
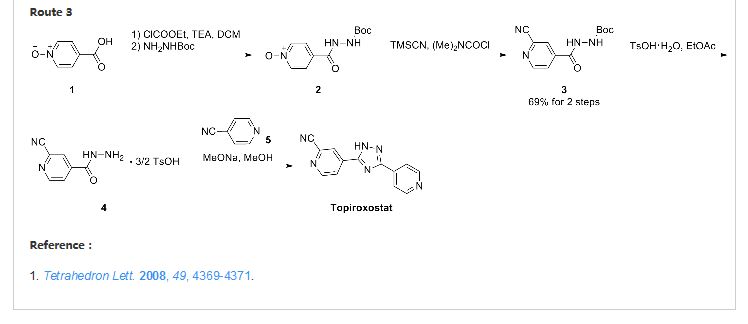
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


Topiroxostat
托匹司他
FUJI YAKUHIN ……..INNOVATOR
Approved in japan PMDA JUNE 28 2013
Xanthine oxidase inhibitor
FOR GOUT AND HYPERURICEMIA
Launched – 2013, Fuji YakuhinSanwa, Topiloric Uriadec
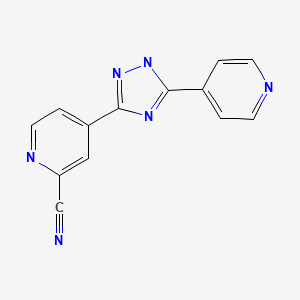
IUPAC Name: 4-(5-pyridin-4-yl-1H-1,2,4-triazol-3-yl)pyridine-2-carbonitrile
CAS Registry Number: 577778-58-6
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1)
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
3-(3-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole
Synonyms: 4-(5-PYRIDIN-4-YL-1H-1,2,4-TRIAZOL-3-YL)PYRIDINE-2-CARBONITRILE,
AC1NRB9T, Topiroxostat (JAN/INN), DB01685, D09786, FYX-051
SK-0910
4-[5-PYRIDIN-4-YL-1H-[1,2,4]TRIAZOL-3-YL]-PYRIDINE-2-CARBONITRILE,
C13H8N6 MF,248.2482 MW
 TOPIROXOSTAT
TOPIROXOSTAT
托匹司他
A xanthine oxidase inhibitor used to treat gout and hyperuricemia.

PATENT EXP 3/12/22, US /EU/CN

FYX-051, TOPIROXOSTAT is a xanthine oxidase inhibitor. This agent was approved in Japan by Fuji Yakuhin and Sanwa for the treatment of gout and hyperuricemia in 2013 and launched at the same year. In 2009, the compound was licensed to Sanwa by Fuji Yakuhin in Japan for the codevelopment and commercialization of gout.
The number of patients with hyperuricemia in Japan is reported to be 1.25 million and the number suffering from asymptomatic hyperuricemia is estimated to reach several millions. Hyperuricemia is becoming a popular disease.
Presently, hyperuricemia and gout due to hyperuricemia are treated by improving the living environment and administering various drug therapies for each period when an attack of gout is predicted to occur (presymptomatic period), when an attack of gout occurs, or when an attack of gout subsides. That is, preventive therapy is conducted in the presymptomatic period by administering colchicines as well as controlling the daily living environment. When an attack occurs, drug therapy using non-steroidal or steroidal anti-inflammatory agents is mainly conducted. After the attack subsides, patients are given guidance to improve their lifestyle. When improvement is judged insufficient, an assessment is made as to whether hyperuricemia is caused by reduced excretion of uric acid or by increased production of uric acid followed by treatment with drugs, which exhibit a uricosuric effect, such as probenecid and benzbromarone, those which inhibit resorption of uric acid, such as sulfinpyrazone, those which improve acidurea conditions, such as citrates, and xanthine oxidase inhibitors which inhibit production of uric acid, such as allopurinol. Colchicine is said to be able to prevent about 90% of attacks through inhibiting chemotaxis and phagocytosis of leukocytes, such as neutrophils, if administration thereof has been completed within a few hours before the attack. Since colchicine has various adverse effects, however, the use thereof is limited to the minimum and it is therefore difficult to timely administer it.
Accordingly, drug therapies are mainly adopted, but only allopurinol is available for the treatment of a disease caused by increased production of uric acid. However, a metabolite of allopurinol, oxypurinol, tends to accumulate and may cause calculi formation. Furthermore, this drug has been reported to induce adverse events such as rash, a decreased renal function and hepatitis, and it is not easy to administer.
Examples of compounds having xanthine oxidase inhibiting activity that can be used for treating gout caused by increased production of uric acid and that are effective for hyperuricemia and gout due to hyperuricemia have been described in J. Medicinal Chemistry, 1975, Vol. 18, No. 9, pp. 895–900, Japanese Patent Publication No. 49-46622 and Japanese Patent Publication No. 50-24315, which disclose some 1,3,5-substituted or 3,5-substituted 1,2,4-triazole compounds.
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1) has a xanthine oxidase inhibitory activity and serum uric acid level known as the agent that reduces (Patent Document 1).

The method for producing the compound (1), for example, 2 by Reissert Henze reaction isonicotinic acid methyl N-oxide – is a cyano isonicotinate, and the hydrazide which is then, 4 – this condensed cyanopyridine After obtaining a hydrazide of isonicotinic acid N-oxide (Patent Document 1, Example 12) and method, a cyano group after introduction, 4 by Reissert Henze reaction – method of condensing a cyano pyridine is known (Patent Document 1, Example 39).Further, 4 – as a starting material cyano-N-oxide, a triazole ring after construction (Patent Document 3), Reissert Henze unprotected or (Patent Document 2) to protect the ring condensed with isonicotinic acid hydrazide method of obtaining the compound (1) by introducing a cyano group by the reaction have also been reported.
The crystalline polymorph, yet the same molecule with the same chemical composition, the molecular arrangement in the crystal are different, and are different crystalline states. The pharmaceutical compounds having crystal polymorphism such the differences in physicochemical properties, affect pharmacological activity, solubility, bioavailability, stability and the like are known.Therefore, when the crystal polymorphism is present in a pharmaceutically useful compound, producing compounds of the crystalline form highly useful from polymorphs thereof is desirable.
WO 2003/064410 discloses WO 2005/009991 discloses Japanese Patent Publication No. 2005-41802
However, 4 of the above Patent Document – no description about the presence of crystalline polymorph on carbonitrile – pyridine-2-[yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol] It has not been, to these manufacturing methods, it is disclosed a method for the purpose of improving the chemical purity and yield, there is no description of the crystallographic plane.
Method of producing topiroxostat, useful for preventing or treating gout; and its intermediates. Picks up from WO2012060308, claiming the use of this topiroxostat for treating renal dysfunction. Along with the concurrently published WO2014017515, claiming crystalline Forms I and II of this compound, which, Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017516
Crystalline Forms I and II of topiroxostat, useful for preventing or treating gout. Along with the concurrently published WO2014017516, claiming a method of producing this compound. Picks up from WO2012060308, claiming a method of treating renal dysfunction using topiroxostat, which Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017515
novel 1,2,4-triazole compounds having an optionally substituted 2-cyanopyridin-4-yl group at 3-position and an optionally substituted aromatic group at 5-position inhibit a xanthine oxidase and are useful for treatment of gout and hyperuricemia, and have previously filed a patent application (Patent Document 1). The compounds can be prepared according to a method shown by the following reaction scheme:
-
 wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
Patent Document 1: JP-A-2002-017825 -
-
A compound represented by formula (1) which is a starting material may be prepared by a method described in, for example, JP-A-47-7120, JP-A-61-152661A, JP-A-62-149673, JP-A-2002-528447, or European Patent Application No. 559363 specification. However, it is preferable to prepare compound (1) according to the following reaction scheme:
-

-


SYNTHESIS



PATENT
- Example 2
-
To the toluene solution obtained in Example 1 (2) was added 2-propanol (700 mL), and the mixture was stirred. To the resulting solution was added p-toluenesulfonic acid monohydrate (151.16 g) and the resulting mixture was stirred for 8 hours at an internal temperature of 80°C. The mixture was brought to room temperature, and the precipitated crystals were taken out and washed with 2-propanol (210 mL×2). The white crystals were dried under reduced pressure at 60°C for 15 hours to give 106.0 g of the captioned compound as white crystals. Subsequently, 90.0 g of the crystals was suspended in a mixture of 2-butanol (49 mL) and water (491 mL) and heated to an internal temperature of 80°C for 1 hour. The internal temperature was brought to room temperature, and the crystals were filtered and washed with a mixture of 2-butanol and water (1:10) (270 mL×3). The resulting crystals were dried under reduced pressure at 60°C for 15 hours to give 75.7 g of the captioned compound in a high purity.
-
1H―NMR(DMSO-d6)δppm:2.29(s,3H), 7.11 (m,2H), 7.48 (dd, 2H, J=6.48, 1.62Hz) , 8.32-8.35(m, 3H) , 8.57(dd, 1H, J=1.62, 0.81Hz) , 8.94-8.98(m, 3H)
- Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonate
Example 3
Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
-
To the white crystals (50.5g) obtained in Example 2 was added 2-propanol (937.5 mL) and water (312.5 mL), and the resulting mixture was heated and dissolved at an internal temperature of 80°C. Immediately thereafter, the solution was filtered and the filtrate was cooled to an internal temperature of 20°C. To the resulting suspension was added dropwise 0.52 mol/l of an aqueous sodium hydrogen carbonate solution (250 mL), and the mixture was stirred at room temperature for 2 hours. Then the crystals were filtered and washed with water (150 mL×3) and 2-butanol (150 mL×2). The crystals were dried under reduced pressure at 80°C for 15 hours to give 29.4 g of the captioned compound as pale yellow crystals.
-
1H―NMR(DMSO-d6)δppm:8.02(dd, 2H, J=4.59, 1.62Hz),8.32(dd, 1H, J=5.13, 1.62Hz), 8.55(dd, 1H, J=1.62, 1.08Hz), 8.80(dd, 2H, J=4.59, 1.62Hz), 8.93 (dd, 1H, J=5.13, 1.08Hz)
SYNTHESIS
Example 12
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of methyl isonicotinate N-oxide
13.9 g of isonicotinic acid N-oxide was added to 209 ml of methylene chloride, 29.7 g of 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline was further added thereto, and the mixture was stirred under argon atmosphere at room temperature for one hour. 32.1 g of methanol was added to this mixture, which was stirred at room temperature for 17 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (3:1) was used as an eluent to yield 11.1 g of a white powder.
1H-NMR (CDCl3) δppm: 3.95 (3H, s), 7.88 (2H, d, J=7.25 Hz), 8.22 (2H, J=7.25 Hz)
2) Production of Methyl 2-cyanoisonicotinate
11.1 g of the crystal obtained in 1) was dissolved in 170 ml of acetonitrile, 14.6 g of triethylamine and 21.5 g of trimethylsilylnitrile were added thereto, and the mixture was refluxed under argon atmosphere for 16 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (95:5) was used as an eluent to yield 8.44 g of a pale yellow powder.
1H-NMR (CDCl3) δppm: 4.01 (3H, s), 8.08 (1H, d, J=5.45 Hz), 8.24 (1H, s), 8.90 (1H, d, J=5.45 Hz)
3) Production of 2-cyanoisonicotinic acid hydrazide
8.44 g of the crystal obtained in 2) was added to 85 ml of methanol, 1.84 g of hydrazine was further added thereto, and the mixture was stirred under argon temperature for 2 hours. After the solvent was evaporated under reduced pressure, chloroform was added to the residue, which was stirred at room temperature for one hour. The precipitated crystal was filtered, washed with chloroform and dried with a vacuum pump to yield 4.15 g of a pale yellow powder.
1H-NMR (DMSO-d6) δppm: 4.72 (2H, s), 8.05 (1H, d, J=5.12 Hz), 8.31 (1H, s),8.90 (1H, d, J=5.12 Hz), 10.23 (1H, s)
4) Production of the Object Compound
2.67 g of 4-cyanopyridine was dissolved in 40 ml of methanol, 0.83 g of sodium methoxide was added thereto, and the mixture was stirred at room temperature for one hour. Then 4.15 g of the crystal obtained in 3) was added and the mixture was refluxed for 37 hours. After the reaction completed, the precipitated solid was filtered, washed with methanol and dried with a vacuum pump to yield 3.66 g of the object compound as a yellow powder.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54, 1.57 Hz), 8.31 (1H, dd, J=5.11, 1.65 Hz), 8.53 (1H, dd, J=1.65, 0.50 Hz), 8.80 (2H, dd, J=4.54, 1.57 Hz), 8.93 (1H, dd, J=5.11, 0.50 Hz)
Example 39
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of isonicotinic acid (N-2-tert-butoxycarbonyl)hydrazide-1-oxide
585 ml of methylene chloride was added to 39.0 g of isonicotinic acid N-oxide, and after 34.0 g of triethylamine was further added thereto, the mixture was cooled under argon atmosphere to −15° C. 33.5 g of ethyl chlorocarbonate in 117 ml of methylene chloride was added dropwise to this mixture, which was stirred at a temperature from −5 to −10° C. for one hour. Then 44.4 g of tert-butyl ester of carbamic acid in 117 ml of methylene chloride was added dropwise to this mixture and it was allowed to slowly rise to room temperature while it was stirred. The precipitated solid was filtered after 15 hours, washed with methylene chloride, and dried with a vacuum pump to yield 49.7 g of white crystal.
1H-NMR (DMSO-d6) δppm: 1.42 (9H, s), 7.82 (2H, d, J=7.09 Hz), 8.33 (2H, d, J=7.09 Hz), 9.02 (1H, s), 10.44 (1H, s)
Production of 2-cyanoisonicotinic acid hydrazine 1½ P-Toluenesulfonic acid salt
228 ml of dioxane was added to 30.4 g of the crystal obtained in 1), and after 13.1 g of trimethylsilyl cyanide and 38.8 g of N,N-dimethylcarbamoyl chloride were further added thereto, the mixture was stirred under argon atmosphere at 60° C. for 5 hours. After the solvent was evaporated under reduced pressure, the residue was dissolved in ethyl acetate and subsequently washed with 1.5 M sodium carbonate aqueous solution and a saturated saline solution and dried over magnesium sulfate. After the magnesium sulfate was filtered off, the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue, 68.5 g of p-toluenesulfonic acid monohydrate was added thereto, and the mixture was stirred at room temperature for 22 hours. The precipitated crystal was filtered, washed with ethyl acetate, and dried with a vacuum pump to yield 40.3 g of white crystal 2).
1H-NMR (DMSO-d6) δppm: 2.28 (4.5H, s), 7.12 (3H, dd, J=7.92 & 0.66 Hz), 7.48 (3H, dd, J=7.92 & 0.66 Hz), 8.10 (1H, dd, J=5.11 & 1.81 Hz), 8.39 (1H, dd, J=1.81 & 0.33 Hz), 8.99 (1H, dd, J=5.11 & 0.33 Hz)
3) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
9.98 g of 4-cyanopyridine was dissolved in 250 ml of methanol, and after 7.77 g of sodium methoxide was added thereto, the mixture was stirred at room temperature for one hour. Then 40.3 g of the crystal obtained in 2) was added and the mixture was refluxed for 24 hours. After the reaction completed, the precipitated crystal was filtered, washed with methanol, and dried with a vacuum pump to yield 16.3 g of yellow crystal.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54 & 1.57 Hz), 8.31 (1H, dd, J=5.11 & 1.65 Hz), 8.53 (1H, dd, J=1.65 & 0.50 Hz), 8.80 (2H, dd, J=4.54 & 1.57 Hz), 8.93 (1H, dd, J=5.11 & 0.50 Hz)
4) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
45 ml of ethanol and 15 ml of 1-methyl-2-pyrrolidone were added to 3.0 g of the crystal obtained in 3), and the mixture was heated and stirred at 80° C. for 19 hours. The crystal was filtered, subsequently washed with a mixture of ethanol and 1-methyl-2-pyrrolidone (3:1) and ethanol, and dried with a vacuum pump to yield 2.71 g of yellow crystal.
5) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonic acid salt
5 ml of ethanol and 30 ml of water were added to 2.48 g of the crystal obtained in 4), and after 3.8 g of p-toluenesulfonic acid monohydrate was further added thereto, the mixture was stirred at room temperature for 5 hours. The precipitated crystal was filtered, subsequently washed with a mixture of ethanol and water (1:6), water and then ethanol, and dried with a vacuum pump to yield 3.5 g of white crystal.
1H-NMR (DMSO-d6) δppm: 2.28 (3H, s), 7.12 (2H, dd, J=7.75 & 0.50 Hz), 7.48 (2H, dd, J=7.75 & 0.50 Hz), 8.33 (1H, dd, J=5.12 & 1.65 Hz), 8.45 (2H, d, J=6.11 Hz), 8.57 (1H, dd, J=1.65 & 0.66 Hz), 8.96˜9.02 (3H, m)
6) Production of the object compound
17 ml of ethanol and 17 ml of water were added to 3.36 g of the crystal obtained in 5), and the mixture was stirred at room temperature for 30 minutes. A solution of sodium carbonate (0.74 g of sodium carbonate in 17 ml of water) was further added, and the mixture was stirred at room temperature for 2 hours. The precipitated crystal was filtered, subsequently washed with water and ethanol, and dried with a vacuum pump to yield 1.89 g of the object compound as a pale yellow crystal.
TOPIROXOSTAT
SYNTHESIS

(First step)
The first step, 4 – is a step of obtaining a compound (3) is reacted in the presence of an alkali metal alkoxide, cyano-N-oxide and (2), and isonicotinic acid hydrazide.
4 used in this reaction – isonicotinic acid hydrazide and (2) a cyano-N-oxide is a known compound both, I can be prepared by known means.
The alkali metal alkoxide is used, 6 alkoxide alkali metal C 1-C are preferred, sodium methylate, sodium ethylate and the like can be given as specific examples. The reaction is preferably carried out in a solvent, as the solvent, alcohol solvents such as methanol, ethanol and the like are preferable.
The reaction is preferably first in a solvent, is treated with an alkali metal alkoxide compound (2) and then to react the isonicotinic acid hydrazide. First, heated to reflux under cooling, at 80 ℃ from 15 ℃ preferably, 30 minutes and 12 hours in general, the reaction temperature in the reaction with an alkali metal alkoxide (2) with the compound is reacted 1-4 hours, preferably about. Under the temperature conditions, using an excess amount or one equivalent of 30 minutes to 12 hours usually, reaction with isonicotinic acid hydrazide Subsequent to reaction for 1 to 5 hours, preferably.
Example 1:
Synthesis 4 oxide (3) – – – (4 – pyridin-carbonyl) -4 – N “pyridine hydrazide imide -1 was suspended in 40mL of methanol cyanopyridine-N-oxide and (2) 5.00g, sodium was added to methylate 22.4mg, and the mixture was stirred for 2 hours under 40 ℃ nitrogen atmosphere. was cooled to room temperature. reaction solution was stirred for 4 hours at 40 ℃ was added isonicotinic acid hydrazide 5.71g at the same temperature, precipitated The filtrated crystals were, washed with methanol 15mL, and dried 15 hours at 80 ℃, N “- to give (3) 9.60g oxide – (4 – pyridin) -4 – pyridine-hydrazide imide -1.
1 H-NMR (DMSO-d 6) δ (ppm): 6.98 (br, 2H), 7.81 (d, 2H, J = 5.77Hz), 7.85 (d, 2H, J = 7 .09 Hz), 8.29 (d, 2H, J = 7.09Hz), 8.73 (d, 2H, J = 5.77Hz), 10.37 (br, 1H)
MS m / z: 256 [M-H] –
(Second step)
The second step is a step of obtaining compound (4) by cyanation agent cyano compound (3).
As the cyanation agent used, trialkyl cyanide alkali metal cyanide, sodium cyanide, potassium cyanide and the like, zinc cyanide, trimethylsilyl cyanide and the like.
The cyanation reaction is preferably, for example, be carried out (Heterocycles, Vol.22, No.5, 1994) by Reissert Henze reaction. This reaction, for example, to give compound (4) by an organic solvent in the compound (3), and after activation with carbamoyl halide, and reacting the cyano agent. The alkylcarbamoyl halide used in the carbamoylation is a first step in Reissert Henze reaction, 6 alkylcarbamoyl halide di C 1-C dimethylcarbamoyl chloride, and di-propyl carbamoyl chloride can be used, preferably, dimethylcarbamoyl is chloride. The solvent used in this reaction, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, tetrahydrofuran and acetonitrile can be used, however, N, N-dimethylformamide is preferred. Further, 15 ~ 60 ℃, more preferably 30 ~ 50 ℃ reaction temperature. The reaction time is preferably 1 to 24 hours, more preferably 1 to 3 hours. As the cyanation agent used in the cyanation reaction followed, cyano agents above can be used, sodium cyanide, potassium cyanide, zinc cyanide, and trimethylsilyl cyanide, and more preferably, it is sodium cyanide . -20 ~ 60 ℃ is preferred, more preferably -10 ~ 40 ℃, reaction temperature is 1-4 hours.
Is a novel compound (4) The compound obtained in this second step, it is useful as an intermediate for the production of compound (1). If through Compound (4) can be synthesized in good yield and easily without the need for purification in the second step is also possible, and can be produced (1) Compound industrially efficiently compound (4).
Synthetic N “hydrazide (4) – (4 – pyridine carbonyl) -4 – pyridine carboxylic acid N’-(carboxylic imidoyloxy – 2 – – cyano-4)
Example 2
4 pyridine hydrazide imide -1 – oxide ( was suspended in N, N-dimethylformamide 48mL and 3) 10.0g, under nitrogen atmosphere, followed by stirring for 1 hour was added dimethylcarbamoyl chloride 9.20g at 40 ℃. was added sodium cyanide 2.48g at the same temperature, After cooling to 5 ℃ below. reaction mixture was stirred for 1 hour, the crystals were collected by filtration. precipitate was successively added dropwise a 5% aqueous sodium bicarbonate solution 100mL, and 100mL water, and washed with water 100mL, at 80 ℃ for 15 h and dried under reduced pressure to give 4 – hydrazide (4) 9.28g of pyridine-carboxylic acid N’-(carboxylic imide yl – 2 – cyano-4).
1 H-NMR (DMSO-d 6) δ (ppm): 7.15 (br, 2H), 7.82 (d, 2H, J = 5.61Hz), 8.14 (d, 1H, J = 5 .11 Hz), 8.37 (s, 1H), 8.75 (d, 2H, J = 5.61Hz), 8.86 (d, 1H, J = 5.11Hz), 10.47 (br, 1H )
MS m / z: 265 [M-H] –

(Third step)
The third step is a step of obtaining a compound (1) by the presence of an acid catalyst, the cyclization reaction of the compound (4).
As the acid, organic phosphoric acid, p-toluenesulfonic acid, such as hydrochloric acid, inorganic acids can be used, inorganic acids, phosphoric acid is particularly preferable. As the reaction solvent, water, 2 – butanol, 2 – mixed solvent of alcohol and water or alcohol, propanol, ethanol and the like can be used, but water and 2 – I was mixed 5:1 to 10:1 butanol solvent. The reaction temperature and time, 60 ~ 100 ℃, preferably 2 to 12 hours at 70 ~ 90 ℃, I want to 8-10 hours, preferably.
Intermediates and compounds of the present invention the method (1) can be isolated and purified from the washed reaction mixture, recrystallization, by means of various conventional chromatography.
Example 3:
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile 4 Synthesis of (1) – pyridine-carboxylic acid N’- (2 – cyano-4 – carboxylic imide yl) water 82mL, 2 hydrazide (4) 9.25g – butanol was added 8.2mL, phosphate 4.00g, was stirred for 8 h at 80 ℃. After cooling to room temperature, the reaction mixture was precipitated crystals were collected by filtration, water: 2 – were washed with a mixed solution of 92.5mL butanol = 10:1. The 13 h and dried under reduced pressure at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl) – 1 H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1 I got a) 7.89g.
Topiroxostat
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
MS m / z: 247 [M-H] –
PATENT
Synthetic water-carbonitrile p-toluenesulfonate – pyridine Example 1: 4 – [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol]: 2 – butanol = was added monohydrate 6.62g p-toluenesulfonic acid in a mixed solution of 55mL of 10:1, 4 at 80 ℃ – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl] pyridine-2 – – triazol-3 was added carbonitrile 7.85g, and the mixture was stirred at the same temperature for 1 hour. After cooling to room temperature, the reaction mixture, and the precipitated crystals were collected by filtration, and water: 2 – were washed with a mixed solution of 40mL of butanol = 10:1. The dried under reduced pressure for 10 hours at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile p-toluene I got a sulfonate 12.6g.
1 H-NMR (DMSO-d 6) δ (ppm): 2.29 (s, 3H), 7.11 (m, 2H), 7.48 (dd, 2H, J = 6.48,1.62 Hz ) ,8.32-8 .35 (m, 3H), 8.57 (dd, 1H, J = 1.62,0.81 Hz) ,8.94-8 .98 (m, 3H)
– [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazole and potassium carbonate 8.22g, 4 in a mixed solution of 80mL of ethanol = 9:1: preparation water of crystal form I: Example 2 I was dissolved carbonitrile p-toluenesulfonate 10.0g – -3 – yl] pyridine-2. After stirring for 5 hours plus 15mL 6M hydrochloric acid at 20 ℃, was the precipitated crystals were collected by filtration, and washed with water 100mL. The 23 h and dried under reduced pressure at 80 ℃, 4 – to obtain carbonitrile 5.78g – pyridin-2 [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazole. Having a DSC as shown in FIG 4 and the powder X-ray diffraction pattern shown in FIG 1, the resulting crystals were type-I crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
N, N carbonitrile 40.0g – preparation of 4 Form II – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl – triazol-3]-2: Example 3 – dimethylformamide was added 300mL, and stirred for 25 min at 150 ℃. After cooling to room temperature the solution, and the precipitated crystals were collected by filtration, and washed twice with water 200mL, 4 and dried under reduced pressure overnight at 80 ℃ the crystal – [5 – (pyridin-4 – yl)-1H-1 , 2,4 – I got carbonitrile 30.4g – yl] pyridine-2 – triazole-3. Having a DSC as shown in FIG 5 and powder X-ray diffraction pattern shown in FIG 2, the resulting crystals were type II crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
The 25 ℃, about 2g carbonitrile, – preparation of the hydrate 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2: Example 4 I was stored for 14 days under conditions of relative humidity 97%. Having a DSC as shown in FIG 7 and the powder X-ray diffraction pattern shown in FIG 3, the obtained crystal was a hydrate.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
Test Example: solubility test Type I crystal by crystal form, II-type crystal, and water solubility of the hydrate was calculated by absorbance measurement method, a saturated solution concentration of each sample. I Figure 8 shows the results.Whereas the 6.2μg/mL water solubility of crystalline Form I, II type crystal 4.2μg/mL, hydrate was 1.9μg/mL.
From Figure 8, the water solubility of Form II and Form I crystals is good, water-soluble type I crystal is particularly good.
NMR
BMCL Volume 19, Issue 21, 1 November 2009, Pages 6225–6229
http://www.sciencedirect.com/science/article/pii/S0960894X09012372?np=y
view compd 39 and ignore rest
 TOPIROXOSTAT, FYX O51
TOPIROXOSTAT, FYX O51
view compd 39 and ignore rest
| 1 | * | Baldwin, J.J., J. Med. Chem.; 1975; 18(9); 895-900, especially p. 898, lines 3-5. |
| 2 | * | Geldard, J.F. et al., J. Org. Chem.; 1965; 30(1); 318-319, especially p. 319, starting line 33. |
| 3 | * | Lever, A.B.P., Inorg. Chem; 1990; 29; 1271-1285, especially p. 1275, line 18 and 19. |
Nucleosides, Nucleotides and Nucleic Acids, 2008 , vol. 27, 6-7 pg. 888 – 893
Inoue, Tsutomu; Sato, Takahiro; Ashizawa, Naoki; Iwanaga, Takashi; Matsumoto, Koji; Nagata, Osamu; Nakamura, Hiroshi
Bioorganic and Medicinal Chemistry Letters, 2009 , vol. 19, 21 pg. 6225 – 6229
WO 2012060308
WO 2007148835
WO 2005009991
| WO2003064410A1 * | Dec 3, 2002 | Aug 7, 2003 | Naoki Ashizawa | Novel 1,2,4-triazole compound |
| US3882134 * | May 21, 1973 | May 6, 1975 | Merck & Co Inc | 1-Substituted-3,5-dipyridyl-1,2,4-triazoles |
| US3947577 * | Jan 8, 1975 | Mar 30, 1976 | Merck & Co., Inc. | Anti-hyperuricemia composition |
| US3984558 * | Nov 29, 1974 | Oct 5, 1976 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds used as bronchodilators |
| US4011218 * | Dec 3, 1974 | Mar 8, 1977 | Merck & Co., Inc. | 1,2,4-triazoles |
| US4104393 * | Sep 2, 1977 | Aug 1, 1978 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds |
| US5571897 * | Dec 5, 1991 | Nov 5, 1996 | Wallac Oy | Luminescent lanthanide chelates |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
US-9199970-B2 |
2015-12-01 |
|
|||
|
2.
US-20150322006-A1 |
2015-11-12 |
|
|||
|
3.
US-20150309021-A1 |
2015-10-29 |
|
|||
|
4.
US-20150291543-A1 |
2015-10-15 |
|
|||
|
5.
EP-2927219-A1 |
2015-10-07 |
EN
|
|
||
|
6.
US-20150274680-A1 |
2015-10-01 |
|
|||
|
7.
EP-2913053-A1 |
2015-09-02 |
EN
|
|
||
|
8.
EP-2511844-B1 |
2015-08-12 |
EN
|
|
||
|
9.
EP-2712861-B1 |
2015-07-29 |
EN
|
|
||
|
10.
US-20150203490-A1 |
2015-07-23 |
|
|||
|
11.
US-20150191463-A1 |
2015-07-09 |
|
|||
|
12.
US-20150166510-A1 |
2015-06-18 |
|
|||
|
13.
EP-2878594-A1 |
2015-06-03 |
EN
|
|
||
|
14.
EP-2878598-A1 |
2015-06-03 |
E
|
|
||
|
15.
EP-2878595-A1 |
2015-06-03 |
EN
|
|
||
|
16.
US-20150126558-A1 |
2015-05-07 |
|
|||
|
17.
US-8987473-B2 |
2015-03-24 |
|
|||
|
18.
EP-2842948-A1 |
2015-03-04 |
EN
|
|
||
|
19.
EP-2776028-A1 |
2014-09-17 |
EN
|
|
||
|
20.
US-20140256748-A1 |
2014-09-11 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-[5-(4-Pyridinyl)-1H-1,2,4-triazol-3-yl]-2-pyridinecarbonitrile
|
|
| Clinical data | |
| Trade names | Topiloric, Uriadec |
| Legal status |
|
| Identifiers | |
| CAS Number | 577778-58-6 |
| ATC code | None |
| PubChem | CID: 5288320 |
| ChemSpider | 4450517 |
| Chemical data | |
| Formula | C13H8N6 |
| Molecular mass | 248.24 g/mol |
/////////////
C1=CN=CC=C1C2=NC(=NN2)C3=CC(=NC=C3)C#N


Reblogged this on MEDCHEMEGYPT.
LikeLike
Reblogged this on DRUG REGULATORY AFFAIRS INTERNATIONAL.
LikeLike