
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Pioglitazone
5-[[4-[2-(5-ethyl-2-pyridinyl)-ethoxy]phenyl] methyl]- 2,4- thiazolidinedione
5-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione


Pioglitazone (Actos) is a prescription drug of the class thiazolidinedione (TZD) with hypoglycemic (antihyperglycemic, antidiabetic) action to treat diabetes. Actos was the tenth-best selling drug in the U.S. in 2008, with sales exceeding $2.4 billion. While pioglitazone does decrease blood sugar levels, studies on the main cardiovascular outcomes has not yielded statistically significant results. Its cardiovascular safety profile compares favorably with rosiglitazone (Avandia), which was withdrawn after concerns about an increased risk of cardiac events. Pioglitazone has been found to be associated with bladder tumors and has been withdrawn in some countries.
Medical uses
Pioglitazone is used for the treatment of diabetes mellitus type 2 either alone or in combination with a sulfonylurea, metformin, orinsulin. There was, however, no statistically significant difference in the main outcomes studied.
Pioglitazone has also been used to treat non-alcoholic steatohepatitis (fatty liver), but this use is presently considered experimental.
| (RS)-5-(4-[2-(5-ethylpyridin-2-yl)ethoxy]benzyl)thiazolidine-2,4-dione | |
| Clinical data | |
|---|---|
| Trade names | Actos |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699016 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. |
|
| Legal status | |
| Routes | oral |
| Pharmacokinetic data | |
| Protein binding | >99% |
| Metabolism | liver (CYP2C8) |
| Half-life | 3–7 hours |
| Excretion | in bile |
| Identifiers | |
| CAS number | 111025-46-8 |
| ATC code | A10BG03 |
| PubChem | CID 4829 |
| IUPHAR ligand | 2694 |
| DrugBank | DB01132 |
| ChemSpider | 4663 |
| UNII | X4OV71U42S |
| KEGG | D08378 |
| ChEBI | CHEBI:8228 |
| ChEMBL | CHEMBL595 |
| Chemical data | |
| Formula | C19H20N2O3S |
| Mol. mass | 356.44 g/mol |

ACTOSView pdf
| US Patent No | Expiry Date | patent use code |
|---|---|---|
| 5965584 | Jun 19, 2016 | U-753 |
| 6150383 | Jun 19, 2016 | U-418 |
| 6150384 | Jun 19, 2016 | U-419 |
| 6166042 | Jun 19, 2016 | U-414 |
| 6166043 | Jun 19, 2016 | U-415 |
| 6172090 | Jun 19, 2016 | U-416 |
| 6211205 | Jun 19, 2016 | U-410 |
| 6271243 | Jun 19, 2016 | U-411 |
| 6303640 | Aug 9, 2016 | U-425 |
| 6329404 | Jun 19, 2016 | U-753 |
DMF
| DMF16635 | STATUSA | TYPEII | SUMISSION6/11/2003 | BIOCON LTD | PIOGLITAZONE HYDROCHLORIDE AS MANUFACTURED IN BALGALORE, INDIA. |
| 16672 | A | II | 6/30/2003 | CIPLA LTD | PIOGLITAZONE HYDROCHLORIDE USP (ESUB) AS MANUFACTURED IN PUNE, MAHARASHTRA, INDIA |
| 16675 | A | II | 7/2/2003 | DR REDDYS LABORATORIES LTD | PIOGLITAZONE HYDROCHLORIDE AS MANUFACTURED IN ANDHRA PRADESH, INDIA. |
| 16682 | A | II | 7/3/2003 | RANBAXY LABORATORIES LTD | PIOGLITAZONE HYDROCHLORIDE AS MANUFACTURED IN PUNJAB, INDIA. |
| 16684 | A | II | 7/8/2003 | USV LIMITED | PIOGLITAZONE HYDROCHLORIDE (ESUB) AS MANUFACTURED IN MUMBAI, INDIA. |
more.clickhttp://www.usactives.com/z1/search.php?zoom_query=PIOGLITAZONE
Synthesis

PATENT
http://www.google.com/patents/WO2004101560A1?cl=en
Many thiazolidinedione derivatives or “glitazones” are known to exhibit hypoglycemic activity and/or blood lipid lowering activity and have been proposed for use in treating, inter alia, diabetes. Some of the more well known and/or studied glitazones include pioglitazone, troglitazone, and rosiglitazone. Pioglitazone, chemically 5-[[4-[2-(5-ethyl-2-pyridinyl)-ethoxy]phenyl] methyl]- 2,4- thiazolidinedione of formula
(1)

is a commercially approved antidiabetic agent. Pharmaceutical compositions comprising pioglitazone, as the hydrochloride salt, are marketed under the brand name ACTOS® (Takeda Chemical Ind.) for treatment of type II diabetes.
Pioglitazone and its hydrochloride have been disclosed in EP 193256 and corresponding U.S. Patent No. 4,687,777. In these patents, the glitazone, such as pioglitazone, can be formed by cyclizing an alpha-bromo acid ester (2) with thiourea. The resulting imino-thiazolidinone (3) is then hydrolyzed to make the corresponding glitazone. For pioglitazone, the reaction can be represented as follows:

The starting alpha-bromo acid ester (2) is taught to be prepared by Meerwein arylation. This process comprises preparing the corresponding aniline (4), diazotation thereof in the presence of hydrobromic acid, and coupling of the product of diazotation with an acrylic acid ester (5) under catalysis by cuprous oxide as shown below:
reduction


(4) (5)
However, forming the alpha-bromo acid ester by the Meerwein arylation reaction can be problematic. The sequence of reactions within this transformation must be controlled precisely. Otherwise the diazo-compound generated during the reaction would react with another nucleophile such as the bromide anion leading to a complicated outcome. Therefore, the reaction often gives a complicated result and lower chemical yield. Furthermore, the preparation of the starting aniline derivative (4) comprises a hydrogenation step that requires a special apparatus, which gives some difficulties when scaling-up.
EP 0 008 203, which is related to U.S. Patent Nos. 4,287,200 and 4,481,141, discloses additional glitazones, i.e., not pioglitazone, that can be formed by several possible methods. In addition to the general scheme described in EP 193256, two more synthetic routes are proposed. One technique comprises a cyclization reaction as shown below to form the intended glitazone:

However, the formation of the starting thiocyano compound is not described. The other technique mentioned in EP 0 008 203 involves coupling the thiazolidine-containing moiety and the substituted alkyl moiety via alkylation of a phenolic oxygen to form the glitazone. If applied to pioglitazone, the reaction would be represented as follows:

(9) (10)

Example 11
Pioglitazone (from 2-thiocyanato-3- {4-[2-(5-ethyl-pyridin-2-yl)-ethoxy]- phenyl} -propionic acid). 0.50 g of 2-thiocyanato-3-{4-[2-(5-ethyl-pyridin-2-yl)-ethoxy]-phenyl}- propionic acid was dissolved in 10 mL of methanesulfonic acid, and the resulting brown solution was stirred overnight. Then, the reaction mixture was poured onto crushed ice upon stirring and sodium hydrogencarbonate was added in portions to neutralize the mixture. Precipitated light brown crystals were filtered off and dried to give 0.70 g of raw product, which was further purified by mixing with 12 % ethanolic solution of hydrogen chloride. The undissolved portion was filtered off, and the filtrate was precipitated with sodium bicarbonate, which gave crystals of pioglitazone
with a m.ρ. of 180 – 184°C.
PATENT
EP 0193256; ES 8705886; JP 1986267580; US 4687777

The condensation of 2-(5-ethyl-2-pyridyl)ethanol (I) with 4-fluoronitrobenzene (II) by means of NaH in DMF gives 4-[2-(5-ethyl-2-pyridyl)ethoxy]nitrobenzene (III), which is reduced with H2 over Pd/C in methanol to yield the corresponding aniline (IV). The reaction of (IV) with NaNO2/HBr and methyl acrylate (V) in acetone/methanol affords the 2-bromopropionate derivative (VI), which is cyclized with thiourea (VII) by means of NaOAc in refluxing ethanol to provide the 2-imino-4-thiazolidinone (VIII). Finally, this compound is hydrolyzed with refluxing 2N HCl.
http://www.drugfuture.com/synth/syndata.aspx?ID=164965
PATENT
EP 0506273

This compound has been obtained by several different methods. The reaction of 2-(5-ethyl-2-pyridyl)ethanol (I) with Ts-Cl and NaOH in THF gives the corresponding tosylate (II), which is condensed with 4-hydroxybenzaldehyde (IV) by means of BnNBu3Cl and NaOH or K2CO3 to yield the aryl ether (V) (1-3). The condensation of (V) with thiazolidine-2,4-dione (VI) in refluxing ethanol affords the 5-benzylidenethiazolidinedione (VII), which is finally reduced with H2 over Pd/C in DMF or dioxane. The reaction of 2-(5-ethyl-2-pyridyl)ethanol (I) with Ms-Cl and TEA in dichloromethane THF gives the corresponding mesylate (III), which is condensed with 4-hydroxybenzaldehyde (IV) by means of K2CO3 to yield the already reported aryl ether (V). The condensation of 2-(5-ethyl-2-pyridyl)ethanol (I) with 4-fluorobenzonitrile (VIII) by means of NaH gives 4-[2-(5-ethyl-2-pyridyl)ethoxy]benzonitrile (IX), which is reduced with RaNi and HCOOH to yield the already reported 4-ethoxybenzaldehyde derivative (V).
http://www.drugfuture.com/synth/syndata.aspx?ID=164965
………………………….
11th Symp Med Chem (Dec 4-5, Tokushima) 1990,Abst P-11

Two new related syntheses of pioglitazone hydrochloride have been described: 1) The condensation of 2-(5-ethylpyridin-2-yl)ethanol (I) with 4-hydroxybenzaldehyde (II) by means of benzyltributylammonium chloride, NaOH and tosyl chloride gives 4-[2-(5-ethylpyridin-2-yl)ethoxy]benzaldehyde (III), which is condensed with thiazolidine-2,4-dione (IV) in basic medium to afford 5-[-4-[2-(5-ethylpyridin-2-yl)ethoxy]benzylidene]thiazolidine-2,4-dione (V). Finally, this compound is hydrogenated in the usual way. 2) The condensation of alcohol (I) with 4-fluorobenzonitrile (VI) by means of NaH gives 4-[2-(5-ethylpyridin-2-yl)ethoxy]benzonitrile (VII), which is reduced with Raney Nickel and formic acid to the aldehyde (III), already obtained.
http://www.drugfuture.com/synth/syndata.aspx?ID=164965
.CLIP
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
Beilstein J. Org. Chem. 2013, 9, 2265–2319.
Pioglitazone is clearly related to rosiglitazone (1.40) with its structure only differing in the substitution pattern of the parent pyridine ring. Its synthesis begins with the hydroxymethylation of 2-methyl-5-ethylpyridine (1.11), a commodity chemical obtained from the condensation of acetaldehyde with ammonium acetate (Scheme 12) [43].
At elevated temperatures and pressures 2-methyl-5-ethylpyridine undergoes a condensation reaction with formaldehyde allowing isolation of the chain extended hydroxyethylpyridine 1.70upon distillation although in poor yield [44].
Following subsequent SNAr reaction aryl ether 1.71 is obtained, which is used as crude material in the subsequent Knoevenagel condensation with thiazolidinedione 1.68. In order to reduce the intermediate benzylidene double bond in this example sodium borohydride is used in the presence of cobalt chloride efficiently delivering pioglitazone in high purity.
![[1860-5397-9-265-i12]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i12.png)
Other syntheses of pioglitazone use related pyridine building blocks and aim to generate late stage intermediates that deliver the target compound upon de novo synthesis of the thiazolidinedione ring system. For instance phenyl ether 1.73 can be obtained via Williamson ether synthesis between mesylate 1.74 and phenol 1.75 (Scheme 13).
Removal of the acetyl protecting group under acidic conditions renders aniline 1.76 which is subsequently subjected to a Meerwein arylation reaction which occurs via diazotisation and subsequent treatment with acrylonitrile in the presence of cuprous oxide [45-48].
![[1860-5397-9-265-i13]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i13.png)
Alternatively, an equivalent acid functional material can be prepared starting from tyrosine (1.78) via a dual protection of the amino acid unit as the methyl ester and the amine as the benzaldehyde imine (Scheme 14). This is then followed by analogous ether formation with the previously generated mesylate 1.74.
Intermediate 1.80 is then hydrolysed to reveal once again the amino acid functionality, which upon diazotisation in the presence of hydrobromic acid selectively forms the α-bromo ester 1.82.
![[1860-5397-9-265-i14]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i14.png)
A more direct SNAr approach utilising 4-fluorobenzonitrile as the acceptor and the sodium alkoxide of hydroxyethylpyridine 1.84 as the nucleophile has been successfully conducted (Scheme 15) [49].
The nitrile unit is partially reduced and hydrolysed to the benzaldehyde 1.71 using Raney-Ni under transfer hydrogenation conditions in wet formic acid. A Darzens reaction between this aldehyde and ethyl chloroacetate in the presence of sodium ethoxide delivers epoxide 1.87.
The material is next subjected to hydrogenolysis using Pd/C in methanol with a 1 bar hydrogen pressure to reductively ring open the epoxide. Finally, the transformation of the alcohol to the mesylate 1.88 occurs under standard conditions.
![[1860-5397-9-265-i15]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i15.png)
In order to complete the syntheses of pioglitazone as outlined in the previous schemes several procedures for the installation of the thiazolidinedione ring system have been reported [50]. For instance the α-bromoester of intermediate 1.82 will render the desired heterocycle upon treatment with sodium isothiocyanate (Scheme 16, A).
Alternatively, the α-amino acid portion of 1.81 can be diazotised under standard conditions and will subsequently deliver the same product when treated with lithium isothiocyanate (Scheme 16, B).
Finally, α-bromonitrile 1.77 can be condensed with thiourea to give pioglitazone after aqueous work-up (Scheme 16, C).
![[1860-5397-9-265-i16]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i16.png?scale=2.0&max-width=1024&background=FFFFFF)
ref for above
43 Frank, R. L.; Pilgrim, F. J.; Riener, E. F. Org. Synth. 1950, 30. doi:10.1002/0471264180.os030.15
44 McGall, G. H.; Barone, A. D. Nucleic Acid Labeling Compounds. U.S. Patent 2001/0018514 A1, Aug 30, 2001.
45 Finch, H.; Fox, C.; Sajad, A. Respiratory Disease Treatment. W.O. Patent WO2010015818, Feb 11, 2010.
46 Adiyaman, M.; Guner, D.; Ridvanoglu, N.; Yurdakul, A. A. Process for the Production of substituted Phenyl Ethers. W.O. Patent WO2004000810, Dec 31, 2003.
47 Nersesian, D. L.; Black, L. A.; Miller, T. R.; Vortherms, T. A.; Esbenshade, T. A.; Hancock, A. A.; Cowart, M. D.Bioorg. Med. Chem. Lett. 2008, 18, 355–359. doi:10.1016/j.bmcl.2007.10.067
48 Halama, A.; Hejtmankova, L.; Lustig, P.; Richter, J.; Srsnova, L.; Jirman, J. Method for obtaining Pioglitazone as an antidiabetic agent. W.O. Patent WO02088120, Nov 7, 2002.
49 Thijs, L.; Zhu, J. Processes for making Pioglitazone and compounds of the processes. W.O. Patent WO2005080387, Sept 1, 2005.
50 Bhanja, C.; Jena, S. J. Chem. Pharm. Res. 2012, 4, 4323–4333.
http://www.google.com/patents/WO2012153312A1?cl=en
Pioglitazone of Formula I, chemically (±)-5-[[4-[2-(5-ethyl-2- pyridinyl)ethoxy]phenyl]methyl]-2,4-thiazolidinedione, is used as an adjunct to diet and exercise to improve glycemic control in patients with type 2 diabetes (non-insulin- dependent diabetes mellitus or “NIDDM”).
The commercially available form of pioglitazone is its monohydrochloride salt.

Dehydropioglitazone (hereinafter referred to as “DHP” or “DHP of Formula II”) is known as an intermediate in the synthesis of pioglitazone or salts thereof.

In addition, DHP is also present as a process impurity or degradation product in pioglitazone or salts thereof. Several reasons are attributed for the generation of DHP in pioglitazone. Some factors linked to the generation of this impurity include the incomplete reduction of DHP and the oxidation of pioglitazone during isolation or while on stability.
Several processes are known in the literature for making pioglitazone or a salt thereof, for example, U.S. Patent Nos. 4,687,777; 5,585,495; 4,812,570; 5,554,758; 5,952,509; and 6,100,403; U.S. Publications 2002/0106762 and 2002/0050563; WO 03/53367; WO 03/80056; WO 2004/07490; WO 2004/024059; WO 2004/101560; WO 2004/101561 , WO 2005/049610; WO 2008/075380; WO 2006/1 17654; WO
2008/142706; WO 2005/058827; WO 2009/104200; and WO 2009/133576.
U.S. Publication 2006/0252803 provides a process for the preparation of pioglitazone which involves dissolving DHP of Formula II in formic acid and hydrogenating with hydrogen in the presence of 10% Palladium on Carbon under 2 atmospheric pressures and a temperature of 80°C. This process reportedly provides pioglitazone having less than about 0.14 % of the impurity at RRT 0.64 (HPLC).
It also provides a process for the preparation of pioglitazone which involves combining DHP of Formula II with dimethylformamide and hydrogenating with hydrogen in the presence of Palladium on Carbon under 3 atmospheric pressures and a temperature of 50°C. It is reported that -68.5% of DHP was converted to pioglitazone, containing about 3.5 % impurities (HPLC).
U.S. Publication 2009/118514 provides various purification methods for pioglitazone. It provides obtaining a solution of pioglitazone with a mixture of dimethylformamide (DMF) and 4% water, DMF and 5% methanol, DMF and 10% methanol, 1 ,4-dioxane or DMF and isopropyl alcohol at a temperature of about 90°C and precipitating pioglitazone from the solution by cooling to a temperature of about 25°C.
U.S. Publication 2007/0078170 provides a process for the preparation of pioglitazone which involves obtaining a clear solution of pioglitazone in
dimethylformamide at 85°C and adding methanol at a temperature of 60°C and further cooling to 10°C to precipitate pioglitazone.
Impurities in pioglitazone or its hydrochloride salt, or any active pharmaceutical ingredient (“API”), are undesirable and, in extreme cases, might even be harmful to a patient being treated with a dosage form containing the API. The purity of an API is critical for commercialization. The product of a chemical reaction is rarely just the target compound. Side products and by-products of the reaction and adjunct reagents used in the reaction will, in most cases, also be present in the product in varying amounts. Extra effort is often required to get pharmaceutically acceptable levels of purity of the target compound.
EXAMPLES
Example 1 : Purification of Pioglitazone
Dimethylformamide (10 mL) was heated to 80°C to 82°C and pioglitazone (2 g) was added to the hot solution and stirred at 80°C to get a clear solution. The solution was cooled to 75 °C and mixture of acetone (10 mL) and methanol (10 mL) was added to the solution in 30 to 45 minutes at 70°C to 75°C. After complete addition, the reaction mass was cooled to 30°C to 35°C slowly in about 2 hours and was filtered. The solid was washed with dimethylformamide/acetone/methanol mixture (6 mL).
DHP impurity content after purification: 0.05%
DHP impurity content before purification: 0.16%
Yield: 90% Example 2: Purification of Pioglitazone
Dimethylformamide (200 mL) was heated to 80°C to 85 °C and pioglitazone (40 g) was added to the hot solution and stirred at 80°C to get a clear solution. The solution was cooled to 75°C and mixture of acetone (200 mL) and methanol (200 mL) was added to this solution in 30 minutes to 45 minutes at 65°C to 75°C. After complete addition, the reaction mass was cooled to 30°C to 35°C slowly in about 2 hours and was filtered. The solid was washed with acetone/methanol mixture (100 mL).
DHP impurity content after purification: 0.04%
DHP impurity content before purification: 0.15%
Yield: 92%
Example 3: Purification of Pioglitazone (Control)
Dimethylformamide (25 mL) was heated to 80°C to 85 °C and pioglitazone (5 g) was added at 85°C. The solution was cooled to 55°C and a mixture of acetone (25 mL) and methanol (25 mL) was added to the solution in 30 minutes to 45 minutes at 55°C to 60°C. After complete addition, the reaction mass was cooled to 35°C to 37°C slowly in about 2 hours and was filtered. The solid was washed with acetone/methanol mixture (10+lOmL).
DHP impurity content after purification: 0.11%
DHP impurity content before purification: 0.12%
Yield: 92%
PATENT
http://www.google.com/patents/US20070078170
EXAMPLE 5
Conversion of Pioglitazone to Pioglitazone Hydrochloride
Pioglitazone hydrochloride was prepared in a similar manner as exemplified in Example 4. To de-ionized water (7 L) heated to 80° C. added the product obtained from Example 4 (1 Kg) under stirring at 80° C. It was further stirred for 10 min at 80 – 82° C. and conc. HCI (0.5 L) was added to it under stirring at 80° C. The resultant mass was further stirred for 10 min at 80-82° C. to obtain a clear solution. The solution was filtered through celite bed directly into RB flask and the bed was washed with IN HCI (1 L) at 80-82° C. It was cooled to 5° C. under stirring at 5-0° C. for 1 hr. The solids were filtered under N2 atmosphere and dried on the Büchner funnel for about 15 minutes. The product obtained was then dried under vacuum at 55-60° C. to afford the title compound.
Yield: 930.0 g (93%)
HPLC Purity: 99.61%
Samples of pioglitazone hydrochloride prepared in examples 4 and 5 were studied for their solubility, as set forth below:
PATENT
US 2014088127
http://www.google.com/patents/US20140088127
5-[4-[2-(5-ethylpyridyl)ethoxy]benzyl-2,4-thiazolidinedione (Pioglitazone hydrochloride, R in 1 is 5-ethyl).

EXAMPLE 55
5-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
To 5 g (0.0121 mol) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione dissolved in 25 mL acetic acid was added 1.62 (0.0243 mol) g zinc in 5 min. Stirring continued for 15 hours at 25-30° C. Reaction mixture was poured in excess water, made alkaline using 10% Na2CO3 and extracted with ethyl acetate. After distilling off ethyl acetate in vacuo, methanol was added to precipitate out 1.13 g (25%) g of the crystalline solid product. The two impurities identified in this reaction were 5-ethyl-2-vinyl-pyridine 0.128 g (10%) and 5-(4-hydroxy benzyl)-thiazolidin-2,4-dione 0.342 g (12%).
The product obtained was characterized by IR, Mass, 13C NMR, and 1H NMR, which are as given below. pioglitazone base
IR spectrum (cm−1): 3417 (N—H str.), 1693, 1743 (C═O str.), 1037 (C—O—C str.)
Mass spectrum (m/z) 357.1 (M+H)+
13C-NMR (DMSO-d6): δ 176.5, 172.5, 157.8, 152.1, 145.9, 142.0, 141.1, 131.2, 129.9, 127.8, 115.2, 66.2, 53.8, 37.0, 33.2, 25.4, 15.5
1H-NMR (DMSO-d6): δ 12.0 (1H, s), 6.84-8.71 (7H, m), 4.86 (1H, dd), 4.38 (2H, t), 3.48 (2H, t), 3.25 (1H, dd), 3.04 (1H, dd), 2.75 (2H,q), 1.21 (3H, t)
Melting point 172-175° C.
The final product was dissolved into 12 mL methanol and 0.05 mL con. HCl was added into it at 25° C. Reaction mixture was refluxed for 30 min. and cooled to 10° C. Precipitated hydrochloride salt was filtered off and dried to yield 1.1 g (22%) of the salt, which was characterized by IR, Mass, 13C NMR and 1H NMR, which are as given below.
pioglitazone hydrochloride
IR spectrum (cm−1): 3257 (N—H str.), 1689, 1743 (C═O str.), 1155, 1244 (C—O—C str.)
Mass spectrum (m/z): 357.1 (M+H)+
13C-NMR (DMSO-d6): δ 175.7, 171.7, 157.0, 151.0, 141.1, 129.0, 145.4, 139.8, 127.2, 130.4, 114.4, 65.4, 53.0, 39.2, 36.2, 24.6, 14.6
1H-NMR (DMSO-d6): δ 12.09 (1H, s), 6.82-8.7 (7H, m), 4.8 (1H, dd), 4.38 (2H, t), 3.5 (2H, t), 3.0 (2H, m), 2.75 (2H,q), 1.21 (3H, t)
Melting point: 190-193° C.
EXAMPLE 565-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
The procedure described in example 55 was repeated except that temperature was maintained 5-10° C., and substantially the same results were achieved. In this particular case, the final product 5-{4-[2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione obtained was 0.911 g (20%), is identical in every respect with the product of example 55. m.p. 172° C.
EXAMPLE 575-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
To a solution of 0.8 g (0.00184 mol) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione dissolved in 10 mL ethyl acetate was added 0.8 mL (0.014 mol) acetic acid and 0.119 g (0.00184 mol) zinc in 5 minutes at 25-30° C. Stirring continued for 9 hr. at 30-35° C. Solid material separated was filtered off and dried to get titled compound. Yield of the product was 0.3 g (41%). m.p. 173° C.
The product obtained was characterized by IR, Mass, 13C NMR, and 1H NMR, which was found to be identical with the product obtained in example 55. The impurity profile was also similar to the product obtained in example 55.
EXAMPLE 585-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
The procedure described in example 57 was repeated except that acetic acid was replaced by 1.038 g (0.014 mol) propionic acid, and substantially the same results were achieved. In this particular case, the final product 5-{4-[2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione obtained was 0.3 g (41%), is identical in every respect with the product of example 55. m.p. 172° C.
EXAMPLE 595-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
The procedure described in example 57 was repeated except that tetrahydrofuran was used as solvent, and substantially the same results were achieved. In this particular case, the final product 5-{4-[2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione obtained was 0.13 g (18%), is identical in every respect with the product of example 55. m.p. 173° C.
EXAMPLE 605-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
To a mixture of 5 mL 35% Con. HCl and 5 mL water, 0.5 g (0.0012 mol) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione was added followed by the addition of 0.039 g (0.0006 mol) zinc (tlc). Reaction mixture was poured into 20 mL water, made alkaline by 10% K2CO3 solution and product was extracted with 25 mL ethyl acetate. Organic layer was separated, dried (magnesium sulfate) and concentrated under reduced pressure. Methanol was added to the residual mass to get 0.1 g (22%) crystalline titled product. m.p. 174° C.
The product obtained was characterized by IR, Mass, 13C NMR, and 1H NMR, which was found to be identical with the product obtained in example 55.
EXAMPLE 615-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
To a mixture of 35 mL ethanol and 7 mL (0.122 mol) glacial acetic acid was dissolved 3.5 g (0.0084 mol) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione followed by addition of 0.819 g (0.0126 mol) zinc. Precipitated product was filtered and dried to yield 1.91 g (60%) product. m.p. 174° C.
The product obtained was characterized by IR, Mass, 13C NMR and 1H NMR, which was found to be identical with the product obtained in example 55.
EXAMPLE 625-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
Into 10 mL (0.175 mol) glacial acetic acid, 1 g (0.0024 mol) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione and 100 mg (0.0018 mol) ammonium chloride were dissolved. Under stirring 0.078 g (0.0012 mol) zinc was added. Subsequent work-up in water yielded 0.6 g (66%) of the desired product. m.p. 175° C.
The product obtained was characterized by IR, Mass, 13C NMR, and 1H NMR, which was found to be identical with the product obtained in example 55.
EXAMPLE 635-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
The procedure described in example 61 was repeated on 5 g scale except that isopropyl alcohol was used as solvent and reaction mixture was allowed to stir for 8 hours. Substantially the same results were achieved. In this particular case, the final product 5-{4-[2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione obtained was 3 g (66%), is identical in every respect with the product of example 55. m.p. 174° C.
EXAMPLE 645-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
0.63 g (0.0097 mol) Zinc dust and 8.93 mL (0.0582 mol) tetramethyl ethylenediamine were added into 80 mL ethanol under nitrogen atmosphere. To this a mixture of 1.18 mL acetic acid and 7.8 g (0.0194 mol)) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione dissolved in 16 mL ethanol was added in 15 min. at 5-10° C. Reaction mixture was allowed to achieve 27° C. and 4.74 mL acetic acid was added in 15 minutes. Reaction mixture was stirred for 15 hr. The product precipitated was filtered off and dried to obtain 3.98 g (56%) of the titled product. m.p. 173° C.
The product obtained was characterized by IR, Mass, 13C NMR, and 1H NMR, which was found to be identical with the product obtained in example 55.
EXAMPLE 655-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
To a mixture of 40 mL ethanol and 10 mL propionic acid, 5 g (0.0117 mol) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione hydrochloride was dissolved. To this was added 1.5 g (0.0234 mol) of zinc and stirred. Reaction mixture was stirred for 12 hr. Product precipitated was filtered off and dried to yield 2 g (48%) of the titled product. m.p. 175° C.
The product obtained was characterized by IR, Mass, 13C NMR, and 1H NMR, which was found to be identical with the product obtained in example 55.
EXAMPLE 665-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
70 g (0.150 mol) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxyl]-benzyl}-2,4-thiazolidene dione dissolved in a mixture methanol and acetic acid. To this was added 21.85 g (0.3 mol) of and stirred. Product precipitated was filtered off and dried to get 51 g (80%) of the desired product. The impurity profile in this reaction was similar to the impurity profile described in example 55. m.p. 174° C.
The product obtained was characterized by IR, Mass, 13C NMR, and 1H NMR, which was found to be identical with the product obtained in example 55.
The final product obtained above was treated with cone. HCl in isopropanol. Precipitated hydrochloride salt was filtered off and dried to yield 50 g of the salt, which was characterized by IR, Mass, 13C NMR and 1H NMR, which was found identical with hydrochloride salt obtained in example 55. m.p. 194-197° C.
EXAMPLE 675-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
A solution of 250 g (0.5854 mol) of 5-{4-[2-chloro-2-(5-ethylpyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione hydrochloride in methanol and 500 mL (8.75 mol) acetic acid was added into a flask fitted with an overhead stirrer and a thermometer. To this was added 76.54 g (1.17 mol) of and stirred. The product precipitated was filtered and dried. Yield of the product was 100 g (48%). The impurity profile in this reaction was similar to the impurity profile described in example 55. m.p. 175° C.
The product obtained was characterized by IR, Mass, 13C NMR, and 1H NMR, which was found to be identical with the product obtained in example 55.
The final product treated with conc. HCl in ethanol. Precipitated hydrochloride salt was filtered off and dried to yield 100 g of the salt, which was characterized by IR, Mass, 13C NMR and 1H NMR, which was found identical with hydrochloride salt obtained in example 55. m.p. 192° C.
EXAMPLE 685-{4-[2-(5-Ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione (1)
10 kg (23.42 mol) of 5-{4-[2-chloro-2-(5-ethyl-pyridin-2-yl)-ethoxy]-benzyl}-2,4-thiazolidene dione hydrochloride was dissolved in a mixture of 80 lit. methanol and 20 lit. acetic acid. To this was added 3.06 kg (46.8 mol) of zinc and stirred (tlc). Product precipitated was filtered off and dried to get 7.5 Kg of the desired product. m.p. 172° C.
The product obtained was converted into its hydrochloride salt as described in example 67 to obtain 7.72 Kg (84%) of the salt, which was found to be identical in all respect with the salt obtained in example 55. The impurity profile in this reaction was similar to the impurity profile described in example 55. m.p. 192° C.
PAPER
see

An efficient six-step synthesis has been developed for the preparation of the thiazolidinedione analogue PNU-91325 (3) from the commercially available olefin 12. This process involves a novel epoxide ring opening with a deactivated phenol under phase-transfer conditions. Significant improvements were made in the oxidation of a secondary alcohol to the ketone and the 1,4-reduction of an enone from a previous process. Overall, this route allows for the preparation of PNU-91325 in 25% yield.
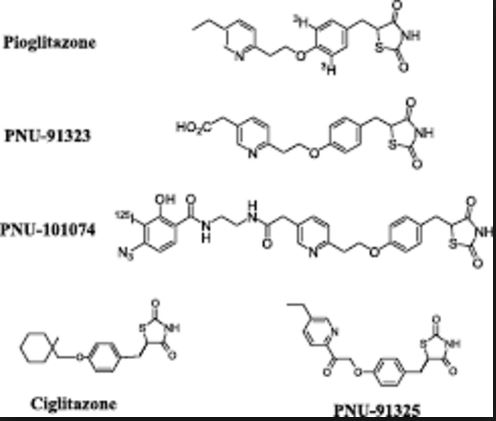

Improved synthetic approach: route A (feasibility) and B (after optimization)

Rakeshwar Bandichhor Ph.D. FRSC, CChem, SSWB,BB,MB
Director at Dr. Reddy’s Laboratories, Vice-Chair, ACS-India Chapter (South India)

They have developed an improved process for pioglitazone which appears to be more compatible with industrial scale and has some advantages over the existing synthesis.
Preparation of Pioglitazone Hydrochloride (1·HCl) salt
An Improved Process for Pioglitazone and Its Pharmaceutically Acceptable Salt†
, * Corresponding author. E-mail: rakeshwarb@drreddys.com. Telephone: +91 4044346000. Fax: +91 4044346285.,
Abstract

An improved process for pioglitazone (1) is described. The process features high-yielding transformations employing inexpensive reagents and recoverable solvents.
link is
http://pubs.acs.org/doi/full/10.1021/op900131m
The original paper in OPRD is interesting example of process improvement
| US8067450 | Sep 15, 2008 | Nov 29, 2011 | Metabolic Solutions Development Company | Thiazolidinedione analogues for the treatment of metabolic diseases |
| US8304441 | Nov 16, 2011 | Nov 6, 2012 | Metabolic Solutions Development Company, Llc | Thiazolidinedione analogues for the treatment of metabolic diseases |
| WO2011017244A1 * | Aug 2, 2010 | Feb 10, 2011 | Metabolic Solutions Development Company | Polymorphs of 5-(4-(2-(5-ethylpyridin-2-yl)-2-oxoethoxy)benzyl)-1,3-thiazolidine-2,4-dione (mitoglitazone) |
| WO2012153312A1 | May 11, 2012 | Nov 15, 2012 | Ranbaxy Laboratories Limited | Process for the purification of pioglitazone |
| WO2003053367A2 | Dec 20, 2002 | Jul 3, 2003 | Ben-Zion Dolitzky | Hydrogenation of precursors to thiazolidinedione antihyperglycemics |
| WO2003080056A2 | Mar 21, 2003 | Oct 2, 2003 | Ben-Zion Dolitzky | Fine particle size pioglitazone |
| WO2004007490A2 | Jul 15, 2003 | Jan 22, 2004 | Cadila Healthcare Ltd | A process to prepare pioglitazone via several intermediates. |
| WO2004024059A2 | Aug 29, 2003 | Mar 25, 2004 | Parag Narayan Gadkari | Improved process for preparation of thiazolidinedione derivatives |
| WO2004101560A1 | May 11, 2004 | Nov 25, 2004 | Frantisek Picha | Processes for making thiazolidinedione derivatives and compounds thereof |
| WO2004101561A1 | May 13, 2004 | Nov 25, 2004 | Frantisek Picha | Pioglitazone salts, such as pioglitazone sulfate, and pharmaceutical compositions and processes using the same |
| WO2005049610A1 | Oct 27, 2004 | Jun 2, 2005 | Sudhir Nambiar | Process for preparing thiazolidinediones |
| WO2005058827A1 | Dec 16, 2004 | Jun 30, 2005 | Janos Fischer | Process for the synthesis of pioglitazone hydrogen chloride |
| WO2006117654A1 | May 3, 2006 | Nov 9, 2006 | Ranbaxy Lab Ltd | Processes for the preparation of pioglitazone or salts thereof |
| WO2008075380A2 | Dec 20, 2007 | Jun 26, 2008 | Ind Swift Lab Ltd | Process for the preparation of thiazolidine derivatives |
| WO2008142706A2 | May 19, 2008 | Nov 27, 2008 | Yogendra Kumar Chauhan | Novel process for the synthesis of pioglitazone and its salts thereof |
| WO2009104200A1 | Apr 8, 2008 | Aug 27, 2009 | Biocon Limited | A method for preparation of thiazolidinedione derivatives |
| WO2009133576A1 | Apr 28, 2008 | Nov 5, 2009 | Erregierre S.P.A. | A process for the preparation of 4-[2-(5-ethyl-2-pyridyl)ethoxy]nitrobenzene and pioglitazone |
| US4687777 | Jan 17, 1986 | Aug 18, 1987 | Takeda Chemical Industries, Ltd. | Thiazolidinedione derivatives, useful as antidiabetic agents |
| US4812570 | Jul 14, 1987 | Mar 14, 1989 | Takeda Chemical Industries, Ltd. | Method for producing thiazolidinedione derivatives |
| US5554758 | Jun 7, 1995 | Sep 10, 1996 | Takeda Chemical Industries, Ltd. | P-/2-/5-ethyl-2-pyridyl/ethoxy/benzaldehyde |
| US5585495 | Dec 4, 1992 | Dec 17, 1996 | The Upjohn Company | Using a cobalt compound, ligand and reducing agent |
| US5952509 | Jun 23, 1997 | Sep 14, 1999 | Takeda Chemical Industries, Ltd. | Reacting p-hydroxybenzaldehyde with a pyridyl-ethyl sulfonate derivative to form 4-(2-(substituted or nonsubstituted-2-pyridyl)ethoxy) benzaldehyde |
| US6100403 | Apr 12, 1999 | Aug 8, 2000 | Takeda Chemical Industries, Ltd. | Production of benzaldehyde compounds |
| US20020050563 | Sep 14, 2001 | May 2, 2002 | Smithkline Beecham P.L.C. | Process for the preparation of pharmaceutically active thiazolidine or oxazolidine compounds by a yeast reductase |
| US20020106762 | Oct 9, 2001 | Aug 8, 2002 | Smith Kline Beecham Plc | Generating preerential compound for use as prophylactic agent; obtain chemical intermediate, incubate with enzyme, recover prophylactic agent |
| US20060252803 | Jan 26, 2006 | Nov 9, 2006 | Ben-Zion Dolitzky | Hydrogenation of precursors to thiazolidinedione antihyperglycemics |
| US20070078170 | Aug 30, 2004 | Apr 5, 2007 | Khanduri Chandra H | Process for the preparation of pioglitazone |
| US20090118514 | Nov 5, 2008 | May 7, 2009 | Raghupathi Reddy Anumula | Processes for preparing pioglitazone and its pharmaceutically acceptable salts |
| WO2012153312A1 | May 11, 2012 | Nov 15, 2012 | Ranbaxy Laboratories Limited | Process for the purification of pioglitazone |
| EP1734036A1 * | Jun 14, 2005 | Dec 20, 2006 | Well-being Biochemical Corp. | Process for preparation of tamsulosin and its derivatives |
| WO1997031907A1 * | Feb 26, 1997 | Sep 4, 1997 | Grady Evan Boswell | Substituted 4-hydroxy-phenylalcanoic acid derivatives with agonist activity to ppar-gamma |
| WO2002088120A1 * | Apr 25, 2002 | Jul 11, 2002 | Ales Halama | Method for obtaining pioglitazone as an antidiabetic agent |
| EP0008203A1 * | Aug 3, 1979 | Feb 20, 1980 | Takeda Chemical Industries, Ltd. | Thiazolidine derivatives, preparing same and pharmaceutical compositions comprising same |
| EP0193256A1 * | Jan 15, 1986 | Sep 3, 1986 | Takeda Chemical Industries, Ltd. | Thiazolidinedione derivatives, their production and use |

