
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
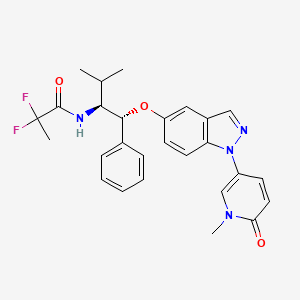
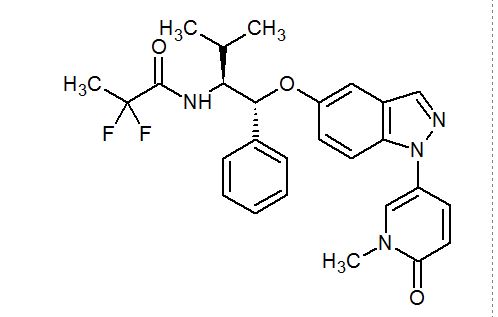
AZD 9567
CAS 1893415-00-3
1893415-64-9 as MONOHYDRATE
2,2-Difluoro-N-[(1R,2S)-3-methyl-1-[[1-(1-methyl-6-oxo-1,6-dihydropyridin-3-yl)-1H-indazol-5-yl]oxy]-1-phenylbutan-2-yl]propanamide
Propanamide, N-[(1S)-1-[(R)-[[1-(1,6-dihydro-1-methyl-6-oxo-3-pyridinyl)-1H-indazol-5-yl]oxy]phenylmethyl]-2-methylpropyl]-2,2-difluoro-
2,2-difluoro-N-[(1R,2S)-3-methyl-1-[1-(1-methyl-6-oxopyridin-3-yl)indazol-5-yl]oxy-1-phenylbutan-2-yl]propanamide
2,2-difluoro- V-[(lR,25)-3-methyl-l-{[l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)-lH-indazol-5-yl]oxy}-l-phenylbutan-2-yl]propanamide
MF C27 H28 F2 N4 O3, MF 494.533
AstraZeneca INNOVATOR
AZD-9567, a glucocorticoid receptor modulator, is in early clinical development at AstraZeneca in healthy male volunteers.
Phase I Rheumatoid arthritis
- Originator AstraZeneca
- Class Antirheumatics
- Mechanism of Action Glucocorticoid receptor modulators
-
- 01 Sep 2016 AstraZeneca completes a phase I trial (In volunteers) in Germany (NCT02512575)
- 24 May 2016 Phase-I clinical trials in Rheumatoid arthritis (In volunteers) in United Kingdom (PO) (NCT02760316)
- 24 May 2016 AstraZeneca initiates a phase I trial in Rheumatoid arthritis (In volunteers) in Germany (PO) (NCT02760316)
| Inventors | Lena Elisabeth RIPA, Karolina Lawitz, Matti Juhani Lepistö, Martin Hemmerling, Karl Edman, Antonio Llinas |
| Applicant | Astrazeneca |

Macclesfield AstraZeneca site in Cheshire

Glucocorticoids (GCs) have been used for decades to treat acute and chronic inflammatory and immune conditions, including rheumatoid arthritis, asthma, chronic obstructive pulmonary disease (“COPD”), osteoarthritis, rheumatic fever, allergic rhinitis, systemic lupus erythematosus, Crohn’s disease, inflammatory bowel disease, and ulcerative colitis. Examples of GCs include dexamethasone, prednisone, and
prednisolone. Unfortunately, GCs are often associated with severe and sometimes irreversible side effects, such as osteoporosis, hyperglycemia, effects on glucose metabolism (diabetes mellitus). skin thinning, hypertension, glaucoma, muscle atrophy. Cushing’s syndrome, fluid homeostasis, and psychosis (depression ). These side effects can particularly limit the use of GCs in a chronic setting. Thus, a need continues to exist for alternative therapies that possess the beneficial effects of GCs, but with a reduced likel ihood of side effects.
GCs form a complex with the GC receptor ( GR ) to regulate gene transcription. The GC-GR complex translocates to the cell nucleus, and then binds to GC response elements (GREs) in the promoter regions of various genes. The resulting GC-GR- GRE complex, in turn, activates or inhibits transcription of proximally located genes. The GC-GR complex also (or alternatively) may negatively regulate gene transcription by a process that does not involve DNA binding. In this process, termed transrepression, the GC-GR complex enters the nucleus and directly interacts (via protein-protein interaction) with other transcription factors, repressing their ability to induce gene transcription and thus protein expression.
Some of the side effects of GCs are believed to be the result of cross-reactivity with other steroid receptors (e.g., progesterone, androgen, mineralocorticoid, and estrogen receptors), which have somewhat homologous ligand binding domains; and/or the inability to selectively modulate gene expression and downstream signaling. Consequently, it is believed that an efficacious selective GR modulator (SGRM), which binds to GR with greater affinity relative to other steroid hormone receptors, would provide an alternative therapy to address the unmet need for a therapy that possesses the beneficial, effects of GCs, while, at the same time, having fewer side effects.
A range of compounds have been reported to have SGRM activity. See, e.g., WO2007/0467747, WO2007/114763, WO2008/006627, WO2008/055709, WO2008/055710, WO2008/052808, WO2008/063116, WO2008/076048,
WO2008/079073, WO2008/098798, WO2009/065503, WO2009/142569,
WO2009/142571, WO2010/009814, WO2013/001294, and EP2072509. Still, there continues to be a need for new SGRMs that exhibit, for example, an improved potency, efficacy, effectiveness in steroid-insensitive patients, selectivity, solubility allowing for oral administration, pharmacokinetic profile allowing for a desirable dosing regimen, stability on the shelf {e.g., hydro lytic, thermal, chemical, or photochemical stability), crystallinity, tolerability for a range of patients, side effect profile and/or safety profile.
PATENT
Scheme 1 below illustrates a general protocol for making compounds described in this specification, using either an Ullman route or an aziridine route.
Scheme 1

In Scheme 1, Ar is 
[182] The amino alcohol reagent used in Scheme 1 may be made using the below Scheme 2.
Scheme 2

The Grignard reagent (ArMgBr) used in Scheme 2 can be obtained commercially, or, if not, can generally be prepared from the corresponding aryl bromide and Mg and/or iPrMgCl using published methods.
[183] The iodo and hydroxy pyridone indazole reagents used in Scheme 1 may be made using the below Scheme 3A or 3B, respectively.
Scheme 3A



[184] Scheme 4 below provides an alternative protocol for making compounds described in this specification.
Scheme 4

Example 1. Preparation of 2,2-difluoro- V-[(lR,2S)-3-methyl-l-{[l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)-lH-indazol-5-yl]oxy}-l-phenylbutan-2-yl]propanamide.

[199] Step A. Preparation of 5-[5-[(te^butyldimethylsilyl)oxy]-lH-indazol-l-yl]-l-methyl-l,2-dihydropyridin-2-one.

Into a 2 L 4-necked, round-bottom flask, purged and maintained with an inert atmosphere of N2, was placed a solution of 5-[(tert-butyldimethylsilyl)oxy]-lH-indazole (805 g, 3.2 mol) in toluene (8 L), 5 -iodo-1 -methyl- 1 ,2-dihydropyridin-2-one (800 g, 3.4 mol) and
K3PO4 (1.2 kg, 5.8 mol). Cyclohexane-l,2-diamine (63 g, 0.5 mol) was added followed by the addition of Cul (1.3 g, 6.8 mmol) in several batches. The resulting solution was stirred overnight at 102°C. The resulting mixture was concentrated under vacuum to yield 3.0 kg of the title compound as a crude black solid. LC/MS: m/z 356 [M+H]+.
[200] Step B. Preparation of 5-(5-hydroxy-lH-indazol-l-yl)-l-methylpyridin-2(lH)-one.

Into a 2 L 4-necked, round-bottom flask was placed 5-[5-[(fert-butyldimethylsilyl)oxy]-lH-indazol-l-yl]-l-methyl-l,2-dihydropyridin-2-one (3.0 kg, crude) and a solution of HCl (2 L, 24 mol, 36%) in water (2 L) and MeOH (5 L). The resulting solution was stirred for 1 hr at 40°C and then evaporated to dryness. The resulting solid was washed with water (4 x 5 L) and ethyl acetate (2 x 0.5 L) to afford 480 g (61%, two steps) of the title product as a brown solid. LC/MS: m/z 242 [M+H]+. 1HNMR (300 MHz, DMSO-d6): δ 3.52 (3H, s),6.61 (lH,m),7.06 (2H,m),7.54 (lH,m), 7.77 (lH,m), 8.19 (2H, m) 9.35 (lH,s).
[201] Ste C. Preparation of tert-butyl((lR,25)-l-hydroxy-3-methyl-l-phenylbutan-2-yl)carbamate.

(S)-tert-butyl 3 -methyl- l-oxo-l-phenylbutan-2-ylcarbamate (1.0 kg, 3.5 mol) was dissolved in toluene (4 L). Afterward, 2-propanol (2 L) was added, followed by triisopropoxyaluminum (0.145 L, 0.73 mol). The reaction mixture was heated at 54-58°C for 1 hr under reduced pressure (300-350 mbar) to start azeothropic distillation. After the collection of 0.75 L condensate, 2-propanol (2 L) was added, and the reaction mixture was stirred overnight at reduced pressure to afford 4 L condensate in total. Toluene (3 L) was added at 20°C, followed by 2M HC1 (2 L) over 15 min to keep the temperature below 28°C. The layers were separated (pH of aqueous phase 0-1) and the organic layer was washed successively with water (3 L), 4% NaHCCte (2 L) and water (250 mL). The volume of the organic layer was reduced from 6 L at 50°C and 70 mbar to 2.5 L. The resulting mixture was heated to 50°C and heptane (6.5 L) was added at 47-53°C to maintain the material in solution. The temperature of the mixture was slowly decreased to 20°C, seeded with the crystals of the title compound at 37°C (seed crystals were prepared in an earlier batch made by the same method and then evaporating the reaction mixture to dryness, slurring the residue in heptane, and isolating the crystals by filtration), and allowed to stand overnight. The product was filtered off, washed with heptane (2 x 1 L) and dried under vacuum to afford 806 g (81%) of the title compound as a white solid. 1HNMR (500 MHz, DMSO-d6): δ 0.81 (dd, 6H), 1.16 (s, 8H), 2.19 (m, 1H), 3.51 (m, 1H), 4.32 (d, 1H), 5.26 (s, 1H), 6.30 (d, 1H), 7.13 – 7.2 (m, 1H), 7.24 (t, 2H), 7.3 – 7.36 (m, 3H).
[202] Step D. Preparation of (lR,2S)-2-amino-3-methyl-l-phenylbutan-l-ol hydrochloride salt.

To a solution of HC1 in propan-2-ol (5-6 N, 3.1 L, 16 mol) at 20°C was added tert-butyl((li?,25)-l-hydroxy-3-methyl-l-phenylbutan-2-yl)carbamate (605 g, 2.2 mol) in portions over 70 min followed by the addition of MTBE (2 L) over 30 min. The reaction mixture was cooled to 5°C and stirred for 18 hr. The product was isolated by filtration and dried to afford 286 g of the title compound as an HC1 salt (61% yield). The mother liquor was concentrated to 300 mL. MTBE (300 mL) was then added, and the resulting precipitation was isolated by filtration to afford additional 84 g of the title compound as a HC1 salt (18% yield). Total 370 g (79%). 1HNMR (400 MHz, DMSO-d6): δ 0.91 (dd, 6H), 1.61 – 1.81 (m, 1H), 3.11 (s, 1H), 4.99 (s, 1H), 6.08 (d, 1H), 7.30 (t, 1H), 7.40 (dt, 4H), 7.97 (s, 2H).
[203] Step E. Preparation of (2S,35)-2-isopropyl-l-(4-nitrophenylsulfonyl)-3-phenylaziridine.

(li?,25)-2-Amino-3-methyl-l-phenylbutan-l-ol hydrochloride (430 g, 2.0 mol) was mixed with DCM (5 L) at 20°C. 4-Nitrobenzenesulfonyl chloride (460 g, 2.0 mol) was then added over 5 min. Afterward, the mixture was cooled to -27°C. Triethylamine (1.0 kg, 10 mol) was slowly added while maintaining the temperature at -18°C. The reaction mixture was cooled to -30°C, and methanesulfonyl chloride (460 g, 4.0 mol) was added slowly while maintaining the temperature at -25 °C. The reaction mixture was then stirred at 0°C for 16 hr before adding triethylamine (40 mL, 0.3 mol; 20 mL ,0.14 mol and 10 mL, 0.074 mol) w at 0°C in portions over 4 hr. Water (5 L) was subsequently added at 20°C, and the resulting layers were separated. The organic layer was washed with water (5 L) and the volume reduced to 1 L under vacuum. MTBE (1.5 L) was added, and the mixture was stirred on a rotavap at 20°C over night and filtered to afford 500 g (70%) of the title product as a solid. 1HNMR (400 MHz, CDCls): δ 1.12 (d, 3H), 1.25 (d, 3H), 2.23 (ddt, 1H), 2.89 (dd, 1H), 3.84 (d, 1H), 7.08 – 7.2 (m, 1H), 7.22 – 7.35 (m, 4H), 8.01 – 8.13 (m, 2H), 8.22 – 8.35 (m, 2H)
[204] Step F. Preparation of V-((lR,2S)-3-methyl-l-(l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)-lH-indazol-5-yloxy)-l-phenylbutan-2-yl)-4-nitrobenzenesulfonamide.

[205] (25′,35)-2-Isopropyl-l-(4-nitrophenylsulfonyl)-3-phenylaziridine (490 g, 1.3 mol) was mixed with 5-(5-hydroxy-lH-indazol-l-yl)-l-methylpyridin-2(lH)-one (360 g, 1.4 mol) in acetonitrile (5 L) at 20°C. Cesium carbonate (850 g, 2.6 mol) was added in portions over 5 min. The reaction mixture was then stirred at 50°C overnight. Water (5 L) was added at 20°C, and the resulting mixture was extracted with 2-methyltetrahydrofuran (5L and 2.5 L). The combined organic layer was washed successively with 0.5 M HC1 (5 L), water (3 x 5L) and brine (5L). The remaining organic layer was concentrated to a thick oil, and then MTBE (2 L) was added. The resulting precipitate was filtered to afford 780 g (purity 71% w/w) of the crude title product as a yellow solid, which was used in the next step without further purification. 1HNMR (400 MHz, DMSO-d6): δ 0.93 (dd, 6H), 2.01 -2.19 (m, 1H), 3.50 (s, 3H), 3.74 (s, 1H), 5.00 (d, 1H), 6.54 (d, 1H), 6.78 (d, 1H), 6.95 -7.15 (m, 4H), 7.23 (d, 2H), 7.49 (d, 1H), 7.69 (dd, 1H), 7.74 (d, 2H), 8.00 (s, 1H), 8.08 (d, 2H), 8.13 (d, 2H).
[206] Step G. Preparation of 2,2-difluoro- V-[(lR,25)-3-methyl-l-{[l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)-lH-indazol-5-yl]oxy}-l-phenylbutan-2-yl]propanamide.

[207] N-((lR,2S)-3-Methyl- 1 -(1 -(1 -methyl-6-oxo- 1 ,6-dihydropyridin-3-yl)- \H-indazol-5-yloxy)-l-phenylbutan-2-yl)-4-nitrobenzenesulfonamide (780 g, 71%w/w) was mixed with DMF (4 L). DBU (860 g, 5.6 mol) was then added at 20°C over 10 min. 2-Mercaptoacetic acid (170 g, 1.9 mol) was added slowly over 30 min, keeping the temperature at 20°C. After 1 hr, ethyl 2,2-difluoropropanoate (635 g, 4.60 mol) was added over 10 min at 20°C. The reaction mixture was stirred for 18 hr. Subsequently, additional ethyl 2,2-difluoropropanoate (254 g, 1.8 mol) was added, and the reaction mixture was stirred for an additional 4 hr at 20°C. Water (5 L) was then slowly added over 40 min, maintaining the temperature at 20°C. The water layer was extracted with isopropyl acetate (4 L and 2 x 2 L). The combined organic layer was washed with 0.5M HC1 (4 L) and brine (2 L). The organic layer was then combined with the organic layer from a parallel reaction starting from 96 g of N-((li?,25)-3-methyl-l-((l-(l-methyl-6-oxo-l,6-dihydropyridin-3-yl)- lH-indazol-5-yl)oxy)- 1 -phenylbutan-2-yl)-4-nitrobenzenesulfonamide, and concentrated to approximate 1.5 L. The resulting brown solution was filtered. The filter was washed twice with isopropyl acetate (2 x 0.5 L). The filtrate was evaporated until a solid formed. The solid was then co evaporated with 99.5% ethanol (1 L), affording 493 g (77%, two steps) of an amorphous solid.
[208] The solid (464 g, 0.94 mol) was dissolved in ethanol/water 2: 1 (3.7 L) at 50°C. The reaction mixture was then seeded with crystals () of the title compound (0.5 g) at 47°C, and a slight opaque mixture was formed. The mixture was held at that temperature for 1 hr. Afterward, the temperature was decreased to 20°C over 7 hr, and kept at 20°C for 40 hr. The solid was filtrated off, washed with cold (5°C) ethanol/water 1 :2 (0.8 L), and dried in vacuum at 37°C overnight to afford 356 g (0.70 mol, 74%, 99.9 % ee) of the title compound as a monohydrate. LC/MS: m/z 495 [M+H]+. ‘HNMR (600 MHz, DMSO-d6) δ 0.91 (dd, 6H), 1.38 (t, 3H), 2.42 (m, 1H), 3.50 (s, 3H), 4.21 (m, 1H), 5.29 (d, 1H), 6.53 (d, 1H), 7.09 (d, 1H), 7.13 (dd, 1H), 7.22 (t, 1H), 7.29 (t, 2H), 7.47 (d, 2H), 7.56 (d, 1H), 7.70 (dd, 1H), 8.13 (d, 1H), 8.16 (d, 1H), 8.27 (d, 1H).
[209] The seed crystals may be prepared from amorphous compound prepared according to Example 2 using 2,2-difluoropropanoic acid, followed by purification on HPLC. The compound (401 mg) was weighed into a glass vial. Ethanol (0.4 mL) was added, and the vial was shaken and heated to 40°C to afford a clear, slightly yellow solution. Ethanol/Water (0.4 mL, 50/50% vol/vol) was added. Crystallization started to
occur within 5 min, and, after 10 min, a white thick suspension formed. The crystals were collected by filtration
/////////////AZD 9567, AstraZeneca, lucocorticoid receptor modulator, Rheumatoid arthritis, phase 1, Lena Elisabeth RIPA, Karolina Lawitz, Matti Juhani Lepistö, Martin Hemmerling, Karl Edman, Antonio Llinas
3rd speaker this afternoon in 1st time disclosures is Lena Ripa of @AstraZeneca on a glucocorticoid receptor modulator #ACSSanFran
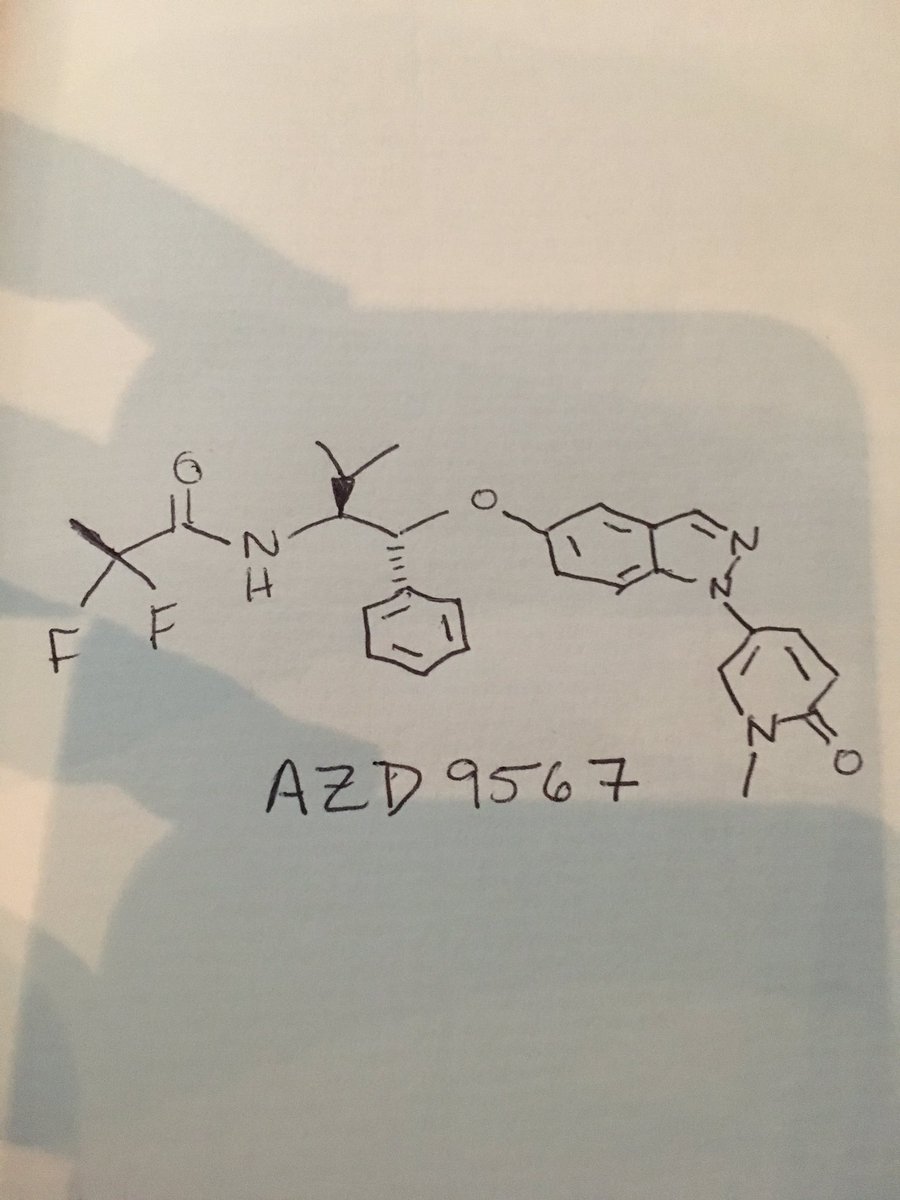
CC(F)(F)C(=O)N[C@@H](C(C)C)[C@H](Oc1cc2cnn(c2cc1)C=3C=CC(=O)N(C)C=3)c4ccccc4

