
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

CCT 245737
CAS:1489389-18-5
M.Wt: 379.34
Formula: C16H16F3N7O
2-Pyrazinecarbonitrile, 5-[[4-[[(2R)-2-morpholinylmethyl]amino]-5-(trifluoromethyl)-2-pyridinyl]amino]-
(R)-5-(4-(Morpholin-2-ylmethylamino)-5-(trifluoromethyl)pyridin-2-ylamino)pyrazine-2-carbonitrile
(+)-5-[[4-[[(2R)-Morpholin-2-ylmethyl]amino]-5-(trifluoromethyl)pyridin-2-yl]amino]pyrazine-2-carbonitrile
Cancer Research Technology Limited INNOVATOR
SAREUM
IND Filed, Sareum FOR CANCER
![]()

Synthesis, Exclusive by worlddrugtracker
5-[[4-[[morpholin-2-yl]methylamino]-5- (trifluoromethyl)-2-pyridyl]amino]pyrazine-2-carbonitrile compounds (referred to herein as “TFM compounds”) which, inter alia, inhibit Checkpoint Kinase 1 (CHK1) kinase function. The present invention also pertains to pharmaceutical compositions comprising such compounds, and the use of such compounds and compositions, both in vitro and in vivo, to inhibit CHK1 kinase function, and in the treatment of diseases and conditions that are mediated by CHK1 , that are ameliorated by the inhibition of CHK1 kinase function, etc., including proliferative conditions such as cancer, etc., optionally in combination with another agent, for example, (a) a DNA topoisomerase I or II inhibitor; (b) a DNA damaging agent; (c) an antimetabolite or a thymidylate synthase (TS) inhibitor; (d) a microtubule targeted agent; (e) ionising radiation; (f) an inhibitor of a mitosis regulator or a mitotic checkpoint regulator; (g) an inhibitor of a DNA damage signal transducer; or (h) an inhibitor of a DNA damage repair enzyme.
Checkpoint Kinase 1 (CHK1)
Progression through the cell division cycle is a tightly regulated process and is monitored at several positions known as cell cycle checkpoints (see, e.g., Weinert and Hartwell,
1989; Bartek and Lukas, 2003). These checkpoints are found in all four stages of the cell cycle; G1 , S (DNA replication), G2 and M (Mitosis) and they ensure that key events which control the fidelity of DNA replication and cell division are completed correctly. Cell cycle checkpoints are activated by a number of stimuli, including DNA damage and DNA errors caused by defective replication. When this occurs, the cell cycle will arrest, allowing time for either DNA repair to occur or, if the damage is too severe, for activation of cellular processes leading to controlled cell death.
All cancers, by definition, have some form of aberrant cell division cycle. Frequently, the cancer cells possess one or more defective cell cycle checkpoints, or harbour defects in a particular DNA repair pathway. These cells are therefore often more dependent on the remaining cell cycle checkpoints and repair pathways, compared to non-cancerous cells (where all checkpoints and DNA repair pathways are intact). The response of cancer cells to DNA damage is frequently a critical determinant of whether they continue to proliferate or activate cell death processes and die. For example, tumour cells that contain a mutant form(s) of the tumour suppressor p53 are defective in the G1 DNA damage checkpoint. Thus inhibitors of the G2 or S-phase checkpoints are expected to further impair the ability of the tumour cell to repair damaged DNA. Many known cancer treatments cause DNA damage by either physically modifying the cell’s DNA or disrupting vital cellular processes that can affect the fidelity of DNA replication and cell division, such as DNA metabolism, DNA synthesis, DNA transcription and microtubule spindle formation. Such treatments include for example, radiotherapy, which causes DNA strand breaks, and a variety of chemotherapeutic agents including topoisomerase inhibitors, antimetabolites, DNA-alkylating agents, and platinum- containing cytotoxic drugs. A significant limitation to these genotoxic treatments is drug resistance. One of the most important mechanisms leading to this resistance is attributed to activation of cell cycle checkpoints, giving the tumour cell time to repair damaged DNA. By abrogating a particular cell cycle checkpoint, or inhibiting a particular form of DNA repair, it may therefore be possible to circumvent tumour cell resistance to the genotoxic agents and augment tumour cell death induced by DNA damage, thus increasing the therapeutic index of these cancer treatments.
CHK1 is a serine/threonine kinase involved in regulating cell cycle checkpoint signals that are activated in response to DNA damage and errors in DNA caused by defective replication (see, e.g., Bartek and Lukas, 2003). CHK1 transduces these signals through phosphorylation of substrates involved in a number of cellular activities including cell cycle arrest and DNA repair. Two key substrates of CHK1 are the Cdc25A and Cdc25C phosphatases that dephosphorylate CDK1 leading to its activation, which is a
requirement for exit from G2 into mitosis (M phase) (see, e.g., Sanchez et al., 1997). Phosphorylation of Cdc25C and the related Cdc25A by CHK1 blocks their ability to activate CDK1 , thus preventing the cell from exiting G2 into M phase. The role of CHK1 in the DNA damage-induced G2 cell cycle checkpoint has been demonstrated in a number of studies where CHK1 function has been knocked out (see, e.g., Liu et ai, 2000; Zhao et al., 2002; Zachos et al., 2003).
The reliance of the DNA damage-induced G2 checkpoint upon CHK1 provides one example of a therapeutic strategy for cancer treatment, involving targeted inhibition of CHK1. Upon DNA damage, the p53 tumour suppressor protein is stabilised and activated to give a p53-dependent G1 arrest, leading to apoptosis or DNA repair (Balaint and Vousden, 2001). Over half of all cancers are functionally defective for p53, which can make them resistant to genotoxic cancer treatments such as ionising radiation (IR) and certain forms of chemotherapy (see, e.g., Greenblatt et al., 1994; Carson and Lois, 1995). These p53 deficient cells fail to arrest at the G1 checkpoint or undergo apoptosis or DNA repair, and consequently may be more reliant on the G2 checkpoint for viability and replication fidelity. Therefore abrogation of the G2 checkpoint through inhibition of the CHK1 kinase function may selectively sensitise p53 deficient cancer cells to genotoxic cancer therapies, and this has been demonstrated (see, e.g., Wang et al., 1996; Dixon and Norbury, 2002). In addition, CHK1 has also been shown to be involved in S phase cell cycle checkpoints and DNA repair by homologous recombination. Thus, inhibition of CHK1 kinase in those cancers that are reliant on these processes after DNA damage, may provide additional therapeutic strategies for the treatment of cancers using CHK1 inhibitors (see, e.g., Sorensen et al., 2005). Furthermore, certain cancers may exhibit replicative stress due to high levels of endogenous DNA damage (see, e.g., Cavalier et al., 2009; Brooks et al., 2012) or through elevated replication driven by oncogenes, for example amplified or overexpressed MYC genes (see, e.g., Di Micco et al. 2006; Cole et al., 2011 ; Murga et al. 2011). Such cancers may exhibit elevated signalling through CHK1 kinase (see, e.g., Hoglund et al., 2011). Inhibition of CHK1 kinase in those cancers that are reliant on these processes, may provide additional therapeutic strategies for the treatment of cancers using CHK1 inhibitors (see, e.g., Cole et al., 2011 ; Davies et al., 2011 ; Ferrao et al., 2011).
Several kinase enzymes are important in the control of the cell growth and replication cycle. These enzymes may drive progression through the cell cycle, or alternatively can act as regulators at specific checkpoints that ensure the integrity of DNA replication through sensing DNA-damage and initiating repair, while halting the cell cycle. Many tumours are deficient in early phase DNA-damage checkpoints, due to mutation or deletion in the p53 pathway, and thus become dependent on the later S and G2/M checkpoints for DNA repair. This provides an opportunity to selectively target tumour cells to enhance the efficacy of ionising radiation or widely used DNA-damaging cancer chemotherapies. Inhibitors of the checkpoint kinase CHK1 are of particular interest for combination with genotoxic agents. In collaboration with Professor Michelle Garrett (University of Kent, previously at The Institute of Cancer Research) and Sareum (Cambridge) we used structure-based design to optimise the biological activities and pharmaceutical properties of hits identified through fragment-based screening against the cell cycle kinase CHK1, leading to the oral clinical candidate CCT245737. The candidate potentiates the efficacy of standard chemotherapy in models of non-small cell lung, pancreatic and colon cancer. In collaboration with colleagues at The Institute of Cancer Research (Professor Louis Chesler, Dr Simon Robinson and Professor Sue Eccles) and Newcastle University (Professor Neil Perkins), we have shown that our selective CHK1 inhibitor has efficacy as a single agent in models of tumours with high replication stress, including neuroblastoma and lymphoma.

The checkpoint kinase CHK2 has a distinct but less well characterised biological role to that of CHK1. Selective inhibitors are valuable as pharmacological tools to explore the biological consequences of CHK2 inhibition in cancer cells. In collaboration with Professor Michelle Garrett (University of Kent, previously at The Institute of Cancer Research), we have used structure-based and ligand-based approaches to discover selective inhibitors of CHK2. We showed that selective CHK2 inhibition has a very different outcome to selective CHK1 inhibition. Notably, while CHK2 inhibition did not potentiate the effect of DNA-damaging chemotherapy, it did sensitize cancer cells to the effects of PARP inhibitors that compromise DNA repair.
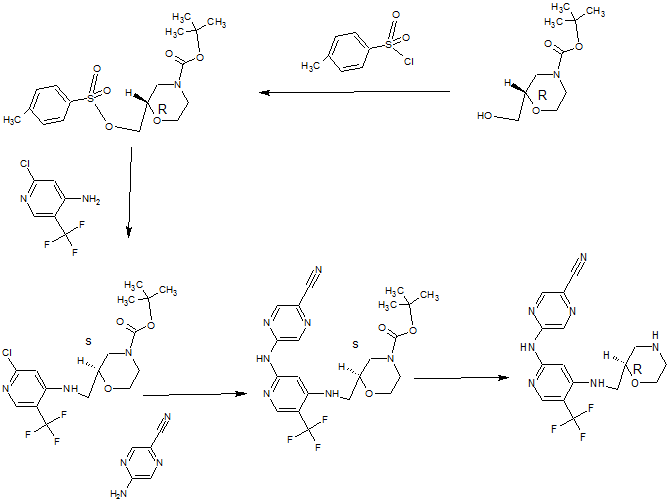
Synthesis
(R)-5-(4-(Morpholin-2-ylmethylamino)-5-(trifluoromethyl)pyridin-2-ylamino)pyrazine-2-carbonitrile
PATENT
http://www.google.com/patents/WO2013171470A1?cl=enSynthesis 1 D
5-[[4-[[(2R)-Morpholin-2-yl]methylamino]-5-(trifluoromethyl)-2-pyridyl]amino]py
carbonitrile (Compound 1)

A solution of (S)-tert-butyl 2-((2-(5-cyanopyrazin-2-ylamino)-5-(trifluoromethyl)pyridin-4- ylamino)methyl)morpholine-4-carboxylate (1.09 g, 2.273 mmol) in dichloromethane (8 mL) was added dropwise over 10 minutes to a solution of trifluoroacetic acid (52.7 mL, 709 mmol) and tnisopropylsilane (2.61 mL, 12.73 mmol) in dry dichloromethane (227 mL) at room temperature. After stirring for 30 minutes, the mixture was concentrated in vacuo. The concentrate was resuspended in dichloromethane (200 mL) and
concentrated in vacuo, then resuspended in toluene (100 mL) and concentrated.
The above procedure was performed in triplicate (starting each time with 1.09 g (S)-tert- butyl 2-((2-(5-cyanopyrazin-2-ylamino)-5-(trifluoromethyl)pyridin-4- ylamino)methyl)morpholine-4-carboxylate) and the three portions of crude product so generated were combined for purification by ion exchange chromatography on 2 x 20 g Biotage NH2 Isolute columns, eluting with methanol. The eluant was concentrated and 10% methanol in diethyl ether (25 mL) was added. The resulting solid was filtered, washed with diethyl ether (30 mL), and dried in vacuo to give the title compound as a light straw coloured powder (2.30 g, 89%). H NMR (500 MHz, CD3OD) δ 2.62 (1 H, J = 12, 10 Hz), 2.78-2.84 (2H, m), 2.95 (1 H, dd, J = 12, 2 Hz), 3.27-3.38 (2H, m), 3.63 (1 H, ddd, J = 14, 9.5, 3 Hz), 3.73-3.78 (1 H, m), 3.91 (1 H, ddd, J = 11 , 4, 2 Hz), 7.26 (1 H, s), 8.18 (1 H, s), 8.63 (1 H, s), 9.01 (1 H, s).
LC-MS (Agilent 4 min) Rt 1.22 min; m/z (ESI) 380 [M+H+]. Optical rotation [a]D 24 = +7.0 (c 1.0, DMF).
Synthesis 2B
(R)-tert- Butyl 2-((2-chloro-5-(trifluoromethyl)pyridin-4-ylamino)methyl)morpholine-

To a solution of 2-chloro-5-(trifluoromethyl)pyridin-4-amine (1 g, 5.09 mmol) in
dimethylformamide (32.6 mL) was added sodium hydride (60% by wt in oil; 0.407 g, 10.18 mmol) portionwise at room temperature followed by stirring for 10 minutes at 80°C. (S)- tert-Butyl 2-(tosyloxymethyl)morpholine-4-carboxylate (2.268 g, 6.1 1 mmol) was then added portionwise and the reaction mixture was stirred at 80°C for 2.5 hours. After cooling, the mixture was partitioned between saturated aqueous sodium
hydrogencarbonate solution (30 mL), water (100 mL) and ethyl acetate (30 mL). The organic layer was separated and the aqueous layer was further extracted with ethyl acetate (2 x 30 mL). The combined organic layers were washed with brine (2 x 70 mL), dried over magnesium sulfate, filtered, concentrated and dried thoroughly in vacuo. The crude material was purified by column chromatography on a 90 g Thomson SingleStep column, eluting with an isocratic mix of 2.5% diethyl ether / 2.5% ethyl acetate in dichloromethane, to give the title compound as a clear gum that later crystallised to give a white powder (1.47 g, 73%). H NMR (500 MHz, CDCI3) δ 1.48 (9H, s), 2.71-2.83 (1 H, m), 2.92-3.05 (1 H, m), 3.18- 3.23 (1 H, m), 3.33-3.37 (1 H, m), 3.56-3.61 (1 H, m), 3.66-3.71 (1 H, m), 3.80-4.07 (3H, m), 5.32 (1 H, broad s), 6.61 (1 H, s), 8.24 (1 H, s). LC-MS (Agilent 4 min) Rt 3.04 min; m/z (ESI) 396 [MH+]. Svnthesis 2C
(R)-tert-Butyl 2-((2-(5-cyanopyrazin-2-ylamino)-5-(trifluoromethyl)pyridin-4-

(R)-tert-Butyl 2-((2-chloro-5-(trifluoromethyl)pyridin-4-ylamino)methyl)morpholine-4- carboxylate (1.44 g, 3.64 mmol), 2-amino-5-cyanopyrazine (0.612 g, 5.09 mmol, 1.4 eq.), tris(dibenzylideneacetone)dipalladium(0) (0.267 g, 0.291 mmol, 0.08 eq.), rac-2,2′- bis(diphenylphosphino)-1 ,1 ‘-binaphthyl (0.362 g, 0.582 mmol, 0.16 eq.) and caesium carbonate (2.37 g, 7.28 mmol) were suspended in anhydrous dioxane (33 ml_) under argon. Argon was bubbled through the mixture for 30 minutes, after which the mixture was heated to 100°C for 22 hours. The reaction mixture was cooled and diluted with dichloromethane, then absorbed on to silica gel. The pre-absorbed silica gel was added to a 100 g KP-Sil SNAP column which was eluted with 20-50% ethyl acetate in hexanes to give the partially purified product as an orange gum. The crude product was dissolved in dichloromethane and purified by column chromatography on a 90 g SingleStep Thomson column, eluting with 20% ethyl acetate in dichloromethane, to give the title compound (1.19 g, 68%). H NMR (500 MHz, CDCI3) δ 1.50 (9H, s), 2.71-2.88 (1 H, m), 2.93-3.08 (1 H, m), 3.27- 3.32 (1 H, m), 3.40-3.44 (1 H, m), 3.55-3.64 (1 H, m), 3.71-3.77 (1 H, m), 3.82-4.11 (3H, m), 5.33 (1 H, broad s), 7.19 (1 H, s), 8.23 (1 H, s), 8.58 (1 H, s), 8.84 (1 H, s). LC-MS (Agilent 4 min) Rt 2.93 min;m/z (ESI) 480 [MH+].
Paper

Multiparameter optimization of a series of 5-((4-aminopyridin-2-yl)amino)pyrazine-2-carbonitriles resulted in the identification of a potent and selective oral CHK1 preclinical development candidate with in vivo efficacy as a potentiator of deoxyribonucleic acid (DNA) damaging chemotherapy and as a single agent. Cellular mechanism of action assays were used to give an integrated assessment of compound selectivity during optimization resulting in a highly CHK1 selective adenosine triphosphate (ATP) competitive inhibitor. A single substituent vector directed away from the CHK1 kinase active site was unexpectedly found to drive the selective cellular efficacy of the compounds. Both CHK1 potency and off-target human ether-a-go-go-related gene (hERG) ion channel inhibition were dependent on lipophilicity and basicity in this series. Optimization of CHK1 cellular potency and in vivo pharmacokinetic–pharmacodynamic (PK–PD) properties gave a compound with low predicted doses and exposures in humans which mitigated the residual weak in vitro hERG inhibition.
Multiparameter Lead Optimization to Give an Oral Checkpoint Kinase 1 (CHK1) Inhibitor Clinical Candidate: (R)-5-((4-((Morpholin-2-ylmethyl)amino)-5-(trifluoromethyl)pyridin-2-yl)amino)pyrazine-2-carbonitrile (CCT245737)
///////////CCT 245737, IND, PRECLINICAL, Cancer Research Technology Limited, SAREUM
N#CC(C=N1)=NC=C1NC2=NC=C(C(F)(F)F)C(NC[C@@H]3OCCNC3)=C2
- Supporting Info SEE NMR OF COMPD 4


Reblogged this on ORGANIC CHEMISTRY SELECT.
LikeLike