

Levetiracetam industrial process


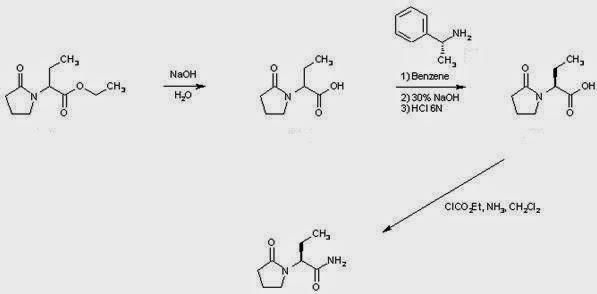
EXAMPLE 1 (a) Preparation of the (R)-alpha-methyl-benzylamine salt of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
EXAMPLE 2 (a) Preparation of ethyl (S)-4-[[1-(aminocarbonyl)propyl]amino]butyrate
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide
EXAMPLE 3 (a) Preparation of (S)-N-[1(aminocarbonyl)propyl]-4-chlorobutanamide
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide
EXAMPLE 4 Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide……levetiracetam
—
(±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide a key levetiracetam intermediate

(±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
methyl (±)-(R,S)-alpha-ethyl-2-oxo-l -pyrrolidine acetate with (+)-(R)-(l-phenylethyl)- amine in toluene in the presence of a base such as sodium hydride or methoxide; crystallization- induced dynamic resolution of the resultant (±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
(R)-(+)-1-Phenylethylamine
33978-83-5
1-Pyrrolidineacetic acid, α-ethyl-2-oxo-, methyl ester

1004767-60-5
1-Pyrrolidineacetamide, α-ethyl-2-oxo-N-[(1R)-1-phenylethyl]-
(±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
Example 1
(±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide.
In a 100 ml reactor equipped with mechanical stirring, thermometer and bubble condenser, 13.4 g of (±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (71.6 mmol), 8.8 g of (+)-(R)-(l-phenylethyl)-amine (72.5 mmol) and 45 ml of tetrahydrofuran were charged. 3.4 g of NaH (60% dispersion in mineral oil, 85.6 mmol) was added in small portions under nitrogen atmosphere. Reaction mixture was maintained at room temperature for about 2 h. Then, it was heated up to 350C and kept under stirring overnight. Reaction was controlled by TLC (Rf = 0.5, AcOEt/silica gel).
7.35-7.19 (1OH, m),
6.49 (2H, br s),
5.09-5.00 (2H, m),
4.41 (IH, dd, J = 8.3, 7.4 Hz),
4.36 (IH, dd, J = 8.6, 7.1 Hz),
3.49 (IH, ddd, J = 9.8, 7.7, 6.6 Hz),
3.41 (IH, ddd, J = 9.8, 7.7, 6.2 Hz),
3.30 (IH, ddd, J = 9.6, 8.3, 5.5 Hz),
3.13 (IH, ddd, 9.7, 8.5, 6.1 Hz), 2.47-2.38 (2H, m), 2.41 (IH, ddd, J = 17.0, 9.6, 6.3 Hz), 2.26 (IH, ddd, 17.0, 9.5, 6.6 Hz), 2.10-1.98 (2H, m), 2.01-1.89 (IH, m), 1.99-1.88 (IH, m), 1.98-1.85 (IH, m), 1.88-1.78 (IH, m), 1.75- 1.62 (IH, m), 1.72-1.59 (IH, m), 1.45 (3H, d, J = 7.1 Hz), 1.44 (3H, d, J = 7.1 Hz), 0.90 (3H, t, J = 7.4 Hz), 0.86 (3H, t, J = 7.4 Hz).


13C NMR (100.62 MHz, CDCl3, 25 0C): δ (ppm, TMS)
176.05 (CO), 176.00 (CO), 169.08 (CO),
168.81 (CO), 143.59 (Cquat),
143.02 (Cquat), 128.66 (2 x CH), 128.55 (2 x CH),
127.33 (CH), 127.19 (CH), 126.05 (2 x CH),
125.80 (2 x CH), 56.98 (CH), 56.61 (CH),
48.90 (CH), 48.84 (CH), 44.08 (CH2),
43.71 (CH2), 31.19 (CH2), 31.07 (CH2), 22.08 (CH3),
22.04 (CH3), 21.21 (CH2), 20.68 (CH2),
18.28 (CH2), 18.08 (CH2), 10.50 (CH3), 10.45 (CH3).


Example 2 (±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide (alternative 1).
At reaction completed, reaction mixture was cooled and when room temperature was reached, 100 ml of water was slowly charged. Aqueous phases were separated and extracted with toluene (2 x 75 ml). Collected organic phases were treated with acid water till neuter pH. Solvent was evaporated and residue was suspended in about 100 ml of heptane for about 30 minutes. Product was isolated by filtration and dried in oven at 400C temperature under vacuum overnight to give 45.2 g of the title compound (164.54 mmol, 83.2% yield, d.e. 0.0%) as white dusty solid.
Example 3
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

 amcrasto@gmail.com
amcrasto@gmail.com
updated
US 7902380, Levetiracetam
 US 7902380, Levetiracetamhttp://www.google.im/patents/US7902380
US 7902380, Levetiracetamhttp://www.google.im/patents/US7902380


Suitable resolving agents include optically pure bases such as alpha-methylbenzylamine and dehydroabietylamine, of which alpha-methylbenzylamine is preferred. (S)-2 can be prepared by forming the salt with (R)-alpha-methylbenzylamine and the (R)-2 can be prepared by forming the salt with (S)-alpha-methylbenzylamine.
NOTE……R)-alpha-methylbenzylamine is desired agent to get levetiracetam



EXAMPLE 1
Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide from (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
EXAMPLE 2
Preparation of (R)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide from (R)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
EXAMPLE 3
Preparation of (S)-alpha-Ethyl-2-oxo-1-pyrrolidineacetic acid (R)-alpha-methylbenzylamine salt
EXAMPLE 4
Recovery and Epimerization of (R)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid from the Mother Liquor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus 
 amcrasto@gmail.com
amcrasto@gmail.com

vietnam
.

 dalat city
dalat city
 hanoi
hanoi


