
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
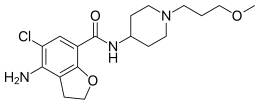
Prucalopride (Resolor)
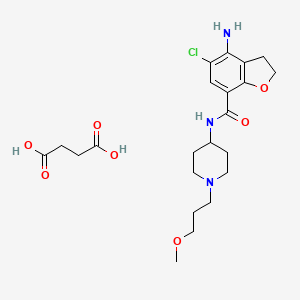
| Prucalopride succinate; 179474-85-2; Resolor; Prucalopride (succinate); UNII-4V2G75E1CK; R-108512; | |
| Molecular Formula: | C22H32ClN3O7 |
|---|---|
| Molecular Weight: | 485.95838 g/mol |
Drug Name:Prucalopride Succinate
Trade Name:Resolor®, MOA:Serotonin (5-HT4) receptor agonist, Indication:Chronic constipation
Company:Shire (Originator) , Johnson & Johnson
APPROVED EU 2009-10-15
CHINA 2014-01-21
Prucalopride (brand name Resolor, developed by Johnson & Johnson and licensed to Movetis) is a drug acting as a selective, high affinity 5-HT4 receptor agonist[1] which targets the impaired motility associated with chronic constipation, thus normalizing bowel movements.[2][3][4][5][6][7] Prucalopride was approved for use in Europe in 2009,[8] in Canada (named Resotran) on December 7, 2011[9] and in Israel in 2014[10] but it has not been approved by the Food and Drug Administration for use in the United States. The drug has also been tested for the treatment of chronic intestinal pseudo-obstruction.[11][12]
Mechanism of action
Prucalopride, a first in class dihydro-benzofuran-carboxamide, is a selective, high affinity serotonin (5-HT4) receptor agonist with enterokinetic activities.[13] Prucalopride alters colonic motility patterns via serotonin 5-HT4 receptor stimulation: it stimulates colonic mass movements, which provide the main propulsive force for defecation.

The observed effects are exerted via highly selective action on 5-HT4 receptors:[13] prucalopride has >150-fold higher affinity for 5-HT4 receptors than for other receptors.[1][14] Prucalopride differs from other 5-HT4 agonists such as tegaserod and cisapride, which at therapeutic concentrations also interact with other receptors (5-HT1B/D and the cardiac human ether-a-go-go K+ or hERG channelrespectively) and this may account for the adverse cardiovascular events that have resulted in the restricted availability of these drugs.[14] Clinical trials evaluating the effect of prucalopride on QT interval and related adverse events have not demonstrated significant differences compared with placebo.[13]
Pharmacokinetics
Prucalopride is rapidly absorbed (Cmax attained 2–3 hours after single 2 mg oral dose) and is extensively distributed. Metabolism is not the major route of elimination. In vitro, human liver metabolism is very slow and only minor amounts of metabolites are found. A large fraction of the active substance is excreted unchanged (about 60% of the administered dose in urine and at least 6% in feces).Renal excretion of unchanged prucalopride involves both passive filtration and active secretion. Plasma clearance averages 317 ml/min, terminal half-life is 24–30 hours,[15] and steady-state is reached within 3–4 days. On once daily treatment with 2 mg prucalopride, steady-state plasma concentrations fluctuate between trough and peak values of 2.5 and 7 ng/ml, respectively.[13]
In vitro data indicate that prucalopride has a low interaction potential, and therapeutic concentrations of prucalopride are not expected to affect the CYP-mediated metabolism of co-medicated medicinal products.[13]
Efficacy
The primary measure of efficacy in the clinical trials is three or more spontaneous complete bowel movements per week; a secondary measure is an increase of at least one complete spontaneous bowel movement per week.[7][16][17] Further measures are improvements in PAC-QOL[18] (a quality of life measure) and PAC-SYM[19] (a range of stool,abdominal, and rectal symptoms associated with chronic constipation). Infrequent bowel movements, bloating, straining, abdominal pain, and defecation urge with inability to evacuate can be severe symptoms, significantly affecting quality of life.[20][21][22][23][24]
In three large clinical trials, 12 weeks of treatment with prucalopride 2 and 4 mg/day resulted in a significantly higher proportion of patients reaching the primary efficacy endpoint of an average of ≥3 spontaneous complete bowel movements than with placebo.[7][16][17] There was also significantly improved bowel habit and associated symptoms, patient satisfaction with bowel habit and treatment, and HR-QOL in patients with severe chronic constipation, including those who did not experience adequate relief with prior therapies (>80% of the trial participants).[7][16][17] The improvement in patient satisfaction with bowel habit and treatment was maintained during treatment for up to 24 months; prucalopride therapy was generally well tolerated.[25][26]
Side effects
Prucalopride has been given orally to ~2700 patients with chronic constipation in controlled clinical trials. The most frequently reported side effects are headache andgastrointestinal symptoms (abdominal pain, nausea or diarrhea). Such reactions occur predominantly at the start of therapy and usually disappear within a few days with continued treatment.[13]
Approval
In the European Economic Area, prucalopride was originally approved for the symptomatic treatment of chronic constipation in women in whom laxatives fail to provide adequate relief.[13] Subsequently, it has been approved by the European Commission for use in adults – that is, including male patients – for the same indication.[27]
Contraindications
Prucalopride is contraindicated where there is hypersensitivity to the active substance or to any of the excipients, renal impairment requiring dialysis, intestinal perforation orobstruction due to structural or functional disorder of the gut wall, obstructive ileus, severe inflammatory conditions of the intestinal tract, such as Crohn’s disease, and ulcerative colitis and toxic megacolon/megarectum.[13]
CLIP
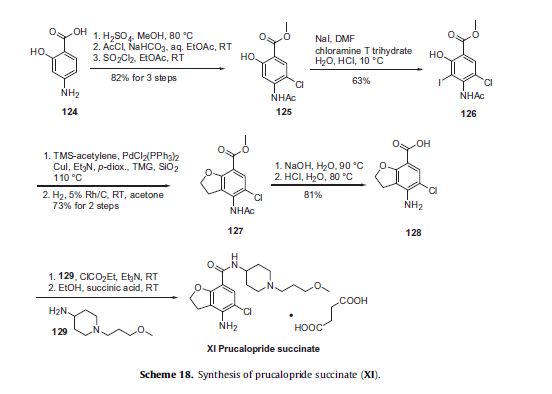
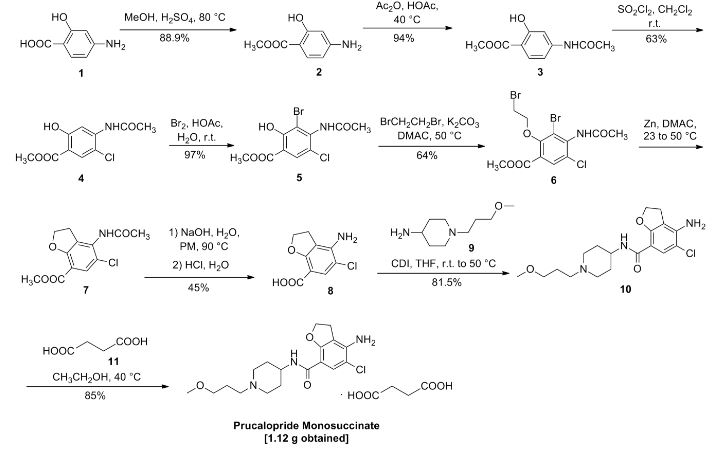
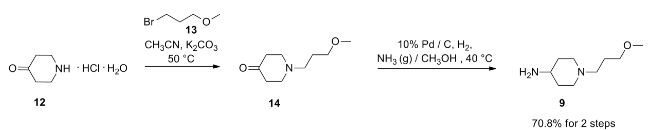
Prucalopride succinate, a first-in-class dihydrobenzofurancarboxamide, is a selective serotonin (5-HT4) receptor agonist.86–94 The drug, marketed under the brand name Resolor, possesses enterokinetic activity and was developed by the Belgian-based pharmaceutical firm Movetis. Prucalopride alters colonic motility patterns via serotonin 5-HT4 receptor stimulation, triggering the central propulsive force for defecation.95–97 The preparation of prucalopride succinate begins with the commercially available salicylic aniline 124 (Scheme 18). Acidic esterification, acetylation of the aniline nitrogen atom, and ambient-temperature chlorination via sulfuryl chloride (SO2Cl2) converted aminophenol 124 to acetamidoester 125 in 83% yield over the course of three steps.98–102 An unique set of conditions involving sodium tosylchloramide (chloramine T) trihydrate and sodium iodide were then employed to convert 125 to o-phenolic iodide 126, which then underwent sequential Sonogashira/cyclization reaction utilizing TMS-acetylene with tetramethylguanidine (TMG) in the presence of silica gel to furnish the benzofuran progenitor of 127.103 Hydrogenation of this intermediate benzofuranyl Sonagashira product saturated the 2,3-benzofuranyl bond while leaving the chlorine atom intact, ultimately delivering dihydrobenzofuran 127 in excellent yield for the two step sequence. Base-induced saponification and acetamide removal gave rise to acid 128. This acid was activated as the corresponding mixed anhydride and treated with commercial piperidine 129 to construct prucalopride which was stirred at room temperature for 24 h in ethanolic succinic acid to provide prucalopride succinate (XI). The yield for the formation of the salt was not provided.
86. Briejer, M. R.; Bosmans, J. P.; Van Daele, P.; Jurzak, M.; Heylen, L.; Leysen, J. E.;Prins, N. H.; Schuurkes, J. A. J. Eur. J. Pharmacol. 2001, 423, 71.
87. Briejer, M. R.; Prins, N. H.; Schuurkes, J. A. J. Neurogastroenterol. Motil. 2001, 13,465.
88. Coggrave, M.; Wiesel, P. H.; Norton, C. Cochrane Database Syst. Rev. 2006.CD002115.
89. Coremans, G.; Kerstens, R.; De Pauw, M.; Stevens, M. Digestion 2003, 67, 82.
90. De Winter, B. Y.; Boeckxstaens, G. E.; De Man, J. G.; Moreels, T. G.; Schuurkes, J.A. J.; Peeters, T. L.; Herman, A. G.; Pelckmans, P. A. Gut 1999, 45, 713.
91. Emmanuel, A. V.; Roy, A. J.; Nicholls, T. J.; Kamm, M. A. Aliment. Pharmacol.Ther. 2002, 16, 1347.
92. Frampton, J. E. Drugs 2009, 69, 2463.
93. Krogh, K.; Bach Jensen, M.; Gandrup, P.; Laurberg, S.; Nilsson, J.; Kerstens, R.;De Pauw, M. Scand. J. Gastroenterol. 2002, 37, 431.
94. Pau, D.; Workman, A. J.; Kane, K. A.; Rankin, A. C. J. Pharmacol. Exp. Ther. 2005,313, 146.
95. De Maeyer, J. H.; Schuurkes, J. A. J.; Lefebvre, R. A. Br. J. Pharmacol. 2009, 156,362.
96. Irving, H. R.; Tochon-Danguy, N.; Chinkwo, K. A.; Li, J. G.; Grabbe, C.; Shapiro,M.; Pouton, C. W.; Coupar, I. M. Pharmacology 2010, 85, 224.
97. Ray, A. M.; Kelsell, R. E.; Houp, J. A.; Kelly, F. M.; Medhurst, A. D.; Cox, H. M.;Calver, A. R. Eur. J. Pharmacol. 2009, 604, 1.
98. Baba, Y.; Usui, T.; Iwata, N. EP 640602 A1, 1995.
99. Fancelli, D.; Caccia, C.; Severino, D.; Vaghi, F.; Varasi, M. WO 9633186 A1,1996.
100. Hirokawa, Y.; Fujiwara, I.; Suzuki, K.; Harada, H.; Yoshikawa, T.; Yoshida, N.;Kato, S. J. Med. Chem. 2003, 46, 702.
101. Kakigami, T.; Usui, T.; Tsukamoto, K.; Kataoka, T. Chem. Pharm. Bull. 1998, 46,42.
102. Van Daele, G. H. P.; Bosmans, J.-P. R. M. A.; Schuurkes, J. A. J. WO 9616060 A1,1996.
103. Candiani, I.; DeBernadinis, S.; Cabri, W.; Marchi, M.; Bedeschi, A.; Penco, S.Synlett 1993, 269.
PAPER
Synlett 1993, 269
https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-1993-22663
PAPER
Chem. Pharm. Bull. 1998, 46,42.
https://www.jstage.jst.go.jp/article/cpb1958/46/1/46_1_42/_article
https://www.jstage.jst.go.jp/article/cpb1958/46/1/46_1_42/_pdf
PATENT
US5948794
http://www.google.co.in/patents/US5948794
EXAMPLE 1
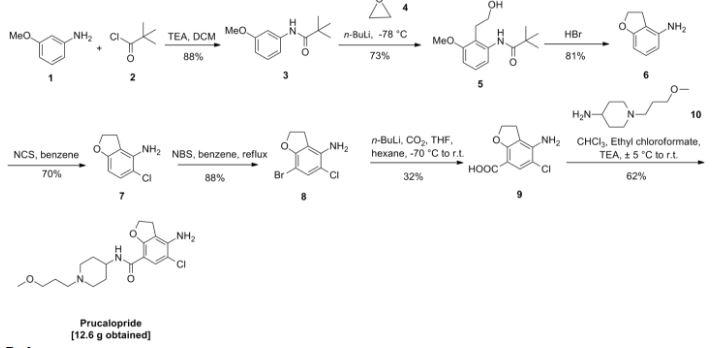
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ±5° C. N,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10° C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10° C. The resulting mixture was added dropwise over a 20-min period to a solution of 1-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCl3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml)+a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50° C.), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-N- 1-(3-methoxypropyl)-4-piperidinyl!-7-benzofurancarboxamide monohydrochloride (comp. 1).
US5854260
http://www.google.co.in/patents/US5854260
EXPERIMENTAL PART EXAMPLE 1
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ±5° C. N,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10° C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10° C. The resulting mixture was added dropwise over a 20-min period to a solution of 1-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCl3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml)+ a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50° C.), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-N- 1-(3-methoxypropyl)-4-piperidinyl!-7-benzofurancarboxamide monohydrochloride (comp. 1).
PATENT
WO199616060A1
http://www.google.co.in/patents/WO1996016060A1?cl=en
EP-0,389,037-A, published on September 26, 1990, N-(3-hydroxy-4-piperidin- yl) (dihydrobenzofuran or dihydro-2H-benzopyran)carboxamide derivatives are disclosed as having gastrointestinal motility stimulating properties. In our EP-0,445,862-A, published on September 11, 1991, N-(4-piperidinyl) (dihydrobenzo¬ furan or dihydro-2H-benzopyran)carboxamide derivatives are disclosed also having gastrointestinal motility stimulating properties.
The compound subject to the present application differs therefrom by showing superior enterokinetic properties.
The present invention concerns a compound of formula

and the pharmaceutically acceptable acid addition salts thereof.
The chemical name of the compound of formula (I) is 4-amino-5-chloro-2,3-dihydro-N- [l-(3-methoxypropyl)-4-piperidinyl]-7-benzofurancarboxamide.

Example 1
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ± 5 °C. H,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10 °C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10°C. The resulting mixture was added dropwise over a 20-min period to a solution of l-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCI3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml) + a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50 °C), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-M-[ 1 -(3-methoxypropyl)-4-piperidinyl]-7- benzofurancarboxamide monohydrochloride (comp. 1).
Example 2
A mixture of 4-amino-5-chloro-2,3-dihydro-N-(4-piperidinyl)-7-benzofuran- carboxamide(O.Olmol), l-chloro-3-methoxypropane (0.012mol), M,M-diethyl- ethanamine (2Jml) and KI (catalytic amount) in N,M-dimethylformamide (75ml) was stirred overnight at 50°C. The reaction mixture was cooled. The solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent: CHCl3/(CH3OH/NH3) 97/3). The pure fractions were collected and the solvent was evaporated. The residue was dissolved in 2-propanol and converted into the hydrochloric acid salt (1:1) with HCl/2-propanol. The precipitate was filtered off and dried (vacuum; 80°C), yielding 1.40g (35%) of 4-amino-5-chloro-2,3-dihydro-N-[l-(3-methoxypropyl)- 4-piperidinyl]-7-benzofurancarboxamide monohydrochloride (comp. 1).
PAPER
Chinese Journal of Pharmaceuticals 2012, 43, 5-8.
CLIP
Chinese Patent CN 103012337 A report is as follows:

PAPER
Pharmaceutical & Clinical Research 2011, 19, 306-307.
CLIP
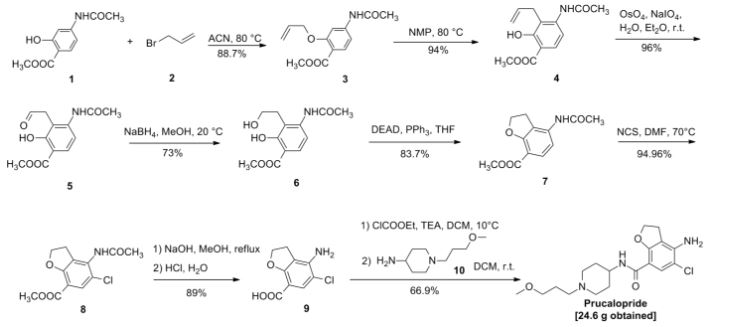
US5374637 (CN1045781, EP389037) and J. Het Chem, 1980,17 (6): 1333-5 reported synthetic route, as follows:

CLIP
Chinese Patent CN 104016949 A synthetic route reported as follows:

PATENT
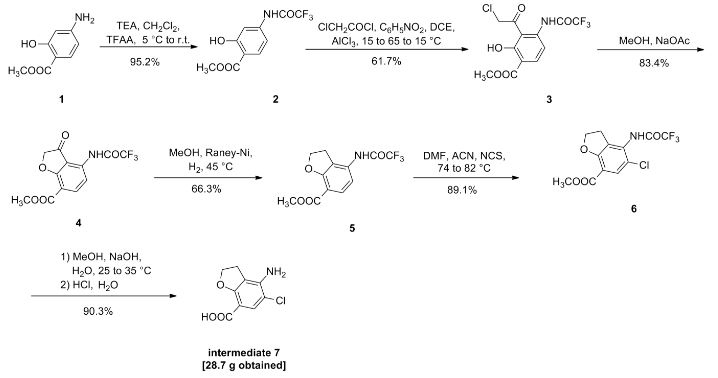
CN104529960A
https://www.google.com/patents/CN104529960A?cl=zh

 .
.

Example 1
1. Preparation of Compound II
Compound I (167. lg, Imol), triethylamine (111. lg, I. Imol) and methylene chloride (KMOg) added to the reaction flask, nitrogen cooled to 5 ° C, was slowly added dropwise trifluoroacetic anhydride (220. 5g, 1.05mol) / methylene chloride (150g) solution, maintaining the temperature throughout 5~15 ° C, dropping was completed, the reaction after 3 hours at room temperature, TLC (DCM = MeOH = 25: 1) The reaction was monitored to complete the reaction; the reaction mixture was slowly poured into ice water (560g) and stirred for 20 minutes, standing layer, the aqueous phase was separated, the organic phase was washed with saturated aqueous sodium bicarbonate (IOOg) wash sash; IM hydrochloric acid (IlOg) wash sash, then with saturated brine (200g) washed sash, magnesium sulfate (40g) dried, filtered and concentrated to give compound II (250. Ig), yield: 952%.
[0066] 2. Preparation of Compound III
[0067] Chloroacetyl chloride (101. 7g, 0. 9mol), nitrobenzene (20g) and dichloroethane (580 g) added to the reaction flask, nitrogen cooled to 5 ° C, was slowly added anhydrous trichloro aluminum powder (359. 2g, 2. 7mol), to keep the whole temperature 5~20 ° C, plus complete, insulation 15~25 ° C for 30 minutes to obtain a mixture A.
[0068] Compound II (. 236. 7g, 0 9mol) and dichloroethane (500g) added to the reaction flask, nitrogen cooled to 15 ° C; the mixture was added Compound II A quick solution, plus complete, rapid heating 65~75 ° C, 1 hours later once every 15 minutes in the control, monitoring TLC (DCM = MeOH = 50: 1) to complete the reaction; the reaction mixture was immediately poured into ice water (800g) and stirred for 30 minutes, controlling the temperature between 15~25 ° C, the organic phase was separated, the organic phase washed with water (180g) was washed with saturated brine (240g), dried over magnesium sulfate (45g) was dried, filtered and concentrated to give crude compound III (303 . 2g).
[0069] Take the crude compound III (291. 3g) / ethanol 1 dichloromethane: 1 solution (1500ml) was dissolved, and then adding activated carbon (14. 5g) was refluxed for one hour, cooled to room temperature filtered and the filtrate concentrated at room temperature to 600~ 650g, stop and concentrated down to 5~10 ° C, filtered to give a yellow solid (204. 7g); the resulting yellow solid (207. 6g) in tetrahydrofuran (510g) was purified, reduced to 10~15 ° C, filtered, The filter cake was washed with tetrahydrofuran (90g) dip, dried under vacuum to give compound III (181. 3g), yield: 61.7% billion
[0070] 3. Preparation of Compound IV
[0071] Compound 111 (! 169.68,0.5 11〇1), methanol (5,801,111) and sodium acetate (123.38,1.5111〇1) was added to the reaction flask. After 6 hours of reaction, began TLC (DCM: MeOH = 30: 1 ) the reaction was monitored to completion of the reaction; the reaction mixture was cooled to room temperature, concentrated, and the residue with ethyl acetate (500g) and water (200g) was dissolved, the organic phase was separated, the organic phase was washed with 2M sodium hydrogen carbonate (120g) was washed, then with saturated brine (IOOg), dried over magnesium sulfate (50g) was dried, filtered and concentrated to 250~280g, cooled to room temperature with stirring was added cyclohexane (200 g of), after stirring for 1 hour and then filtered and dried to obtain compound IV (126. 7g), yield: 83.4% billion
[0072] 4. Preparation of Compound V
[0073] Compound IV (12L 2g, 0. 4mol), methanol (380g) and Raney-Ni (12. 5g) added to the autoclave, purged with nitrogen, hydrogen is introduced (3. Ompa), the reaction was heated to 45 ° C after 8 hours, TLC (DCM = MeOH = 30: 1) to monitor the reaction, to complete the reaction, cooled to room temperature and pressure, and then purged with nitrogen, the reaction solution was filtered and concentrated to give crude compound V (103. 7g), taking compound V crude product (103g) was refluxed with ethyl acetate (420g) (1 hour) was purified, cooled to room temperature and stirred for 30 minutes and filtered to give a yellow solid was dried in vacuo to give compound V (76 8g.), yield: 663 %.
[0074] 5. Preparation of Compound VI
[0075] Compound ¥ (57.88,0.2111〇1), 1 ^ dimethylformamide (4.58) and acetonitrile (30 (^) was added to the reaction flask and heated 74~76 ° C; solution of N- chlorosuccinimide imide (. 26. 7g, 0 2mol) and acetonitrile (45g) was added dropwise over 30 minutes and maintaining the temperature finished 76~82 ° C, dropping was completed, the reaction was kept, after one hour the reaction started TLC (DCM: MeOH = 30: 1) to monitor the reaction, the reaction is complete the reaction solution cooled to 5~8 ° C, the filter cake was washed with water (210g) washed stirred, filtered, and dried in vacuo to give compound VI (57. 6g), yield. rate of 89.1%.
6. Preparation of Compound VII
Compound VI (48. 5g, 0. 15mol) and methanol (80g) added to the reaction flask, stirring at room temperature was added dropwise 4M aqueous sodium hydroxide (HOg), dropwise complete, for the reaction, 25 ° C~35 after 4 hours of reaction ° C, samples of about 7:00 adjust PH TLC (DCM = MeOH = 30: 1) to monitor the reaction, until the reaction was complete, down to 5~10 ° C, with 6M hydrochloric acid solution PH ~ 7. 5, half the solution was concentrated, then 2M hydrochloric acid solution PH ~ 7, reduced to 15~20 ° C was stirred for 30 minutes, filtered, the filter cake with methyl tert-butyl ether (70g) beating, filtration, and dried in vacuo to give compound VII (28. 7g), yield: 903%.
PAPER
Chem Pharm Bull 46 (1), 42-52 (1998) and Pharmaceutical and clinical study based on 2011 (4) 306-307 reported synthetic route is as follows:

Biological Activity
| Description | Prucalopride is a selective, high affinity 5-HT4 receptor agonist, inhibiting human 5-HT(4a) and 5-HT(4b) receptor with Ki value of 2.5 nM and 8 nM, respectively. | |||||
|---|---|---|---|---|---|---|
| Targets | 5-HT4A [1] | 5-HT4B [1] | ||||
| IC50 | 2.5 nM(Ki) | 8 nM(Ki) | ||||
| In vitro | Prucalopride induces contractions in a concentration-dependent manner with pEC50 of 7.5. Prucalopride (1 mM) significantly amplifies the rebound contraction of the guinea-pig proximal colon after electrical field stimulation. Prucalopride induces relaxation of the rat oesophagus preparation of rat oesophagus tunica muscularis mucosae with pEC50 of 7.8, yielding a monophasic concentration–response curve. [1] Prucalopride (0.1 μM) concentration-dependently increases the amplitude of submaximal cholinergic contractions and of acetylcholine release induced by electrical field stimulation in pig gastric circular muscle, and the effect is induced and enhanced IBMX (10 μM). [2] Prucalopride (1 μM) significantly enhances the electrically induced cholinergic contractions in pig descending colon, and the facilitating effect is significantly enhanced by Rolipram. [3] | |||||
| In vivo | Prucalopride alters colonic contractile motility patterns in a dose-dependent fashion by stimulating high-amplitude clustered contractions in the proximal colon and by inhibiting contractile activity in the distal colon of fasted dogs. Prucalopride also causes a dose-dependent decrease in the time to the first giant migrating contraction (GMC); at higher doses of prucalopride, the first GMC generally occurres within the first half-hour after treatment. [4] | |||||
| Features | ||||||
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
| Rat dose (mg/kg) = mouse dose (22.4 mg/kg) × | mouse Km(3) | = 11.2 mg/kg |
| rat Km(6) |
1
References
[1] Briejer MR, et al. Eur J Pharmacol, 2001, 423(1), 71-83.
[2] Priem E, et al. Neuropharmacology, 2012, 62(5-6), 2126-2135.
Clinical Trial Information( data from http://clinicaltrials.gov, updated on 2016-07-23)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT02806206 | Not yet recruiting | Gastrointestinal Hemorrhage|Crohn Disease|Celiac Disease|Intestinal Diseases|Inflammatory Bowel Diseases | University of British Columbia | July 2016 | Phase 4 |
| NCT02781493 | Not yet recruiting | Prucalopride Plus Polyethylene Glycol in Bowel Preparation for Colonoscopyp | Shandong University|Binzhou Peoples Hospital|Taian People …more | June 2016 | Phase 4 |
| NCT02538367 | Recruiting | Functional Constipation | Yuhan Corporation | August 2015 | Phase 1|Phase 2 |
| NCT02228616 | Recruiting | Constipation | Xian-Janssen Pharmaceutical Ltd. | October 2014 | Phase 4 |
| NCT02425774 | Recruiting | Postoperative Ileus | Katholieke Universiteit Leuven|Universitaire Ziekenhuizen …more | July 2014 | Phase 4 |
References
- Briejer, M. R.; Bosmans, J. P.; Van Daele, P.; Jurzak, M.; Heylen, L.; Leysen, J. E.; Prins, N. H.; Schuurkes, J. A. (2001). “The in vitro pharmacological profile of prucalopride, a novel enterokinetic compound”. European Journal of Pharmacology 423 (1): 71–83.doi:10.1016/S0014-2999(01)01087-1. PMID 11438309.
- Clinical trial number [1] for “NCT00793247” at ClinicalTrials.gov
- Emmanuel, A. V.; Kamm, M. A.; Roy, A. J.; Kerstens, R.; Vandeplassche, L. (2012).“Randomised clinical trial: The efficacy of prucalopride in patients with chronic intestinal pseudo-obstruction – a double-blind, placebo-controlled, cross-over, multiple n = 1 study”.Alimentary Pharmacology & Therapeutics 35 (1): 48–55. doi:10.1111/j.1365-2036.2011.04907.x. PMC 3298655. PMID 22061077.
- Smart, C. J.; Ramesh, A. N. (2011). “The successful treatment of acute refractory pseudo-obstruction with Prucalopride”. Colorectal Disease: no. doi:10.1111/j.1463-1318.2011.02929.x.
- Jump up^ Bouras, E. P.; Camilleri, M.; Burton, D. D.; McKinzie, S. (1999). “Selective stimulation of colonic transit by the benzofuran 5HT4 agonist, prucalopride, in healthy humans”. Gut44 (5): 682–686. doi:10.1136/gut.44.5.682. PMC 1727485. PMID 10205205.
- Jump up^ Bouras, E. P.; Camilleri, M.; Burton, D. D.; Thomforde, G.; McKinzie, S.; Zinsmeister, A. R. (2001). “Prucalopride accelerates gastrointestinal and colonic transit in patients with constipation without a rectal evacuation disorder”. Gastroenterology 120 (2): 354–360.doi:10.1053/gast.2001.21166. PMID 11159875.
- ^ Jump up to:a b c d Tack, J.; Van Outryve, M.; Beyens, G.; Kerstens, R.; Vandeplassche, L. (2008). “Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives”. Gut 58 (3): 357–365. doi:10.1136/gut.2008.162404.PMID 18987031.
- European Medicines Agency -EPAR
- Health Canada, Notice of Decision for Resotran
- Digestive Remedies in Israel
- Briejer, M. R.; Prins, N. H.; Schuurkes, J. A. (2001). “Effects of the enterokinetic prucalopride (R093877) on colonic motility in fasted dogs”. Neurogastroenterology and motility : the official journal of the European Gastrointestinal Motility Society 13 (5): 465–472. doi:10.1046/j.1365-2982.2001.00280.x. PMID 11696108.
- Oustamanolakis, P.; Tack, J. (2012). “Prucalopride for chronic intestinal pseudo-obstruction”. Alimentary Pharmacology & Therapeutics 35 (3): 398–9. doi:10.1111/j.1365-2036.2011.04947.x. PMID 22221087.
- SmPC. Summary of product characteristics Resolor (prucalopride) October, 2009: 1-9.
- De Maeyer, JH; Lefebvre, RA; Schuurkes, JA (Feb 2008). “5-HT(4) receptor agonists: similar but not the same”. Neurogastroenterol Motil 20 (2): 99–112. doi:10.1111/j.1365-2982.2007.01059.x. PMID 18199093.
- Frampton, J. E. (2009). “Prucalopride”. Drugs 69 (17): 2463–2476.doi:10.2165/11204000-000000000-00000. PMID 19911858.
- Camilleri, M.; Kerstens, R.; Rykx, A.; Vandeplassche, L. (2008). “A Placebo-Controlled Trial of Prucalopride for Severe Chronic Constipation”. New England Journal of Medicine 358 (22): 2344–2354. doi:10.1056/NEJMoa0800670. PMID 18509121.
- ^ Jump up to:a b c Quigley, E. M. M.; Vandeplassche, L.; Kerstens, R.; Ausma, J. (2009). “Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation – a 12-week, randomized, double-blind, placebo-controlled study”.Alimentary Pharmacology & Therapeutics 29 (3): 315–328. doi:10.1111/j.1365-2036.2008.03884.x. PMID 19035970.
- Marquis, P.; De La Loge, C.; Dubois, D.; McDermott, A.; Chassany, O. (2005). “Development and validation of the Patient Assessment of Constipation Quality of Life questionnaire”. Scandinavian Journal of Gastroenterology 40 (5): 540–551.doi:10.1080/00365520510012208. PMID 16036506.
- Frank, L.; Kleinman, L.; Farup, C.; Taylor, L.; Miner Jr, P. (1999). “Psychometric validation of a constipation symptom assessment questionnaire”. Scandinavian journal of gastroenterology 34 (9): 870–877. doi:10.1080/003655299750025327.PMID 10522604.
- Johanson, JF; Kralstein, J (2007). “Chronic constipation: a survey of the patient perspective.”. Alimentary pharmacology & therapeutics 25 (5): 599–608. doi:10.1111/j.1365-2036.2006.03238.x. PMID 17305761.
- Koch, A.; Voderholzer, W. A.; Klauser, A. G.; Müller-Lissner, S. (1997). “Symptoms in chronic constipation”. Diseases of the colon and rectum 40 (8): 902–906.doi:10.1007/BF02051196. PMID 9269805.
- McCrea, G. L.; Miaskowski, C.; Stotts, N. A.; MacEra, L.; Paul, S. M.; Varma, M. G. (2009). “Gender differences in self-reported constipation characteristics, symptoms, and bowel and dietary habits among patients attending a specialty clinic for constipation”.Gender Medicine 6 (1): 259–271. doi:10.1016/j.genm.2009.04.007. PMID 19467522.
- Pare, P.; Ferrazzi, S.; Thompson, W. G.; Irvine, E. J.; Rance, L. (2001). “An epidemiological survey of constipation in Canada: definitions, rates, demographics, and predictors of health care seeking”. The American Journal of Gastroenterology 96 (11): 3130–3137. doi:10.1111/j.1572-0241.2001.05259.x. PMID 11721760.
- Wald, A.; Scarpignato, C.; Kamm, M. A.; Mueller-Lissner, S.; Helfrich, I.; Schuijt, C.; Bubeck, J.; Limoni, C.; Petrini, O. (2007). “The burden of constipation on quality of life: results of a multinational survey”. Alimentary Pharmacology & Therapeutics 26 (2): 227–236. doi:10.1111/j.1365-2036.2007.03376.x. PMID 17593068.
- Camilleri, M; Beyens, G; Kerstens, R; Vandeplassche, L (2009). “Long-term follow-up of safety and satisfaction with bowel function in response to oral prucalopride in patients with chronic constipation [Abstract]”. Gastroenterology 136 (Suppl 1): 160. doi:10.1016/s0016-5085(09)60143-8.
- Van Outryve, MJ; Beyens, G; Kerstens, R; Vandeplassche, L (2008). “Long-term follow-up study of oral prucalopride (Resolor) administered to patients with chronic constipation [Abstract T1400]”. Gastroenterology 134 (4 (suppl 1)): A547. doi:10.1016/s0016-5085(08)62554-8.
- https://www.shire.com/newsroom/2015/june/resolor-eu-male-indication-press-release
External links
| EP0389037A1 * | 13 Mar 1990 | 26 Sep 1990 | Janssen Pharmaceutica N.V. | N-(3-hydroxy-4-piperidinyl)(dihydrobenzofuran, dihydro-2H-benzopyran or dihydrobenzodioxin)carboxamide derivatives |
| EP0445862A2 * | 22 Feb 1991 | 11 Sep 1991 | Janssen Pharmaceutica N.V. | N-(4-piperidinyl)(dihydrobenzofuran or dihydro-2H-benzopyran)carboxamide derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999058527A2 * | 13 May 1999 | 18 Nov 1999 | EGIS Gyógyszergyár Rt. | Benzofuran derivatives, pharmaceutical composition containing the same, and a process for the preparation of the active ingredient |
| WO1999058527A3 * | 13 May 1999 | 27 Jan 2000 | Bela Agai | Benzofuran derivatives, pharmaceutical composition containing the same, and a process for the preparation of the active ingredient |
| WO2000030640A1 * | 16 Nov 1999 | 2 Jun 2000 | Janssen Pharmaceutica N.V. | Use of prucalopride for the manufacture of a medicament for the treatment of dyspepsia |
| WO2000066170A1 * | 20 Apr 2000 | 9 Nov 2000 | Janssen Pharmaceutica N.V. | Prucalopride oral solution |
| WO2003059906A1 * | 13 Jan 2003 | 24 Jul 2003 | Janssen Pharmaceutica N.V. | Prucalopride-n-oxide |
| WO2012116976A1 | 28 Feb 2012 | 7 Sep 2012 | Shire – Movetis Nv | Prucalopride oral solution |
| WO2013024164A1 | 17 Aug 2012 | 21 Feb 2013 | Shire Ag | Combinations of a 5-ht4 receptor agonist and a pde4 inhibitor for use in therapy |
| US6413988 | 20 Apr 2000 | 2 Jul 2002 | Janssen Pharmaceutica N.V. | Prucalopride oral solution |
| US8063069 | 30 Oct 2007 | 22 Nov 2011 | Janssen Pharmaceutica N.V. | Prucalopride-N-oxide |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016082123 | 2016-03-24 | Hydrogel-Linked Prodrugs Releasing Tagged Drugs |
| US2015202317 | 2015-07-23 | DIPEPTIDE-BASED PRODRUG LINKERS FOR ALIPHATIC AMINE-CONTAINING DRUGS |
| US2014323402 | 2014-10-30 | Protein Carrier-Linked Prodrugs |
| US2014296257 | 2014-10-02 | High-Loading Water-Soluable Carrier-Linked Prodrugs |
| US2014243254 | 2014-08-28 | Polymeric Hyperbranched Carrier-Linked Prodrugs |
| US2013053301 | 2013-02-28 | DIPEPTIDE-BASED PRODRUG LINKERS FOR ALIPHATIC AMINE-CONTAINING DRUGS |
| US2012220630 | 2012-08-30 | PRUCALOPRIDE ORAL SOLUTION |
| US2012156259 | 2012-06-21 | Biodegradable Polyethylene Glycol Based Water-Insoluble Hydrogels |
| US6413988 | 2002-07-02 | Prucalopride oral solution |
| US6310077 | 2001-10-30 | Enterokinetic benzamide |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-Amino-5-chloro-N-[1-(3-methoxypropyl)piperidin-4-yl]-2,3-dihydro-1-benzofuran-7-carboxamide
|
|
| Clinical data | |
| Trade names | Resolor, Resotran |
| AHFS/Drugs.com | International Drug Names |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 179474-81-8 |
| ATC code | A06AX05 (WHO) |
| PubChem | CID 3052762 |
| IUPHAR/BPS | 243 |
| ChemSpider | 2314539 |
| UNII | 0A09IUW5TP |
| Chemical data | |
| Formula | C18H26ClN3O3 |
| Molar mass | 367.870 g/mol |
//////////Prucalopride succinate, Resolor, R-093877, R-108512, Resolor®, Resolor, Resotran, UNII:0A09IUW5TP, 179474-81-8 , R-093877, R-108512, Shire , Johnson & Johnson, 179474-85-2, UNII-4V2G75E1CK, SHIRE, 2010, LAUNCHED, JANNSEN , PHASE 3, IRRITABLE BOWL SYNDROME
COCCCN1CCC(CC1)NC(=O)C2=CC(=C(C3=C2OCC3)N)Cl

