
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


Omacetaxine mepesuccinate 高三尖杉酯碱
Alkaloid from Cephalotaxus harringtonia; FDA approved orphan drug status for Ceflatonin in the treatment of chronic myeloid leukemia due to being an inducer of apoptosis in myeloid cells and inhibitor of angiogenesis.
26833-87-4 CAS NO
1-((1S,3aR,14bS)-2-Methoxy-1,5,6,8,9,14b-hexahydro-4H-cyclopenta(a)(1,3)dioxolo(4,5-h)pyrrolo(2,1-b)(3)benzazepin-1-yl) 4-methyl (2R)-2-hydroxy-2-(4-hydroxy-4-methylpentyl)butanedioate
1-((11bS,12S,14aR)-13-methoxy-2,3,5,6,11b,12-hexahydro-1H-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-d]cyclopenta[b]pyrrolo[1,2-a]azepin-12-yl) 4-methyl 2-hydroxy-2-(4-hydroxy-4-methylpentyl)succinate
Also known as: NSC-141633,
- BRN 5687925
- Ceflatonin
- CGX-635
- Homoharringtonine
- Myelostat
- NSC 141633
- Omacetaxine mepesuccinate
- Omapro
- Synribo
- UNII-6FG8041S5B
- 高三尖杉酯碱
CGX-635-14 (formulation), CGX-635, HHT, ZJ-C, Myelostat, Ceflatonin
USFDA on 26th October 2012 APPROVED
| Formula | C29H39NO9 |
|---|---|
| Mol. mass | 545.62 g/mol |
The US Food and Drug Administration has now issued full approval for Israeli drugmaker Teva’s Synribo (omacetaxine mepesuccinate) for chronic myeloid leukaemia (CML).
Synribo is indicated for adult patients with chronic phase (CP) or accelerated phase (AP) CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs).
Read more at: http://www.pharmatimes.com/Article/14-02-17/US_green_light_for_Teva_s_CML_drug_Synribo.aspx#ixzz2tdkbGFcw
Homoharringtonine is an angiogenesis-inhibiting and apoptosis-inducing alkaloid which was approved in October 2012 by the FDA for the treatment of adult patients with chronic or accelerated phase chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKI). In November 2012, the product was commercialized as Synribo(R) on the U.S. market by Teva.
The original developer, ChemGenex, selected homoharringtonine for the combination trials due to its complementary mechanism of action that can reduce Bcr-Abl protein expression associated with resistance to imatinib mesylate.
In 2004, the compound received orphan drug designation from the EMEA for the treatment of AML and CML. Orphan drug designation was granted by the FDA for the treatment of CML in 2006 and for the treatment of myelodysplasia in 2009. Fast track designation was assigned to homoharringtonine for CML in 2006. In 2009, the product was licensed to Hospira by ChemGenex Pharmaceuticals for development and marketing in Europe, the Middle East and parts of Africa.
Homoharringtonine, AKA HHT or omacetaxine mepesuccinate, is a cephalotaxine ester and protein synthesis inhibitor with established clinical activity as a single agent in hematological malignancies. Homoharringtonine is synthesized from cephalotaxine, which is an extract from the leaves of the plant, Cephalotaxus species. In October 2005, homoharringtonine received Orphan Drug designation from the EMEA for the treatment of chronic myeloid leukemia (CML). Then in March 2006, homoharringtonine received Orphan Drug status from the FDA for the treatment of CML. In November 2006, homoharringtonine, for the treatment of CML, was granted Fast Track designation by the FDA. Most recently, in October 2012, homoharringtonine was marketed under the brand name Synribo” and FDA approved for patients who are intolerant and/or resistant to two or more tyrosine kinase inhibitors used to treat accelerated or chronic phase CML
Omacetaxine mepesuccinate is administered subcutaneously and acts differently from TKIs. It may have a therapeutic advantage for patients who have failed TKIs. Omacetaxine is currently in global phase 2/3 clinical trials for CML and has been granted Orphan Drug designations by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMEA) as well as Fast Track status by the FDA. In vitro and animal model trails are promising and recent results showed that omacetaxine has potential to treat resistant leukemia mainly CML and ALL.
| PATENT | ||
|---|---|---|
|
3-25-2011
|
CEPHALOTAXUS ESTERS, METHODS OF SYNTHESIS, AND USES THEREOF
|
Tetrahedron Letters,Vo1.23,No.34,pp 3431-3434 … – Brock University
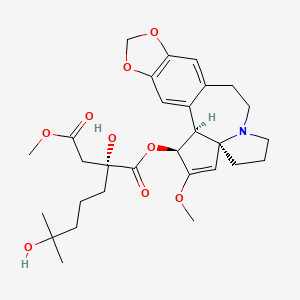
Omacetaxine mepesuccinate (INN, trade name Synribo) is a semi-synthetic analogue of an alkaloid from Cephalotaxus harringtonia that is indicated for treatment of chronic myelogenous leukemia (CML). It was approved by the US FDA in October 2012 for the treatment of adult patients with CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs).[1]
Omacetaxine mepesuccinate is a semisynthetic derivative of the cytotoxic plant alkaloid homoharringtonine isolated from the evergreen tree Cephalotaxus with potential antineoplastic activity. Omacetaxine mepesuccinate binds to the 80S ribosome in eukaryotic cells and inhibits protein synthesis by interfering with chain elongation. This agent also induces differentiation and apoptosis in some cancer cell types. Omacetaxine mepesuccinate (INN, or homoharringtonine, trade name Synribo) is an alkaloid from Cephalotaxus harringtonia that is indicated for treatment of Chronic Myelogenous Leukemia. It was approved by the USFDA on 26th October 2012 for the treatment of adult patients with chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs)
Omacetaxine is indicated for use as a treatment for patients with chronic myeloid leukaemia who are intolerant of tyrosine kinase inhibitors.[2][3]
In June 2009, results of a long-term open label Phase II study were published, which investigated the use of omacetaxine infusions in CML patients. After twelve months of treatment, about one third of patients showed a cytogenetic response.[4] A study in patients who had failed imatinib and who had the drug resistant T315I mutation achieved cytogenetic response in 28% of patients and haematological response in 80% of patients, according to preliminary data.[5]
Phase I studies including a small number of patients have shown benefit in treating myelodysplastic syndrome (MDS, 25 patients)[6] and acute myelogenous leukaemia (AML, 76 patients).[7] Patients with solid tumors did not benefit from omacetaxine.[8]
Omacetaxine is a protein translation inhibitor. It inhibits protein translation by preventing the initial elongation step of protein synthesis. It interacts with the ribosomal A-site and prevents the correct positioning of amino acid side chains of incoming aminoacyl-tRNAs. Omacetaxine acts only on the initial step of protein translation and does not inhibit protein synthesis from mRNAs that have already commenced translation.[9]
Omacetaxine mepesuccinate
SYNRIBO contains the active ingredient omacetaxine mepesuccinate, a cephalotaxine ester. It is a protein synthesis inhibitor. Omacetaxine mepesuccinate is prepared by a semi-synthetic process from cephalotaxine, an extract from the leaves of Cephalotaxus sp. The chemical name of omacetaxine mepesuccinate is cephalotaxine, 4-methyl (2R)-hydroxyl-2-(4-hydroxyl-4-methylpentyl) butanedioate (ester).
Omacetaxine mepesuccinate has the following chemical structure:
 |
The molecular formula is C29H39NO9 with a molecular weight of 545.6 g/mol. SYNRIBO for injection is a sterile, preservative-free, white to off-white, lyophilized powder in a single-use vial. Each vial contains 3.5 mg omacetaxine mepesuccinate and mannitol.
SYNRIBO is intended for subcutaneous administration after reconstitution with 1.0 mL of 0.9% Sodium Chloride Injection, USP. The pH of the reconstituted solution is between 5.5 and 7.0.
…………………………………..
INTRODUCTION
Harringtonines 3 are particular cephalotaxanes formed by attachement of a branched hydroxyacyloxy side-chain at the 3-position of various cephalotaxines moieties. Harringtoriines are natural esters of cephalotaxines exhibiting generally a strong cytotoxic activity. However the lost only one atom of this minimal structure lead to a dramatic lost of activity (see below). Some example of harringtonines are harringtonine
3a, homoharringtonine 3b, drupangtonine 3c, anhydroharringtonine 3d and neoharringtonine 3e.
SCHEME 1 DEFINITION NOMENCLATURE AND NUMBERING OF CEPHALOTAXANES


Examples of harringtonines

Examples of cephalotaxines

Harringtonine 3a (n = 2) Anhydroharringtonine 3d Homoharringtonine 3b (n = 3)

(-)-Cephalotaxine 2a


Drupacine 2b Drupangtonine 3c Neoharringtonine 3e (n = 2)
…………………………………
The term “cephalotaxanes” refers to compounds or salts thereof which have a basic skeleton of formula

where p is equal to 1 or 2 (it being possible for the two units to be identical or different and linked via a single bond or an oxygen atom), which can contain various oxygenated substituents (aliphatic or aromatic ethers, free or esterified alcohols, substituted or free enols and/or phenols, bridged ethers, and more generally any substituent usually encountered in the natural state on compounds of this type).
Harringtonines are alkaloids which are of high interest in anticancer chemotherapy, in particular on certain haematosarcomas which are multi-resistant to the existing therapies. The selectivity of harringtonines, which is based on a novel mechanism of action relating to protein synthesis, is such that this series is favoured with a great future in anticancer therapy.
Several literature compilations give a seemingly exhaustive review of all of the knowledge relating to cephalotaxanes, these compilations being, chronologically: [C. R. Smith, Jr, R. G. Powell and K. L. Mikolajczack, Cancer Treat. Rep., Vol. 60, 1157 (1976); C. R. Smith, Jr, L. Kenneth, K. L. Mikolajczack and R. G. Powell in “Anticancer Agent Based on Natural Product Model”, 391 (1980); Liang Huang and Zhi Xue in “The Alkaloids”, Vol. XXIII (A. Brossi Ed.), 157 (1984); M. Suffness and G. A. Cordell in “The Alkaloids, Chemistry and Pharmacology” (A. Brossi Ed.), Vol. 25, 57-69, 295-298 (1’987); P. J. O’Dwyer, S. A. King, D. F. Hoth, M. Suffness and B. Leyland-Jones, Journal of Clinical Oncology, 1563 (1986); T. Hudlicky, L. D. Kwart and J. W. Reed, in “Alkaloid: Chemical and Biological Perspectives” (S. W. Pelletier Ed.), Vol. 5, 639 (1987); M. A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, 199 (1998)].
Antiparasitic activities, in particular on the haematozoon of malaria, have also been recognized [J. M. Whaun and N. D. Brown, Ann Trop. Med. Par., Vol. 84, 229 (1990)].
Homo-harringtonine (HHT), the most active member of the series, is active at and above daily doses of 2.5 mg/m2 of body area per 24 hours, i.e., as a guide, at doses twenty times lower than that for Taxol. HHT has already undergone fourteen phase I and II clinical trials and it is the only known product capable of a 70% reinduction of full haematological remissions in patients suffering from chronic myeloid leukaemias that have become resistant to alpha-interferon [S. O’Brien, H. Kantarjian, M. Keating, M. Beran, C. Koler, L. E. Robertson, J. Hester, M. Rios, M. Andreeff and M. Talpaz, Blood, 332 (1995); Leukemia Insights, Vol. 3, No. 1 (1998)].
Harringtonines were extracted over 35 years ago from an exclusively Asiatic cephalotaxacea known as Cephalotaxus harringtonia, following the programme of research into novel anticancer agents in the plant kingdom developed by the National Cancer Institute. In fact, the Cephalotaxus alkaloids consist essentially (at least 50%) of cephalotaxine, a biosynthetic precursor of the harringtonines, the latter individually representing only a few percent of the total alkaloids.
Besides their low concentration in the natural state in plant starting material, harringtonines are mixed with many congeners which have very similar chemical structures. Thus, in a high resolution high performance liquid chromatography (HPLC) chromatogram of a semi-purified alkaloid extract, no less than several tens of cephalotaxine esters are counted.
Numerous antileukemia drugs have been investigated but so far, there is no single drug that is effective and safe. As discussed in U.S. 3,497,593, an alkaloid from Tylophora plant is said to have antitumor activity against mouse leukemia (L-1210). U.S. 3,928,584 discloses an organic composition derived from tree sap and is said to have activity against mouse leukemia P-388. Also U.S. 4,431,639 discloses that an extract of Rhisoma Stractylis promotes the production of lymphocytes in the circulating blood, consequently eliminating cancer growth
-
Harringtonine or Homoharringtonine, hereinafter referred to as HH, has been known to be effective against acute chronic granulocytic and monocytic leukemia (Journal of Chinese Internal Medicine 3:162-164, 1978). However, it is highly toxic and causes damage to heart and hematopoietic organs. The results of experiments in animals, such as mice, rabbits and dogs, indicate that most of them die from cardiotoxicity after receiving the drug. Therefore, there is a need to improve the HH drug for safe use against leukemia. This drug is of special importance in that all known antileukemia drugs are effective against lymphatic leukemia and there are no effective drugs for treating nonlymphatic leukemia
All the literature from 1972 to the present date [Mikolajczack et al., Tetrahedron, 1995 (1972); T. Hudlicky, L. D. Kwart and J. W. Reed in “Alkaloid: Chemical and Biological Perspectives” (S. W. Pelletier Ed.), Vol. 5, 639 (1987); M. A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, p. 236 (1998)] mention the impossibility hitherto of esterifying the highly sterically hindered secondary hydroxyl of cephalotaxane 2a with the tertiary carboxyl of the alkanoyl chain of harringtonic acid 3 totally preformed to give a harringtonine 4b, i.e. the conversion 2a+3e(4b as described in the example featured in the scheme below

- ……………………………………………………..
SYNTHESIS
Tetrahedron Lett 1982,23(34),3431, J Org Chem 1983,48(26),5321

The oxidation of 2-methyl-1-cyclopentene-1-carbaldehyde (I) with O3 and Ag2O gives 2,6-dioxoheptanoic acid (II), which is esterified with cephalotaxine (III) by means of (COCl)2, yielding the ester (IV). Reformatsky reaction of (IV) with methyl bromoacetate (V) and Zn affords the adduct (VI), which is treated with an excess of methylmagnesium iodide to provide the target homoharringtonine (as a single diastereomer), along with some starting cephalotaxine that is separated by chromatography.
………………………………
SYNTHESIS
EP 1064285; FR 2776292; WO 9948894, Tetrahedron Lett 1999,402931

The intermediate (racemic)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (VIII) has been obtained by several related methods: 1. The Grignard condensation of 4-methyl-3-pentenyl bromide (I) with diethyl oxalate (II) in HF gives the 2-oxoheptenoate (III), which is condensed with methyl acetate (IV) by means of LiHMDS in THF to yield 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V).
The cyclization of (V) by means of Ts-OH in hot toluene or by means of hot aqueous formic acid affords 2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid ethyl ester (VI), which is hydrolyzed with KOH in boiling water to provide the corresponding dicarboxylic acid (VII). Finally, this compound is regioselectively monoesterified by means of BF3/MeOH in methanol to furnish the intermediate (racemic)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (VIII). 2.
The reaction of 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V) with HCl in hot methanol gives 3-(ethoxycarbonyl)-3,7-dihydroxy-7-methyloctanoic acid methyl ester (IX), which is then cyclized by means of ZnCl2 in hot dichloroethane to yield the previously described intermediate (VIII). 3. The hydrolysis of 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V) with KOH in refluxing methanol/water gives the corresponding diacid (X), which is regioselectively monoesterified by means of BF3/MeOH in methanol to yield 3-carboxy-3-hydroxy-7-methyl-6-octenoic acid methyl ester (XI).
Finally, this compound is cyclized by means of Ts-OH in hot toluene to afford the previously described carboxylic intermediate (VIII). The racemic acid (VIII) is submitted to optical resolution by esterification with quinine (XII) by means of 2,4,6-trichlorobenzoyl chloride and TEA or DCC to give a diastereomeric mixture of esters (XIII) that is separated by preparative HPLC to obtain the desired diastereomer (XIV).
The hydrolysis of (XIV) with KOH in refluxing ethanol/water gives the corresponding chiral dicarboxylic acid (XV), which is regioselectively monoesterified with BF3/MeOH in methanol to yield the chiral (R)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (XVI).
The esterification of (XVI) with cephalotaxine (XVII) by means of 2,4,6-trichlorobenzoyl chloride and TEA in toluene affords the corresponding ester (XVIII), which is treated with HBr in dichloromethane/HOAc, providing the bromoester (XIX). Finally, this compound is treated with NaHCO3, CaCO3 or BaCO3 in acetone/water to give the target hydroxyester.
………………………………………….
EXTRACTION
-
Throughout the specification, the concentration of the solvent is the same as first given unless stated otherwise. Redeuced pressure means about 2,27 kPa (17 mm Hg. abs), l is liter, kg is kilogram. ml is milliliter. Yield in weight %.
Example 1. HH is extracted from the skins, stems, leaves and seeds of Cephalotaxus fortunel Hook and other related species, such as Cephalotaxus sinensis Li, C. hainanensis, and C. wilsoniana, including C.oliveri mast and C.harringtonia. -
1 kg of finely ground Cephalotaxus fortunel Hook is extracted with 8 l of 90% ethanol at room temperature for 24 hrs. The solution is filtered to yield a filtrate A and filtercake. The filtercake is percolated with ethanol and filtered again to yield filtrate B. A and B are combined and distilled under reduced pressure to recover ethanol and an aqueous residue. To this residue, 2% HCl is added to adjust the pH to 2.5. The solids are separated from the solution by filtration to yield a filtrate C. The solids are washed once with 2% HCl and filtered to yield a filtrate D. C and D are combined and the pH adjusted to 9.5 by adding saturated sodium carbonate solution. The alkaline filtrate is extracted with chloroform and the chloroform layer separated from the aqueous layer. This extration process is repeated five times. All the chloroform extracts are combined and distilled at reduced pressure to recover chloroform and alkaloid as a solid residue respectively.
-
The solid alkaloid is then dissolved in 20 ml. of 6% citric acid in water. The solution is divided into three equal portions. These are adjusted to pH 7,8 and 9 by adding saturated sodium carbonate solution.
-
The portions having pH 8 and 9 are combined and extracted with chloroform. The chloroform extracts are distilled under reduced pressure, whereby chloroform is removed and recovered and a solid residue of crude Harringtonine is obtained.
-
The crude Harringtonine is dissolved in pure ethanol i.e. alkaloid : anhydrous ethanol 1:10 , and crystallized. The crystals are refined by recrystalliation in diethyl ether. Overall yield of Harringtonine is about 0.1% including yield from mixed HH from the subsequent process.
Harringtonine has the following chemical structure:
wherein R is

- melting point:
- 135° – 137°C
- crystal:
- colorless
- infrared spectrum:
- 3750, 1660, 1505, 1490, 1050, and 945 cm⁻¹.

-
The portion having a pH of 7 and the mother liquors from the foregoing crystallization of Harringtonine are combined and passed through a liquid chromatographic column of diameter to height ratio 1:50 packed with alumina. The column is finally flushed with chloroform and followed by chloroform-methanol of 9:1 mixture. The resulting alkaloids are mixture of HH. The mixed HH is then separated from each other by countercurrent distribution employing chloroform and pH 5 buffer. The first fraction of the countercurrent distribution is Homoharringtonine and the last fraction of the countercurrent distribution is Harringtonine. Homoharringtonine is purified by crystallization in methyl alcohol.
Homoharringtonine has the following chemical structure:
wherein R is

- yield:
- 0.02%
- melting point:
- 144° – 146°C
- infrared spectrum:
- 3500∼3400, 1750, 1665, 1030 and 940 cm⁻¹.







…………………………………………………………………………..
EXTRACTION
All the literature from 1972 to the present date [Mikolajczack et al.,Tetrahedron, 1995 (1972); T. Hudlicky, L.D. Kwart and J.W. Reed in “Alkaloid: Chemical and Biological Perspectives” (S.W. Pelletier Ed.), Vol. 5, 639 (1987); M.A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, p. 236 (1998)] mention the impossibility hitherto of esterifying the highly sterically hindered secondary hydroxyl of cephalotaxine 2a with the tertiary carboxyl of the alkanoyl chain of harringtonic acid 3e totally preformed to give a harringtonine 4b , i.e. the conversion 2a + 3e ( 4b as described in the example featured in the scheme below

Example 46
Preparation of purified (-) cephalotaxine from total alkaloidic extract of Cephalotaxus sp
-
[0319]

-
Partially racemized cephalotaxine [H. Wenkui; L. Yulin; P. Xinfu, Scientia Sinica,; 23; 7; 835 (1980)]
-
1H NMR of two batches of cephalotaxine (extracted in the same conditions as above) with the optically active NMR shift reagent europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate (1 éq) showed the following results:
- Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine) ; 5.99 (1H, OCH2O (-)-cephalotaxine) and 5.76 (1H, OCH2O (-)-cephalotaxine).
Presence of 11 ± 5 % de (+)-cephalotaxine.
[α]22 = -134,0° (c = 0,214; CHCl3) : calculated rate 25 ± 5 % - Batch B: slightly racemized (1%)
[α]19 = -173,3° (c = 0,208; CHCl3)
- Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine) ; 5.99 (1H, OCH2O (-)-cephalotaxine) and 5.76 (1H, OCH2O (-)-cephalotaxine).

Enantiomeric enrichment of the natural cephalotaxine:
-
Crude chromatographied cephalotaxine (20g) was dissolved at 55°C in dry methanol (100 ml). Crystallization occurs by cooling with rotary evaporator and after filtration the product thus obtained showed 99.9% of HPLC purity.
[α]20 D =-130° (C1, CHD3) corresponding to 10 % of racemization. The crystallized product thus obtained (20g) was dissolved again in hot methanol (100 ml).
Slowly cooling the solution allows translucent prisms composed of pure enantiomeric (-)-cephalotaxine [α]20 D= -185°(C1,CHCl3).
After filtration, the mother liquors was allowed to slowly evaporate at room temperature and crystals in the form of macled needles exclusively composed of racemic cephalotaxine [α]D 20 = 0,5° (C1 ; CHCl3) were obtained.
After filtration, the second mother liquors allowed prisms composed of (-)-cephalotaxine identical to this obtained at the first crystallization.
After filtration, the third mother liquors still allowed macled needles (urchins) composed of (±)-cephalotaxine.
The cycle is repeated three times. The combined prismatic crystals was recrystallized once to give enantiomerically pure (-)-cephalotaxine, while the combined macled needles treated in the same way gives 100% racemic cephalotaxine.
Chemical evaluation of the enantiomeric purity of natural cephalotaxine:
-
A sample of partially racemized natural cephalotaxine was inserted in the process, which sequence is described in the Examples 1,2,3,4,5,6,15,19 and 21, by using a pure (2R)-homoharrintonic acid resulting from Example 19.
The HPLC analysis of the diastereomeric mixture of anhydro-homoharrintonine thus obtained showed a significant enantio-epi-homoharringtonine rate (11% ± 3%) corresponding to the (+)-cephalotaxine content in the racemic mixture of origin (it has been demonstrated that the two antipodes of the homoharringtonic acid react in a stoechiometric way comparable to the pure enantiomeric cephalotaxine).
Example 47Preparation of homoharringtonine, from anhydro-homoharringtonine:

1)° Method A
-
A commercial solution of hydrobromic acid in acetic acid (17.4 ml, 86.6 mmol, HBr 30% w/w) was added to a stirred solution of anhydrohomoharringtonine resulting from Example 21 (50.8 g, 9.63 mmol) in anhydrous dichloromethane (25.6 ml) at -10°C. After stirring at -10°C for 3 hours was added water (240 ml) and the reaction mixture was become viscous. The temperature was allowed to rise to room temperature and after stirring for 2.5 hours was added sodium carbonate 0.76M (406 ml) to pH 8. The resulting aqueous layer was saturated with sodium chloride, then was extracted with dichloromethane (3 × 230 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness to afford a foam. After phase reverse chromatography below-mentioned were obtained 4.03g of homoharringtonine (77%). The product thus obtained showed identical characteristics to this resulting from Example 25.
2°) Method B
-
To a stirred solution of anhydrohomoharringtonine resulting from Example 21 (214 mg, 0.406 mmol) in anhydrous dichloromethane (1.1 ml) was added at -10°C a commercial solution of hydrobromic acid in acetic acid (0.728 ml, 3.6 mmol, HBr 30% w/w). After stirring at -10°C for 3 hours, was added water (13 ml) and then the temperature was raised to 20°C. After stirring at 20°C for 3 hours, was added a sodium carbonate solution (0.76M; 31.5 ml) up to pH 8. The resulting aqueous layer, after saturation with sodium chloride, was extracted with dichloromethane (3 × 20 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by phase reverse chromatography below-mentioned to provide homoharringtonine (166 mg, 75%). The product thus obtained showed identical characteristics to this resulting from Example 25.




……………………
SEMISYNTHESIS
EXAMPLE 27 Preparation of homoharringtonine as a pharmaceutical use from crude semi-synthetic homoharringtonine resulting from example 25 by preparative high-performance liquid chromatography

1°) Method A
Crude homoharringtonine (35 g) is dissolved in buffer (triethylamine (1.55/1000) in deionised water and orthophosphoric acid to adjust pH to 3. The solution was filtered then injected on a preparative high-performance liquid chromatograph equipped with axial compression and high pressure pump (stationary phase: n-octadecylsilane, 15 μm, porosity 100, 1 kg; mobile phase; buffer/tetrahydrofurane 85/15). Elution was performed at a flow rate of 0.2 l/min. Fractions contain was monitored by U.V. detector and TLC. Retained fraction were finally checked by HPLC then combined, alkalinised with 2.5% aqueous ammonia and extracted with dichloromethane (4×400 ml). After concentration under reduced pressure homoharringtonine is obtained as a pale yellow resin which on trituration in a 8/2 water-methanol mixture gave pure homoharringtonine as a white crystalline solid (mp=127° C.), HPLC purity was higher than 99.8%.
2°) Method B
Same procedure of purification as method A was performed but mobile phase buffer/methanol (68/32) was used instead buffer/tetrahydrofurane.
3°) Method C
Same procedure of purification as method A was performed but mobile phase buffer/acetonitrile (85/15) was used instead buffer/tetrahydrofurane.
EXAMPLE 28 Preparation of homoharringtonine as a pharmaceutical use from semi-purified natural cephalotaxine
Crude homoharringtonine, prepared according to Example 25 from a partially racemized natural cephalotaxine and purified by chromatography and crystallisation according to the method A of Example 27, gave an homoharringtonine showing a non natural enantiomeric epi-homoharringtonine content less than 0.05%.
EXAMPLE 46 Preparation of purified (−) cephalotaxine from total alkaloidic extract of cephatotaxus sp

Partially racemized cephalotaxine [H. Wenkui; L. Yulin; P. Xinfu, Scientia Sinica; 23; 7; 835 (1980)]
1H NMR of two batches of cephalotaxine (extracted in the same conditions as above) with the optically active NMR shift reagent europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate (1éq) showed the following results:
Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine); 5.99 (1H, OCH2O (−)-cephalotaxine) and 5.76 (1H, OCH2O (−)-cephalotaxine). Presence of 11±5% de (+)-cephalotaxine. [α]22=−134,0°(c=0,214; CHCl3): calculated rate 25±5%
Batch B: slightly racemized (1%) [α]19=−173,3°(c=0,208; CHCl3)
Enantiomeric Enrichment of the Natural Cephalotaxine:
Crude chromatographied cephalotaxine (20 g) was dissolved at 55° C. in dry methanol (100 ml). Crystallization occurs by cooling with rotary evaporator and after filtration the product thus obtained showed 99.9% of HPLC purity, [α]20 D=−130°(C1, CHD3) corresponding to 10% of racemization. The crystallized product thus obtained (20 g) was dissolyed again in hot methanol (100 ml).
Slowly cooling the solution allows translucent prisms composed of pure enantiomeric (-−)-cephalotaxine [α]20 D=−185°(C1, CHCl3).
After filtration, the mother liquors was allowed to slowly evaporate at room temperature and crystals in the form of macled needles exclusively composed of racemic cephalotaxine [α]D 20=0,5°(C1; CHCl3) were obtained.
After filtration, the second mother liquors allowed prisms composed of (−)-cephalotaxine identical to this obtained at the first crystallization.
After filtration, the third mother liquors still allowed macled needles (urchins) composed of (±)-cephalotaxine.
The cycle is repeated three times. The combined prismatic crystals was recrystallized once to give enantiomerically pure (−)-cephalotaxine, while the combined macled needles treated in the same way gives 100% racemic cephalotaxine.
Chemical Evaluation of the Enantiomeric Purity of Natural Cephalotaxine:
A sample of partially racemized natural cephalotaxine was inserted in the process, which sequence is described in the Examples 1,2,3,4,5,6,15,19 and 21, by using a pure (2R)-homoharrintonic acid resulting from Example 19. The HPLC analysis of the diastereomeric mixture of anhydro-homoharrintonine thus obtained showed a significant enantio-epi-homoharringtonine rate (11%±3%) corresponding to the (+)-cephalotaxine content in the racemic mixture of origin (it has been demonstrated that the two antipodes of the homoharringtonic acid react in a stoechiometric way comparable to the pure enantiomeric cephalotaxine).
EXAMPLE 47
Preparation of homoharringtonine, from anhydro-homoharringtonine

1°) Method A
A commercial solution of hydrobromic acid in acetic acid (17.4 ml, 86.6 mmol, HBr 30% w/w) was added to a stirred solution of anhydrohomoharringtonine resulting from Example 21 (50.8 g, 9.63 mmol) in anhydrous dichloromethane (25.6 ml) at −10° C. After stirring at −10° C. for 3 hours was added water (240 ml) and the reaction mixture was become viscous. The temperature was allowed to rise to room temperature and after stirring for 2.5 hours was added sodium carbonate 0.76M (406 ml) to pH 8. The resulting aqueous layer was saturated with sodium chloride, then was extracted with dichloromethane (3×230 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness to afford a foam. After phase reverse chromatography below-mentioned were obtained 4.03 g of homoharringtonine (77%). The product thus obtained showed identical characteristics to this resulting from Example 25.
2°) Method B
To a stirred solution of anhydrohomoharringtonine resulting from Example 21 (21.4 mg, 0.406 mmol) in anhydrous dichloromethane (1.1 ml) was added at −10° C. a commercial solution of hydrobromic acid in acetic acid (0.728 ml, 3.6 mmol, HBr 30% w/w). After stirring at −10° C. for 3 hours, was added water (13 ml) and then the temperature was raised to 20° C. After stirring at 20° C. for 3 hours, was added a sodium carbonate solution (0.76M; 31.5 ml) up to pH 8. The resulting aqueous layer, after saturation with sodium chloride, was extracted with dichloromethane (3×20 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by phase reverse chromatography below-mentioned to provide homoharringtonine (166 mg, 75%). The product thus obtained showed identical characteristics to this resulting from Example 25.
…………………………………
EXTRACTION
The remarkable clinical efficacy of Homoharringtonine (HHT) resulting in lot of observations of complete remission of leukemia and other solid cancer in human being since 1988. Recently, research articles reported that the HHT efficacy in glaucoma, inhibition of Hepatities B virus replication and using in bone marrow transplantation. For example, the University of Texas M.D. Anderson Cancer Center and National Cancer Institute reported that “Ninety-two percent of patients achieved CHR with HHT.” [Susan O’Brien, at al.; Sequential homoharringtonine and interferon-α in the treatment of early chronic phase chronic myelogenous leukemia; Blood, Vol 93, No 12 (June 15), 1999: pp 4149-4153]. Another article reported that “the median number of days on HHT per month was 2 days with a median follow-up of 26 months; the estimated 2-year survival rate was 90%.” (Susan O’Brien, at al.; Simultaneous homoharringtonine and interferon-α in the treatment of patients with chronic-phase chronic myelogenous leukemia; American Cancer Society; Apr. 1, 2002, Vol 94, No. 7).
On Nov. 8, 1988, U.S. Pat. No. 4,783,454 titled Process for producing harringtonine and homoharringtonine disclosed the technique of isolation of a purified HHT from bark of Cephalotaxus. However, the natural source ofCephalotaxus is very limited. Trees of Cephalotaxus grow slowly. Bark ofCephalotaxus has very low content of HHT. Extracting HHT from bark ofCephalotaxus the yield was about 0.02% only. More important to harvest bark ofCephalotaxus will kill and destroy trees. Supply of HHT is very short now. Therefore, it is necessary to find a new manufacturing method.
DETAILED DESCRIPTION
Great progress has been made in research on Homoharringtonine (HHT) production and on future generation HHT drug since 1988. For example, the University of Texas M.D. Anderson Cancer Center and National Cancer Institute reported that “Ninety-two percent of patients achieved CHR with HHT.” Another article reported that “the median number of days on HHT per month was 2 days with a median follow-up of 26 months; the estimated 2-year survival rate was 90%.”
The good clinical results of HHT in treating cancer brought to the major problem, which is the supply of HHT both short term and long term. It is apparent that a huge amount of bark of Cephalotaxus is needed for collection, extraction and purification of HHT. It is clear that due to the slow growth of the trees ofCephalotaxus, which is a nature source of HHT, and the killing of trees by harvesting bark is not a sustainable resource for HHT production.
Present invention disclosed new methods for producing HHT. The new methods of producing HHT are shown as follows.
1. Tissue Culture (Plant Cell Culture):
Culture manipulation to promote secretion of HHT is a new way for an extracellular product HHT. The biosynthetic methods can yield more HHT through precursor of HHT feeding. The production of HHT increased significantly after the addition of the precursors and special biochemical agents. Content of precursor of HHT abounds in tree and it is very cheap. The present methods include several significant developments in technique of culture plant tissues that are
-
- (a) yields of HHT selected from rapid growth, resistance to infections organisms; and
- (b) HHT can excrete into media.
Traditional method of plant culture is very difficult to overcome the problem of high cost. Therefore, traditional method appears too long to have commercial value. HHT is secondary metabolite of Cephalotaxus. Secondary compound acts in defense against the harmful effects of toxins, carcinogens or mutagens found in the plant. In fact, traditional method is very difficult to increase HHT contenting in plant tissues. The present new method uses a special biochemical agent for increasing content of HHT and more easily to purify HHT from other metabolites.
More important is that the key of the present new technique for producing high content of HHT in plant cell culture is to increase production of HHT by directed fermentation through precursor of HHT feeding. The present new methods are used special metabolite of Cephalotaxus for markedly enhance production of HHT. Therefore, the present invention disclosed a new source for the long term of producing HHT.
2. Using Precursor of HHT:
Recent research’s results have established that direct production of HHT from its precursor and advances in biosynthetic understanding for HHT metabolism. Biosynthesis or semisynthesis of HHT from major nonactivity ingredients is well established through great advances in special biochemistry reactions. Using precursor of HHT for semisynthesis and increase of production in plant cell culture are new developing methods for producing HHT.
3. Using Leaves:
Our new method use leaves of tree of Cephalotaxus not use the bark. So far, the extraction of HHT is used bark. The leaves are harvested from the trees ofCephalotaxus, which grow in mountains of South China. The natural source of leaves is very abundance. The new methods do not use bark. Therefore, it can avoid destroy trees. The natural source of Cephalotaxus tree is very limited and slow growing. In fact, bark of Cephalotaxus has very low yield of HHT. The yield of HHT from Cephalotaxus bark is about 50-100 mg/kg of dried bark. The present new method, therefore, has a great economic and environmental value.
4. Semisynthesis:
HHT has received important chemical studies particularly in regard to structure and anticancer activity relationship and semisynthesis.
A great progress in biochemistry allows semisynthesis to use precursor of HHT from leaves of Cephalotaxus and to produce HHT. The total chemical synthesis of HHT appears too long to have commercial value too. Semisynthesis method can yield a high efficient conversion of precursor to HHT. It is other better biological source for manufacturing HHT. This new method uses closing chemical analogues to convert to HHT. This analogue is produced from leaves or other organ of Cephalotaxus. The present invention disclosed that new methods and techniques of manufacturing HHT could avoid chopping down Cephalotaxus trees which governmental environmentalists are trying to have declared a threatened species.
5. Using Taxol Residual
The anticancer drug Taxol is the most promising new chemotherapeutic agents that developed for cancer treatment in the past twenty years. Taxol has a unique mechanism of action. It has been shown to promote tubulin polymerization and stabilize microtubules against depolymerization. The FDA approved the clinical use of Taxol for several types of cancer. So far, annual sales of Taxol are more than $2 billion in market. Taxol is extracted from bark or leaves of an evergreen tree named Taxus species including Taxus brevifolia (or called Pacific yew). After Taxol has been extracted from bark or leaves, all residual materials of Taxus brecifolia named Taxus residual, which are waste.
Both taxol and HHT can be extracted from yew tree. The content of taxol is less than 0.01% in yew tree. The content of HHT in yew tree is about 0.01% -0.22%. The content of HHT is much higher than content of Taxol. Taxol extracted from bark of yew is difficult and expensive. One reason is that the presences of closely related congeners are similar to Taxol. A major congener is Cephalomannine (CPM), which is a waster of process in manufacturing of Taxol.
The chemical and physical characters are very close between Taxol and Cephalomannine (CPM).
CPM characterized by the same ring structure as Taxol and distinguishes from them only in C-13 ester structure. The present invention disclosed that CPM and related derivative are used to produce HHT.
The following specific examples will provide detailed illustrations of methods of producing relative drugs, according to the present invention and pharmaceutical dosage units containing demonstrates its effectiveness in treatment of cancer cells. These examples are not intended, however, to limit or restrict the scope of the invention in any way, and should not be construed as providing conditions, parameters, reagents, or
EXAMPLE 1
Production of HHT by Culture Cells
So far, HHT is extracted from bark and skins of Cephalotaxus species. However, growth of Cephalotaxus species is very slow and concentration of HHT in plant is extremely low. Furthermore, it is difficult to harvest the plants because of their low propagation rate and the danger of drastic reduced in plant availability. Also, cost of total chemical synthesis of HHT is very expensive and is not available for commerce now. For the reasons given above it is more difficult to obtainCephalotaxus on a large scale for long time. Therefore, Cephalotaxus cell cultures are one of best methods for obtaining HHT. In this present invention, special elicitation is disclosed and it will significantly increase production of HHT.
The methods of cell and tissue culture are disclosed as below.
Parts of bark, stems, leaves, or roots of Cephalotaxus species were surface disinfected by treatment in 70% ethanol for 10 minutes and followed by 0.1 HgCl2for 3 minutes. Plant materials were washed five times for 10 minutes each by sterilized water. Parts of plant were cut into small pieces (0.5-1 mm) and put pieces to Murashige and Skoog’s (MS) medium and supplemented with derivative of new active ingredient of phylum mycota (IPM), precursor of HHT which is a derivative of Cephalotaxus (CEP), tyrosine (TYR) naphthaleneacetic acid (NAA), Kinetin (3 mg/L), and 3% sucrose (w/v). PH of medium was adjusted to 5.7˜5.8. Agar (10 g/L) added to medium. Callus tissues are collected from agar media and suspension cultured cells were harvested by filtration and cultured in MS medium.
The cultures were kept in a culture room at 26° C.±1° C. Friable callus tissues were obtained. The callu was inoculated into 4 L of MS liquid medium containing sucrose, derivative of CEP, PHE, TYR, NAA and Kinetin. Then callus tissues were cultivated 26° C. for 35 days on rotary shaker operated at 120 rpm in the dark. Cells were subcultured into fresh medium of same composition every 2 weeks and maintained at 120 rpm at 26°±1° C. Packed cell volume (PCV), fresh weight (FW), dry weight (DW), concentration of HHT and concentration of sugar were determined every 5th day. The cells were harvested and dried.
In general, callus and suspension cultures of cephalotaxus species grow very slow and no production of free or esterified HHT. However, according to the present invention, addition of IPM to cultures cause a drastic increasing in HHT after 30 days of incubation. For example, in control group (no IPM), HHT in cultured cells is 0.020 mg/g dry weight, but in treatment group (addition of IPM) HHT is about 0.050 mg/g dry weight. Therefore, IPM can increase 250% of content of HHT. It has resulted in plant cell culture systems that producing HHT at concentration higher than those produced by the mother plant. The production of HHT increases significantly after the addition of precursors (CEP). Addition of CEP can increase HHT. Obviously, the present invention provided a new commercial and economic method for producing HHT. The IPM and precursors (CEP) play key role in cultured cells.
EXAMPLE 2
Semi-Synthesis of HHT
HHT shows a significant inhibitory activity against leukemia and other cancer. Concentration of HHT, however, has only 0.01% in natural sources. Cephalotazine (CEP) is major alkaloids present in plant extracts and the concentration ofCephalotaxus has about 1%. Therefore, concentration of CEP is about 100 times higher then HHT in nature plant sources. But CEP is inactive. For the reason given above, semisynthesis of HHT from CEP will increase huge natural sources of HHT.
-
- (1) Extraction of CEP
10 kg of dried stems or leaves or roots of Cephalotaxus species were milled, placed in a percolator, along 80 L of 95% of ethanol, and allowed to stand 24 hours. The ethanol was recovered under reduced pressure (below 40° C.). 20 L of 5% tartaric acid was added to concentrated ethanol solution. The ammonia water was added to the acidic solution and adjusted pH to 9. The solution of pH 9 was filtered and yielded a filtrate. The filtrate was extracted with CHCl3. CHCl3 was recovered under reduced pressure and residue was obtained. The residue was chromatographed packed with alumna and eluted by CHCl3-MeOH (9:1). Eluate was concentrated under reduced pressure. Residue was dried under vacuum. The product is CEP.
-
- (2) Semisynthesized HHT from CEP
Materials and Methods
Melting points were determined on a Fisher-Johns apparatus. Infrared spectra were obtained on a Perkin-Elmer 567 infrared spectrophotometer or on a Beckman 4230 IR spectrophotometer. Peak positions were given in cm−1. The IR spectra of solid samples were measured as potassium bromide dispersions, and the spectra of liquids were determined in chloroform or carbon tetrachloride solutions. NMR spectra were measured on a Varian A-60, Perkin-Elmer R-32, Varian EM-390, or Brüker WH-90 NMR spectrometer. Chemical-shift values were given in parts per million downfield from Me4Si as an internal standard. Mass spectra were run on an AE1 MS-12 Finnigan 3300, or CEC21-110B mass spectrometer.
Preparative thin-layer chromatography was accomplished using 750-μm layers of aluminum oxide HF-254 (type E), aluminum oxide 60 PF-254 (type E), silica gel HF-254 (type 60 PF-254), or silica gel GF-254. Visualization was by short-wave ultraviolet light. Grace silica gel, Grade 923, and Woelm neutral aluminum oxide, activity III, were used for column chromatography. Analytical thin-layer chromatography was run on plastic sheets precoated with aluminum oxide F-254 neutral (type T), 200-μm thick, and on Polygram Sil G/UV254 (silica gel), 250 μm on plastic sheets. Visualization was usually by short-wave ultraviolet light, phosphomolybdic acid, or iodoplatinate.
Preparation of α-Ketoester-Harringtonine
1 g of Benzene-α-acetone Na was put into 10 L of benzene. Mixture was stirred at room temperature then was dissolved in 10 L of pyridine and stirred at 0° C. Oxalic chloride was added from a dropping funnel to solution of pyridine. Stirring was continued while the solution warmed to room temperature and stand overnight. Excess reagent was removed. This solution was dissolved in CH2Cl2and cooled to near 0° C. in an ice water bath. 5 g of CEP, 2.5 L of CH2Cl2 and 2.5 L of pyridine were added to cold CH2Cl2 solution. Manipulations were done in a dry N2 atmosphere and all glassware heat-dried just before use. The suspension was stirred at room temperature and overnight. The mixture was washed with 10% Na2CO3 and saturated aqueous NaCl, then dried with auhydrous magenesium sulfate, and filtered and the solvents were removed in vacuo. Evaporation provided as an amorphous solid α-ketoester-harringtonine (mp 143˜145° C.).
Semi-Synthesis of HHT
10 L of CH3CHBrCOOEt and activated zin dust and THF were added to the α-ketoester-harringtonine (at −78° C.) for 6 hours followed by slow warming to room temperature with stirred. The reaction mixture was diluted with 10 L CHCl3 and 10 L H2O and solid Na2CO3 was added. CHCl3 was evaporated under reduced pressure and residue was obtained.
The residue was purified by chromatography on alumina. The column was flushed with chloroform and followed by chloroform-methanol (9:1). The solvents were recovered under reduced pressure to provide as a solid. Solid was dissolved in pure ethanol and crystallized. The crystals were refined by recrystalization in diethyl ether. The crystals dried under vacuum. The product is HHT, which has the following characters:
[α]D −119° (C=0.96),
MSm/e (%): 689 (M+, 3), 314 (3), 299 (20), 298 (100), 282 (3), 266 (4), 20 (3), 150 (8), 131 (12), 73 (18)
EXAMPLE 3
HHT Extracted from Plant Tissue
Extraction of HHT has several major methods which including extraction by organic solvent, chromatograph and adjust pH.
HHT was extracted from plant tissue culture, plant cells or leaves of Cephalotaxusspecies.
1 kg of ground Cephalotaxus fortunei Hook was extracted with 10 liters of water at room temperature for 24 hrs. To filtered the solution to yield a filtrate. Ten liters of 90% ethanol added to filtrate. The mixture was Centrifugalized to yield a sediment. Percolated the sediment with ethanol and filter again to yield filtrate, combined filtrates, and distilled under reduced pressure to recover ethanol and an aqueous residue. To this residue, added 10% of HCl to adjust the pH to 2.5. To separated the solids from the solution by filtration to yield a filtrate (1). Washed the solids once with 2% HCl and filtered to yield a filtrate (2). Combined (1) and (2) and adjusted the pH to 9.5 by adding saturated sodium carbonate solution. Extracted the alkaline filtrate with chloroform and separated the chloroform layer from the aqueous layer. To repeated this extraction process five times. Combined all the chloroform extracts and distilled at reduced pressure to recover chloroform and alkaloid as a solid residue obtained. The solid alkaloid was then dissolved in 6% citric acid in water. The solution was divided into three equal portions. These were adjusted to pH 7, 8 and 9 by adding saturated sodium carbonate solution. The portions having pH 8 and 9 were combined and extracted with chloroform. The chloroform extracts were distilled under reduced pressure, whereby chloroform was removed and recovered and crude HHT was obtained. The crude HHT was dissolved in pure ethanol and crystallized. The crystals were refined by recrystallization in diethyl ether. The crude HHT obtained.
The portion having a pH of 7 passed through a liquid chromatographic column packed with alumina of diameter to height 1:50. The column was finally flushed with chloroform and followed by chloroform-methanol of 9:1 mixture. The resulting alkaloids were mixture crude of HHT. Combined crude HHT and then separated from each other by countercurrent distribution employing chloroform and pH 5 buffers. The first fraction of the countercurrent distribution was HHT. HHT was purified by crystallization in methyl alcohol. The crystallization was purified by recrystallization in methyl alcohol and dried under vacuum.
…………………….
Example 1 : Preparation of harringtonine drug substance by purification of commercial natural harringtonine
A. Analytical profile of starting product
By combination of HPLC analysis with UV detection (see Figure 6) and mass spectrometry detection (see figure 7 and 8) a total of 6.5% of related compound (identified as b,c: position isomer of harringtonine = 3.4%; d: homoharringtonine = 3%; e: 4′-demethyl harringtonine = 0.01%; f: drupacine derivative: 0.05%) are found in the starting product.
B. Chromatography of natural harringtonine
Natural harringtonine (5 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 80 mm; length: 1000 mm) containing 1000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi). Unwanted fractions are discarded based upon in-line UV spectrophotometric detection. Kept fractions are collected in 16 separate containers which each are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase). During the development phase, a dual in-line UV-MS detection is used. After discarding of the fractions representing more than 0.5 % of the total content of harringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure. Then crude concentrated solution of harringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 1.5 %. C. Crystallization of raw harringtonine
Under a laminar flow hood, the above raw harringtonine (4.1 grams) is dissolved in methanol (5ml), at 30°C. The resulting alcoholic solution was filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized rotary flask. Then, desionized water (50mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of harringtonine is kept under vacuum and rotation is continued during appearance of white crystals of pure harringtonine. The stirring is continued until no more crystal occurs. Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water (10 mL x 2). The white translucent crystals are then dried using high vacuum at 40°C for 24 hours. Overall yield is 76%. All operations were documented prior to start the process and full current Good Manufacturing Practices were applied. This clinical batch corresponds to 400 therapeutic units dosed at 10mg.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 7 and 9 compare HPLC chromatogram before and after purification in using this process. Table II shows the comparison of the corresponding related compound content.

For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC) thermogravimetry, 2D NMR, solid NMR and X-ray powder diffractometry.
Infrared Spectrometry:
Identical IR spectra were obtained by either the KBr pellet and/or mineral oil mull preparation technique. Figure 5 shows typical infrared spectrum (KBr) for unambiguous identification at the solid state of the crystalline harringtonine obtained by this process. A series of sharp absorption bands are noted at 615, 654, 674, 689, 709, 722, 750, 761 805, 850, 928, 989, 1022, 1033, 1062, 1083, 1112, 1162, 1205, 1224, 1262, 1277, 1308, 1340, 1364, 1382, 1438 1486, 1508, 1625, 1656, 1725, 1745, 2883, 2936, 2972, 3079, 3353, 3552 and 3647 cm“1
Differential Scanning Calorimetry (DSC) And Thermogravimetry (TG) Measurement of DSC and TG were obtained on a Mettler Toledo STAR System. Approximately 12 mg of harringtonine drug substance were accurately weighed (12.4471 mg) into a DSC pan. The sample was heated from 25°C to 200°C at a rate of 10°C/min. The DSC data were obtained following a standard method in the art. The DSC curve of crystalline harringtonine drug substance ((Figure 4), exhibits a melting endotherm at 79.5 °C . No subsequent decomposition occurred under the upper tested temperature 200°C. Simultaneous TG measurement, indicated a loss on drying of 1.3 % which did not correspond to a lost of structural molecule of solvent or water.
Example 2: Preparation of homoharringtonine drug substance by purification of raw semi- synthetic (hemi-synthetic) homoharringtonine
A. Analytical profile of starting product
Crude reaction mixture of raw homoharringtonine contains a potential of 250 grams of homoharringtonine DS together with process impurities such as catalyst, unchanged starting product (anhydro-homo-harringtonine), and some related side product. HPLC analysis with UV detection (see left-side chromatogram on Figure 10) indicated a total of 9 % of related impurities. B. Chromatography of semi-synthetic homoharringtonine
Raw semi-synthetic homoharringtonine (550 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 450 mm; length: 1000 mm) containing 48,000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi, flow-rate 540 L/hour). Unwanted fractions are discarded based upon by- passed in-line UV spectrophotometric detector. Kept fractions are collected in 30 separate stainless steel containers (20 or 50 L each) which are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase) and equipped with a diode array detector. After discarding of the fractions representing more than 0.5 % of the total content of homoharringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure in using a mechanically stirred thin film evaporator. Then crude concentrated solution of homoharringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 0.5 % (see rigth-side chromatogram on Figure 10)
C. Crystallization of homoharringtonine DS
In a controlled clean room, under a laminar flow hood, the above raw homoharringtonine DS (210 grams) is dissolved in methanol (240 mL), at 30°C. The resulting alcoholic solution is filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized pilot rotary flask. Then, desionized water (2400mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated pilot rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of homoharringtonine DS is kept under vacuum and rotation is continued during appearance of white crystals of pure homoharringtonine. The stirring is continued until no more crystal occurs. Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water (450 mL x 2). The white cryitals are then dried using high vacuum at 60°C for 48 hours. Overall yield is 88% from potential content of homoharringtonine in raw semi-synthetic homoharringtonine. All operations were documented prior to start the process and full current Good Manufacturing Practices were applied. This clinical batch corresponds to 40,000 therapeutic units dosed at 5mg.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 11 shows HPLC chromatogram before and after crystallization. Total of related impurities of homoharringtonine DS is 0.03%.
For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC), thermogravimetry (TD), 2D NMR, solid NMR and X-ray powder diffractometry.
Infrared Spectrometry:
Identical IR spectra were obtained by either the KBr pellet and/or mineral oil mull preparation technique. Figure 3 shows typical infrared spectrum (KBr) for unambiguous identification at the solid state of the crystalline homoharringtonine obtained by this process. A series of sharp absorption bands are noted at 612, 703, 771 , 804, 826, 855, 879, 932, 1029, 1082, 1119,
1135, 1161 , 1191 , 1229, 1274, 1344, 1367, 1436, 1457, 1488, 1505, 1653, 1743, 2814, 2911 ,
2958, 3420, and 3552 cm“1
Differential Scanning Calorimetry (DSC) And Thermogravimetry (TG)
Measurement of DSC and TG were obtained on a Mettler Toledo STAR System. Approximately 11 mg of homoharringtonine drug substance were accurately weighed (10.6251 mg) into a DSC pan. The sample was heated from 25°C to 250°C at a rate of 5°C/min. The
DSC data were obtained following a standard method in the art. The DSC curve of crystalline homoharringtonine drug substance (Figure 1), exhibits a melting endotherm at 145.6 °C.
Melting range performed by the capillary method (Bucchi Apparatus) gave 143-145°C. Literature indicated 144-146°C [Anonymous, Acta Bot. Sin. 22, 156 (1980) cited by L. Huang and Z. Xue, Cephalotaxus Alkaloids, in “The Alkaloids”, vol. XXIII, pp157, (1988).
Crystallization medium was not published. This is the only literature reference regarding melting point of a crystalline form of HHT] X-Ray Powder Diffraction
X-ray powder diffraction pattern was collected on a INEL microdiffractomer, model
DIFFRACTINEL. Powdered homoharringtonine DS was packed in a glass capillary tube and was analyzed according to a standard method in the art. The X-ray generator was opered at 45 kV and 40 mA, using the copper Kalpha line as the radiation source. The sample was rotated along the chi axis and data was collected between 0 and 120 deg 2-theta. A collection time of 1200 sec was used. As showed on Figure 2, the x-ray powder diffraction for this crystalline form of homoharringtonine shows a typical pattern including major reflection peaks at approximately 7.9, 9.2, 10.9, 14.9 16.0, 17.7, 19.5, 19.7, 21.78, 23.1 , 25.3, 25.4 and 25.7 deg 2-theta.
Example 3: Preparation of homoharringtonine drug substance by purification of a commercial sample of impure homoharringtonine from Chinese source
A. Analytical profile of starting product
Analytical HPLC chromatogram of natural homoharringtonine (China National Pharmaceutical) is displayed on Figure 12 (bottom left).
B. Chromatography of Natural Homoharringtonine
Natural homoharringtonine (25 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 200 mm; length: 1000 mm) containing 12,000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi, flow-rate 120 IJhour). Unwanted fractions are discarded based upon bypassed in-line UV spectrophotometric detector. Kept fractions are collected in 22 separate stainless steel containers which are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase) and equipped with a diode array detector. After discarding of the fractions representing more than 0.5 % of the total content of homoharringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure in using a mechanically stirred thin film evaporator. Then crude concentrated solution of homoharringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 0.5 %.
C. Crystallization of homoharringtonine DS
In a controlled clean room, under a laminar flow hood, the above chromatographied homoharringtonine DS (18 grams) is dissolved in methanol (35 mL), at 30°C. The resulting alcoholic solution is filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized pilot rotary flask. Then, desionized water (300 mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated pilot rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of homoharringtonine DS is kept under vacuum and rotation is continued during appearance of white crystals of pure homoharringtonine. The stirring is continued until no more crystal occurs.
Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water
(50 mL x 2). The white crystals are then dried using high vacuum at 60°C for 48 hours. Overall yield is 84% from potential content of homoharringtonine in raw semi-synthetic homoharringtonine. All operations were documented prior to start the process and full current
Good Manufacturing Practices were applied.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 12 (bottom right) shows HPLC chromatogram after crystallization. Total of related impurities of homoharringtonine DS is 0.05%.
For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC), thermogravimetry (TD), 2D NMR, solid NMR and X-ray powder diffractometry. Infrared Spectra, Differential Scanning Calorimetry (DSC) and X-Ray Powder Diffraction gave patterns strictly superimposable to the one of example 2 obtained from semi-synthetic homoharringtonine (Figure 3, 1 , and 2, respectively).
………………………………….
KOREAN PAPER.. LINK
Title: 한국산 개비자(Cephalotaxus koreans)에서의 Harringtonine과 Homoharringtonine의 확인 및 함량 분석
Author: 박호일 ; 이연 (한국생물공학회)
Source: 한국생물공학회지 = Korean journal of biotechnology and bioengineering; ISSN:1225-7117 @ 1225-7117 @ ; VOL.11; NO.6; PAGE.689-695; (1996)
Pub.Country: Korea
Language: Korean
Abstract: Harringtonine and homoharringtonine known as anti-cancer agents were isolated from Korean native plumyew(Cephalotaxus koreana) using column chromatography(CHCl3:MeOH=19:1, Rf=0.28). The structure of the mixture of two compounds was characterized by 1H-NMR. Comparison of our spectra of harringtonine and homoharringtonine with previously reported ones indicated that the two are identical. The contents of harringtonine and homoharringtonine in the needles, stems, and roots of Korean native plumyew were determined by high performance liquid chromatography(HPLC). The contents of both compounds varied with the site of location and the part of plant. The content of harringtonine was higher in needles and roots than in stems, whereas the content of homoharringtonlne was lower than harringtonine. Homoharringtonine contents in needles at Mt. Palgong, Mt. Dukyu, Mt. Baekyang, Mt. Jiri, and Namhae were higher than in stems and roots. But homoharringtonine contents in needles al Mt. Jokye and Jindo were lower than in stems and roots.
http://img.kisti.re.kr/originalView/originalView.jsp
……………………………………………………………………………….
SYNTHESIS OF HOMOHARRINGTONINE AND SEPARATION OF ITS STEREOMERS
-
[PDF]
Chapter 1 Drug Discovery from Plants – Springer
LC-NMR-MS and LC-SPE-NMR to accelerate their future discovery. Keywords …..Ceflatonine (34), a synthetic version of homoharringtonine produced by.
…………………………………………………………………………….
References
- “Synribo (omacetaxine) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 18 February 2014.
- “SYNRIBO (omacetaxine mepesuccinate) injection, powder, lyophilized, for solution [Cephalon, Inc.]”. DailyMed. Cephalon, Inc. October 2012. Retrieved 18 February 2014.
- Sweetman, S, ed. (14 November 2012). Omacetaxine Mepesuccinate. “Martindale: The Complete Drug Reference”. Medicines Complete(Pharmaceutical Press).
- Li, Y. F.; Deng, Z. K.; Xuan, H. B.; Zhu, J. B.; Ding, B. H.; Liu, X. N.; Chen, B. A. (2009). “Prolonged chronic phase in chronic myelogenous leukemia after homoharringtonine therapy”. Chinese medical journal122 (12): 1413–1417. PMID 19567163. edit
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J. (2009). “Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009”. Cancer 115 (23): 5382–5393.doi:10.1002/cncr.24601. PMID 19739234. edit
- Wu, L.; Li, X.; Su, J.; Chang, C.; He, Q.; Zhang, X.; Xu, L.; Song, L.; Pu, Q. (2009). “Effect of low-dose cytarabine, homoharringtonine and granulocyte colony-stimulating factor priming regimen on patients with advanced myelodysplastic syndrome or acute myeloid leukemia transformed from myelodysplastic syndrome”. Leukemia & Lymphoma50 (9): 1461. doi:10.1080/10428190903096719. edit
- Gu, L. F.; Zhang, W. G.; Wang, F. X.; Cao, X. M.; Chen, Y. X.; He, A. L.; Liu, J.; Ma, X. R. (2010). “Low dose of homoharringtonine and cytarabine combined with granulocyte colony-stimulating factor priming on the outcome of relapsed or refractory acute myeloid leukemia”.Journal of Cancer Research and Clinical Oncology 137 (6): 997–1003.doi:10.1007/s00432-010-0947-z. PMID 21152934. edit
- Kantarjian, H. M.; Talpaz, M.; Santini, V.; Murgo, A.; Cheson, B.; O’Brien, S. M. (2001). “Homoharringtonine”. Cancer 92 (6): 1591–1605.doi:10.1002/1097-0142(20010915)92:6<1591::AID-CNCR1485>3.0.CO;2-U. PMID 11745238. edit
- Wetzler M, Segal D. Omacetaxine as an Anticancer Therapeutic: What is Old is New Again. Current Pharmaceutical Design 2011;17:59-64
- Concise total synthesis of (±)-cephalotaxine via a transannulation strategy: Development of a facile reductive oxy-nazarov cyclization
Org Lett 2011, 13(13): 3538 - The first semi-synthesis of enantiopure homoharringtonine via anhydrohomoharringtonine from a preformed chiral acyl moiety
Tetrahedron Lett 1999, 40: 2931 - Synthesis of homoharringtonine and its derivative by partial esterification of cephalotaxine
Tetrahedron Lett 1982, 23(34): 3431 - Construction of chiral tertiary alcohol stereocenters via the (2,3)-Meisenheimer rearrangement: Enantioselective synthesis of the side-chain acids of homoharringtonine and harringtonine
J Org Chem 2013, 78(2): 339 - Studies in Cephalotaxus alkaloids. Stereospecific total synthesis of homoharringtonine
J Org Chem 1983, 48(26): 5321 - Chemistry – A European Journal, 2008 , vol. 14, 14 pg. 4293 – 4306
| WO2000040269A2 * | Jan 5, 2000 | Jul 13, 2000 | Clarence C Lee | Pharmaceutical compositions for treatment of diseased tissues |
| WO2002032904A1 * | Oct 17, 2000 | Apr 25, 2002 | Oncopharm Corp | New cephalotaxanes, their method of preparation and their use in treatment of cancers, leukemias, parasites including thus resistant to usual chemotherapeutic agents and as reversal agents |
| EP0393575A1 * | Apr 17, 1990 | Oct 24, 1990 | G.D. Searle & Co. | Neoplasia treatment compositions containing antineoplastic agent and side-effect reducing protective agent |
| USH271 * | Dec 18, 1985 | May 5, 1987 | The United States Of America As Represented By The Secretary Of The Army | Treatment of malaria with esters of cephalotaxine |
| US7169774 | Jun 25, 2004 | Jan 30, 2007 | Stragen Pharma S.A. | Cephalotaxane derivatives and their processes of preparation and purification |
| US7842687 | May 25, 2006 | Nov 30, 2010 | Chemgenex Pharmaceuticals, Inc. | Cephalotaxane derivatives and their processes of preparation and purification |
| US8466142 | Mar 3, 2009 | Jun 18, 2013 | Sloan-Kettering Institute For Cancer Research | Cephalotaxus esters, methods of synthesis, and uses thereof |
| Reference | ||
|---|---|---|
| 1 | * | KANTARJIAN H.M. ET AL: “Chronic myelogenous leukemia – Progress at the M. D. Anderson Cancer Center over the past two decades and future directions: First Emil J Freireich Award Lecture.” CLINICAL CANCER RESEARCH, (1997) 3/12 II (2723-2733). , XP001095529 |
| 2 | * | LEVY, VINCENT (1) ET AL: “Subcutaneous homoharringtonine (SQ HHT ): 1. Pharmacokinetic study in dogs and HHT determination in blood in using LC-MS method.” BLOOD, (NOVEMBER 16, 2001) VOL. 98, NO. 11 PART 2, PP. 179B. HTTP://WWW.BLOODJOURNAL.ORG/. PRINT. MEETING INFO.: 43RD ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY, PART 2 ORLANDO, FLORIDA, USA DECEMBER 07-11, 2001 , XP001095449 |
| 3 | * | LEVY, VINCENT (1) ET AL: “Subcutaneous homoharringtonine (SQ HHT ): 2. Tolerance in humans and case report of a refractory patient with AML treated by very small dose of SQ HHT.” BLOOD, (NOVEMBER 16, 2001) VOL. 98, NO. 11 PART 2, PP. 202B. HTTP://WWW.BLOODJOURNAL.ORG/. PRINT. MEETING INFO.: 43RD ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY, PART 2 ORLANDO, FLORIDA, USA DECEMBER 07-11, 2001 , XP001095450 |
| 4 | * | WHAUN J M ET AL: “TREATMENT OF CHLOROQUINE -RESISTANT MALARIA WITH ESTERS OF CEPHALOTAXINE HOMOHARRINGTONINE.” ANN TROP MED PARASITOL(1990) 84(3), 229-237, XP008006193 |
1H NMR

13 CNMR

HPLC



Reblogged this on DRUG REGULATORY AFFAIRS INTERNATIONAL.
LikeLike