantiomerically-enriched chlorobenzyl intermediate Treat a solution of enantiomerically-enriched (4R,5R)- 5- (4′ -hydoxypheny1) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (5.0 g, 10.9 mmol, obtained from Step 10) in acetone (100 mL) at 25 °C under N2 with powdered 5 K2C03 (2.3 g, 16.3 mmol, 1.5 eq.) and a, a’ -dichloro-p-xylene (6.7 g, 38.1 mmol, 3.5 eq.) . Stir the resulting solution at 65 °C for about 48 hours. Cool the reaction mixture to 25 °C and concentrate to 1/5 of original volume. Dissolve the residue in EtOAc (150 mL) and wash with water (2 x 150 mL) .
10 Extract the aqueous layer with EtOAc (2 x 150 mL) and wash the combined organic extracts with saturated aqueous NaCI (2 x 150 mL. Dry the combined extracts with MgS04 and concentrate in vacuo to provide the crude product . Purification by flash chromatography (5.4 x 45 cm silica,
1525-40% EtOAc/hexane) will afford the enantiomerically- enriched chlorobenzyl intermediate .
Step 12: Preparation of enantiomerically-enriched (4R.5R)- 1- r [4- [ [4- [3 , 3-Dibutyl-7- (dimethylamino) -2,3 , 4 , 5-tetrahvdro-
Treat a solution of the enantiomerically-enriched chlorobenzyl intermediate (4.6 g, 7.7 mmol, obtained from
25 above in Step 11) in acetonitrile (100 mL) at 25 °C under N2 with diazabicyclo [2.2.2] -octane (DABCO, 0.95 g, 8.5 mmol, 1.1 eq.) and stir at 35 °C for 2 hours. Collect the precipitated solid and wash with CH3CN. Recrystallization from CH3OH/Et20 will give the desired title compound (XXVII) .
ANY ERROR, EMAIL amcrasto@gmail.com, +919323115463
Guanfacine (brand name Tenex, and the extended release Intuniv) is a sympatholytic. It is a selective α2A receptor agonist. These receptors are concentrated heavily in the prefrontal cortex and the locus coeruleus, with the potential to improve attention abilities via modulating post-synaptic α2A receptors in the prefrontal cortex. Guanfacine lowers both systolic and diastolic blood pressure by activating the central nervous system α2A norepinephrine autoreceptors, which results in reduced peripheral sympathetic outflow and thus a reduction in peripheral sympathetic tone. Its side-effects are dose dependent, with practically no dryness of the mouth at doses of 2 mg and less
April 26, 2013
Shire has come to an agreement with drugmakers Actavis and Watson that lays to rest all pending litigation over their attempts to launch a generic form of the attention-deficit hyperactivity drug (ADHD) Intuniv (guanfacine hydrochloride) in the US.
Under the settlement, Shire has granted Actavis a license to make and market its version of Intuniv in the US from December 1 next year, in return for a 25% royalty on gross profit during the 180 day period of exclusivity.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....