
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
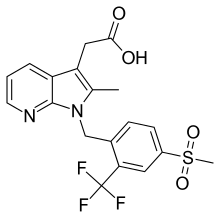
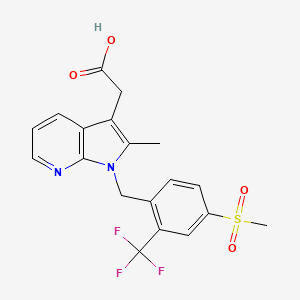
FEVIPIPRANT
| Molecular Formula: | C19H17F3N2O4S |
|---|---|
| Molecular Weight: | 426.41 g/mol |
UNII-2PEX5N7DQ4; 2PEX5N7DQ4; NVP-QAW039; QAW039;
CAS 872365-14-5
Product patent WO2005123731A2, NOVARTIS
| Inventors | Kamlesh Bala, Catherine Leblanc, David Andrew Sandham, Katharine Louise Turner, Simon James Watson, Lyndon Nigel Brown, Brian Cox, |
| Applicant | Novartis Ag, Novartis Pharma Gmbh |
Jun 17, 2004 priority expiry 2014
Synthesis 
![]()
2-[2-methyl-1-[[4-methylsulfonyl-2-(trifluoromethyl)phenyl]methyl]pyrrolo[2,3-b]pyridin-3-yl]acetic acid
- 2-Methyl-1-[[4-(methylsulfonyl)-2-(trifluoromethyl)phenyl]methyl]-1H-pyrrolo[2,3-b]pyridine-3-acetic acid
- [1-(4-((Methane)sulfonyl)-2-trifluoromethylbenzyl)-2-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl]acetic acid
Fevipiprant (INN; code name QAW039) is a drug being developed by Novartis which acts as a selective, orally available antagonistof the prostaglandin D2 receptor 2 (DP2 or CRTh2).[1][2][3]
As of 2016, it is in Phase III[4] clinical trials for the treatment of asthma.[5]
Novartis is developing fevipiprant, a prostaglandin D2 receptor (PD2/CRTh2) antagonist, as an oral capsule formulation for treating asthma and moderate to severe atopic dermatitis.

| Inventors | Kamlesh Bala, Catherine Leblanc, David Andrew Sandham, Katharine Louise Turner, Simon James Watson, Lyndon Nigel Brown, Brian Cox |
| Applicant | Novartis Ag, Novartis Pharma Gmbh |
PATENT
WO2005123731
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2005123731
PATENT


The invention discloses a Fevipiprant and Fevipiprant intermediate preparation method. The method is characterized in that 2-amino-3-bromopyridine and 4-mesyl-2-trifluoromethylbenzaldehyde to condensation reaction to obtain a Schiff base intermediate, then performing reduction reaction to obtain 3-bormo-N-(4-(mesyl)-2-(trifluoromethyl)phenyl)-pyridine-2-amine, subjecting the 3-bormo-N-(4-(mesyl)-2-(trifluoromethyl)phenyl)-pyridine-2-amine to ullmann ring closing reaction with methyl levulinate or ethyl levulinate, and performing saponification reaction or decarboxylic reaction to obtain Fevipiprant namely N[1-(4-((methane)sulfonyl)-2-trifluoromethylphenyl-2-methyl-1H-pyrrolo[2,3-b]pyridine-3-yl] acetic acid. The Fevipiprant and Fevipiprant intermediate preparation method which is a brand new method is short in step, technically convenient in operation, easy in product purification and large-scale production, high yield can be achieved, and Fevipiprant industrial production can be realized easily.
Example 5: Ν [1- (4 – ((methane) sulfonyl) -2-trifluoromethyl-phenyl) -2-methyl -1H- pyrrolo [2,3-b] P ratio preparation of 3-yl] acetic acid (1).
[0056] 3-Bromo -N- (4- (methylsulfonyl) -2- (trifluoromethyl) phenyl) – pyridin-2-amine (40 · 9g, 0 · lmo 1) and levulinic acid A ester (13.0g, 0. lmo 1) was added 300 mL N, N- dimethylformamide, was added copper iodide (1 · 9g, 0 · 0lmo 1) and N, N- dimethylglycine (1.0g , 0.01 mol), after nitrogen substitution, the reaction temperature was raised to 120 degrees 12h, water was added 200mL of saturated sodium chloride solution was cooled and extracted with ethyl acetate, the organic phase was washed with water, dried and concentrated to give a pale yellow powder, was added 100mL ethanol and 100mL water , was added sodium hydroxide (8g, 0.2mol) the reaction temperature was raised to 60 degrees 2h, cooled to 0 ° C, acidified with 4N hydrochloric acid was added dropwise to pH 2, was filtered and the solid washed with ethanol to give the crude product after recrystallization from ethanol in pure 34.5g, yield 81%.
[0057] · ΜΚ (300ΜΗζ, (16-0Μ50) δ: 12 · 3 (ΐ3Γ, 1Η, α) 2Η), 8.24 (s, lH, PhH), 8.11 ~ 8.12 (d, lH, PhH), 8.00 ~ 8.02 (d, lH, PyH), 7.91 ~ 7.93 (d, lH, PyH), 7.09 ~ 7.10 (d, lH, PhH), 6.46 ~ 6.48 (d, lH, PhH), 5.73 (s, 2H, NCH2) , 3.70 (s, 2H, q ^ C〇2H), 3.30 (s, 3H, CH 3).
[0058] HPLC: 99.9%.
[0059] Example 6: N [l- (4 – ((methane) sulfonyl) -2-trifluoromethyl-phenyl) -2-methyl -1H- pyrrolo [2,3-b] P ratio preparation of 3-yl] acetic acid (1).
[0060] 3-Bromo -N- (4- (methylsulfonyl) -2- (trifluoromethyl) phenyl) – (. 40.9g, 0 lmo 1) pyridin-2-amine and acetyl malonate methyl ester (18.8g, 0. lmol) was added 300 mL N, N- dimethylformamide, was added copper iodide (1.9g, O.Olmol) and N, N- dimethylglycine (1. (^ , 0.01111〇1), after nitrogen substitution, the reaction temperature was raised to 120 degrees 1211, 200mL saturated brine was added after cooling, and extracted with ethyl acetate, the organic phase was washed with water, dried and concentrated to give a pale yellow powder, was added 100mL ethanol and 100mL of water, was added sodium hydroxide (8g, 0.2mol) the reaction temperature was raised to 60 degrees 2h, cooled to 0 ° C, acidified with 4N hydrochloric acid was added dropwise to pH 2, was filtered and the solid washed with ethanol, a crude product was obtained from ethanol crystallized to give pure 34. lg, 80% yield.
[0061] HPLC: 99.8%.
[0062] Example 7: Ν [1- (4 – ((methane) sulfonyl) -2-trifluoromethyl-phenyl) -2-methyl -1H- pyrrolo [2,3-b] P ratio preparation of 3-yl] acetic acid (1).
[0063] 3-Bromo -N- (4- (methylsulfonyl) -2- (trifluoromethyl) phenyl) – pyridin-2-amine (40 · 9g, 0 · lmo 1) and levulinic acid A ester (13. (^, 0.1111〇1) was added ^ 3,001,111, 1-dimethyl formamide, was added copper iodide (3.88,0.02111〇1) and N, N- dimethylglycine (2. (^, 0.02111〇1), after nitrogen substitution, the reaction temperature was raised to 120 degrees 1211, 200mL saturated brine was added after cooling, and extracted with ethyl acetate, the organic phase was washed with water, dried and concentrated to give a pale yellow powder, was added 100mL ethanol and 100mL water , was added sodium hydroxide (8g, 0.2mol) the reaction temperature was raised to 60 degrees 2h, cooled to 0 ° C, acidified with 4N hydrochloric acid was added dropwise to pH 2, was filtered and the solid washed with ethanol to give crude product was recrystallized from ethanol to give pure 34. lg, 80% yield billion
[0064] HPLC: 99.9%.
[0065] Example 8: Ν [1- (4 – ((methane) sulfonyl) -2-trifluoromethyl-phenyl) -2-methyl -1H- pyrrolo [2,3-b] P ratio preparation of 3-yl] acetic acid (1).
[0066] 3-Bromo -N- (4- (methylsulfonyl) -2- (trifluoromethyl) phenyl) – pyridin-2-amine (40.9 8,0.1111〇1) was added 300mL N, N- two after dimethylformamide, was added copper iodide (1.9g, 0.01mol) and 2,2,6,6-tetramethyl-heptane-3,5-dione (3.6g, 0.02mo 1), purged with nitrogen , the reaction temperature was raised to 120 degrees 12h, water was added 200mL of saturated sodium chloride solution was cooled and extracted with ethyl acetate, the organic phase was washed with water, dried and concentrated to give a pale yellow powder, was added 100mL ethanol and 100mL water was added sodium hydroxide (8g , 0.2 mol) the reaction temperature was raised to 60 degrees 2h, cooled to 0 ° C, acidified with 4N hydrochloric acid was added dropwise to pH 2, was filtered and the solid washed with ethanol to give crude product was recrystallized from ethanol to give pure product 30.2 g, yield 71%.
[0067] HPLC: 99.6%.
[0068] Example 9: Ν [1- (4 – ((methane) sulfonyl) -2-trifluoromethyl-phenyl) -2-methyl -1H- pyrrolo [2,3-b] P ratio preparation of 3-yl] acetic acid (1).
[0069] 3-Bromo -N- (4- (methylsulfonyl) -2- (trifluoromethyl) phenyl) – pyridin-2-amine (40.9 8,0.1111〇1) was added 1’1 ^ 3,001,111, 1 ‘| – dimethylformamide, was added copper iodide (1.98,0.011] 1〇1) and proline (1.28,0.011] 1〇1), after nitrogen substitution, the reaction temperature was raised to 120 degrees 12h, after cooling, 200mL saturated brine, and extracted with ethyl acetate, the organic phase was washed with water, dried and concentrated to give a pale yellow powder, was added 100mL ethanol and 100mL water was added sodium hydroxide (8g, 0.2mol) was heated to 60 degrees reaction 2h, cooled to 0 ° C, acidified with 4N hydrochloric acid was added dropwise to pH 2, was filtered and the solid washed with ethanol to give crude product was recrystallized from ethanol to give pure product 33.2 g, 78% yield.
[0070] HPLC: 99.8%.
[0071] Example 10: N [1- (4 – ((methane) sulfonyl) -2-trifluoromethyl-phenyl) -2-methyl -1H- pyrrolo [2,3-b] pyridin – preparation of 3- yl] acetic acid (1).
[0072] 3-Bromo -N- (4- (methylsulfonyl) -2- (trifluoromethyl) phenyl) – pyridin-2-amine (40.9 8,0.1111〇1) was added 300mL N, N- two after dimethylformamide, was added copper iodide (1.9g, 0. Olmol) and N, N- dimethylglycine (1.0g, 0.01 mo 1), after nitrogen substitution, the reaction temperature was raised to 120 degrees 12h, cooled was added 200mL saturated brine, and extracted with ethyl acetate, the organic phase was washed with water, dried and concentrated to give a pale yellow powder, was added 100mL of acetic acid and 100mL of concentrated hydrochloric acid was heated to reflux for 6h, cooled to 0 ° C, was added 100mL water analysis crystal was filtered and the solid washed with ethanol to give crude product was recrystallized from ethanol to give pure product 33.2 g, 78% yield.
[0073] HPLC: 99.1%.
PATENT
Example 3b: Preparation of Compound A

Production of C8: Compound C6, (3-[2-({[4-Methanesulfonyl-2-(trifluoromethyl)-phenyl]methyl}amino)pyridin-3-yl]prop-2-yn-l-ol) (20 g, 52 mmol) was dissolved in a mixture of methyl isobutyl ketone (MIBK, 125 g), 25.3 g (156 mmol) of 1 , 1 , 1 -triethoxy ethane, and acetic acid (0.625 g, 10 mmol). The mixture was heated within 40 minutes to 140 °C under a N2 over-pressure of 1 – 4 bar. During the reaction ethanol was formed and removed from the vessel by a pressure-regulated valve. After 3.5 h a second portion of acetic acid (0.625g) was added and the mixture was heated for 3.5 h at 140 °C under a N2 over-pressure of 1 – 4 bar. The resultant product was a solution of Ethyl 2-(l- {[4-methanesulfonyl-2-(trifluoromethyl)phenyl]methyl}-2-methyl-lH-pyrrolo[2,3-¾]pyridin-3-yl)acetate and the conversion rate was measured at 98% and the yield 90%. The solution was filtered and 40 g MIBK was added. The solution was heated to IT=80 °C and cooled down within 3 h to
IT=20 °C. At an IT of 65 °C seed crystals were added. At IT 20 °C intermediate C8 was isolated and washed with 40 g MIBK and dried in the oven at IT=60°C/20mbar.
Conversion to Compound A: The intermediate C8 was concentrated under vacuum at
100 °C/200 mbar and water (6000ml). A sodium hydroxide solution (1734 g, 30%, 13 mol) was added to the mixture and heated for 4 h at 50 °C. The solution was distilled again at 100 °C/100 mbar. The phases were separated at 50 °C and the water phase was extracted with methyl isobutyl ketone (2000 ml). Again the phases were separated and the water phase was filtered at 50 °C. To the filtrate methyl isobutyl ketone (5000 ml) was added and the aqueous solution neutralized in 2 portions with hydrochloric acid (963 g, 37%, 9.8 mol) to pH 4 – 4.5. The phases were heated to 80 °C and the organic phases separated. Water (1000 ml) was added to wash the organic phase and after phase separation the organic phase was cooled down to 70 °C. Seed crystals of Compound A were added along with heptane (1000 ml). The resulting suspension was stirred for 30 minutes before cooling further down to 0 °C within 3 h. The suspension was stirred for 3 h at 0 °C and then filtered through a nutsche. The filter cake was washed first with pre-cooled HPTF/methyl isobutyl ketone (1000 g, 5: 1), then with acetone/water (1000 g, 1 :2) and finally with water (1000 g). Wet Compound A was dried in the oven at 60 °C for 8 h under vacuum to isolate 804 g of compound A. The conversion was calculated to be 99%; the yield was 79%.
Example 3 c: Alternative Preparation of Compound A

Molecular Weight: 426 41
Exact Mass: 384.08 Molecular Weight: 453.48
5 g of (3-[2-({[4-Methanesulfonyl-2-(trifluoromethyl)-phenyl]methyl}amino)pyridin-3-yl]prop-2-yn-l-ol), methyl isobutyl ketone (MIBK, 50 ml), and 1 , 1 -dimethoxy-N,N-dimethylethanamine were put together in a 200 ml reactor and stirred for 15 h at 100 °C. The mixture was acidified by addition of hydrochloric acid (15 ml) and kept stirring for 15 h at 100 °C. Then water (25 ml) was added, and the temperature was decreased to 50 °C. Caustic soda (about 15 ml) was added to set the pH around 12. Then, after phase split and a second extraction with water (10 ml), the combined aqueous phases were diluted with methyl isobutyl ketone (25 ml) and acidified at 80 °C to pH 4 with hydrochloric acid. The mixture was cooled to 70 °C, seeded and cooled to 0 °C within 2 h. After 2 h aging at 0 °C, the crystalline solid was collected by filtration, washed with methyl isobutyl ketone (10 ml) and water (10 ml), and dried under vacuum at 60 °C until constant weight. Yield 2.93 g.
PATENT
WO-2017210261
Novel deuterated analogs of pyrrolo[2,3-b]pyridine compounds, particularly fevipiprant and their salts and compositions and combination comprising them are claimed. Also claims is their use for treating asthma, allergic rhinitis and atopic dermatitis. Compounds are claimed to be a prostaglandin D2 receptor 2 antagonist. Represents first PCT filing from CoNCERT Pharmaceuticals and the inventor on this API.
PAPER
ACS Medicinal Chemistry Letters (2017), 8(5), 582-586
Discovery of Fevipiprant (NVP-QAW039), a Potent and Selective DP2Receptor Antagonist for Treatment of Asthma
 , Lucy Barker, Lyndon Brown, Zarin Brown, David Budd, Steven J. Charlton, Devnandan Chatterjee, Brian Cox, Gerald Dubois, Nicholas Duggan, Edward Hall, Julia Hatto, Janet Maas, Jodie Manini, Rachael Profit, Darren Riddy, Catherine Ritchie, Bindi Sohal, Duncan Shaw, Rowan Stringer, David A. Sykes, Matthew Thomas, Katharine L. Turner, Simon J. Watson, Ryan West, Elisabeth Willard, Gareth Williams, and Jennifer Willis
, Lucy Barker, Lyndon Brown, Zarin Brown, David Budd, Steven J. Charlton, Devnandan Chatterjee, Brian Cox, Gerald Dubois, Nicholas Duggan, Edward Hall, Julia Hatto, Janet Maas, Jodie Manini, Rachael Profit, Darren Riddy, Catherine Ritchie, Bindi Sohal, Duncan Shaw, Rowan Stringer, David A. Sykes, Matthew Thomas, Katharine L. Turner, Simon J. Watson, Ryan West, Elisabeth Willard, Gareth Williams, and Jennifer WillisAbstract

Further optimization of an initial DP2 receptor antagonist clinical candidate NVP-QAV680 led to the discovery of a follow-up molecule 2-(2-methyl-1-(4-(methylsulfonyl)-2-(trifluoromethyl)benzyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)acetic acid (compound 11, NVP-QAW039, fevipiprant), which exhibits improved potency on human eosinophils and Th2 cells, together with a longer receptor residence time, and is currently in clinical trials for severe asthma.
RM sodium methanesulfinate and 4-fluoro-2-(trifluoromethyl)benzaldehyde
Step 1:
4-(methylsulfonyl)-2-(trifluoromethyl)benzaldehyde
A suspension of sodium methanesulfinate (29.6 g, 290 mmol) and 4-fluoro-2-(trifluoromethyl)benzaldehyde (50 g, 260 mmol) in DMSO (200 ml) was heated at 90˚C overnight. The thick yellow suspension was poured onto crushed ice (ca 800 g), diluted with water and the solid reside collected by filtration, washed with water and dried in vacuo to afford 4- (methylsulfonyl)-2-(trifluoromethyl)benzaldehyde as an off-white solid (50.7 g, 77%). LRMS mass ion not detected. 1H NMR (CDCl3) 3.14 (3H s), 8.30 (1H d J=7.5), 8.36 (1H d J=7.5), 8.40 (1H s), 10.49 (1H s).
Step 2:
(4-(methylsulfonyl)-2-(trifluoromethyl)phenyl)methanol
(4-(methylsulfonyl)-2-(trifluoromethyl)phenyl)methanol as an off-white solid (50.7 g, 99%). LRMS mass ion not detected. 1H NMR (CDCl3) 3.11 (3H s), 5.02 (2H s), 8.09 (1H d J=7.5), 8.19 (1H d J=7.5), 8.25 (1H s).
STEP 3
1-(bromomethyl)-4-(methylsulfonyl)-2-(trifluoromethyl)benzene
1-(bromomethyl)-4-(methylsulfonyl)-2- (trifluoromethyl)benzene (47.1 g, 74%) as a white solid. LRMS mass ion not detected. 1H NMR (CDCl3) 3.11 (3H s), 4.67 (2H s), 7.86 (1H d J=7.5), 8.14 (1H d J=7.5), 8.25 (1H s).
STEP 4
methyl 2-(2-methyl-1-(4-(methylsulfonyl)-2-(trifluoromethyl)benzyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)acetate
(83:17) of methyl 2-(2-methyl-1-(4-(methylsulfonyl)-2- (trifluoromethyl)benzyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)acetate (N-1 product) and methyl 2-(2-methyl-7-(4- (methylsulfonyl)-2-(trifluoromethyl)benzyl)-7H-pyrrolo[2,3-b]pyridin-3-yl)acetate (N-7 product) as a white solid (22.5 g 42%). LRMS C20H19F3N2O4S requires M+ 440.4 found [MH]+ m/z 441. 1H NMR (CDCl3) 2.27 (3H s), 3.06 (3H s N-1 product), 3.11 (3H s N-7 product), 3.72 (3H s), 3.77 (2H s), 5.03 (2H s N-7 product), 5.82 (2H s N-1 product), 6.66 (1H d J=8.2), 7.16 (1H dd J=7.8, 4.8), 7.91 (1H d, J=8.3), 7.95 (1H d J=7.7), 8.12 (1H d J=7.8 N-7), 8.19 (1H d J=8.1 N-7), 8.17 (1H s N-7), 8.27 (1H d J=3.6), 8.30 (1H s).
FINAL
2-(2-methyl-1-(4-(methylsulfonyl)-2-(trifluoromethyl)benzyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)acetic acid 11 as needles, m.p. 208˚C (16.3 g, 44%). HRMS C19H18F3N2O4S requires [MH]+ 427.0939 found [MH]+ 427.093. 1H NMR (500 MHz, DMSO-d6) 2.28 (3H s), 3.28 (3H s), 3.73 (2H s), 5.76 (2H s), 6.49 (1H d J=8.3), 7.12 (1H dd J=7.7, 4.8), 7.95 (1H d J=7.8), 8.04 (1H d, J=8.3), 8.14 (1H d J=4.7), 8.26 (1H s), 12.28 (1H br s ). Elemental analysis calcd. for C19H17F3N2O4S: C, 53.52; H, 4.02; N, 6.57; S, 7.52%. Found C, 53.90 ± 0.04; H, 4.28 ± 0.06; N, 6.43 ± 0.02; S, 7.76 ± 0.09%.
PAPER
Drug Metabolism & Disposition (2017), 45(7), 817-825
References
- ^ Jump up to:a b Erpenbeck VJ, Vets E, Gheyle L, Osuntokun W, Larbig M, Neelakantham S, et al. (2016). “Pharmacokinetics, Safety, and Tolerability of Fevipiprant (QAW039), a Novel CRTh2 Receptor Antagonist: Results From 2 Randomized, Phase 1, Placebo-Controlled Studies in Healthy Volunteers”. Clin Pharmacol Drug Dev. 5 (4): 306–13. doi:10.1002/cpdd.244. PMC 5071756
 . PMID 27310331.
. PMID 27310331. - Jump up^ Sykes DA, Bradley ME, Riddy DM, Willard E, Reilly J, Miah A, Bauer C, Watson SJ, Sandham DA, Dubois G, Charlton SJ. Fevipiprant (QAW039), a Slowly Dissociating CRTh2 Antagonist with the Potential for Improved Clinical Efficacy. Mol Pharmacol. 2016 May;89(5):593-605. doi: 10.1124/mol.115.101832 PMID 26916831
- Jump up^ Erpenbeck VJ, Popov TA, Miller D, Weinstein SF, Spector S, Magnusson B, et al. (2016). “The oral CRTh2 antagonist QAW039 (fevipiprant): A phase II study in uncontrolled allergic asthma”. Pulm Pharmacol Ther. 39: 54–63. doi:10.1016/j.pupt.2016.06.005. PMID 27354118.
- Jump up^ https://clinicaltrials.gov/ct2/show/NCT02555683
- Jump up^ Gonem S, Berair R, Singapuri A, Hartley R, Laurencin M, Bacher G, et al. (2016). “Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial”. Lancet Respir Med. 4: 699–707. doi:10.1016/S2213-2600(16)30179-5
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | Unaffected by food[1] |
| Metabolism | Hepatic glucuronidation |
| Biological half-life | ~20 hours |
| Excretion | Renal (≤30%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C19H17F3N2O4S |
| Molar mass | 426.41 g/mol |
| 3D model (JSmol) | |
////////////////FEVIPIPRANT, QAW039, PHASE 3, asthma, UNII-2PEX5N7DQ4,2PEX5N7DQ4, NVP-QAW039, QAW039, 872365-14-5,
CC1=C(C2=C(N1CC3=C(C=C(C=C3)S(=O)(=O)C)C(F)(F)F)N=CC=C2)CC(=O)O

