
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Zanamivir
139110-80-8
APPROVED 26-7-96……. GSK NDA 021036
A guanido-neuraminic acid that is used to inhibit neuraminidase.
Zanamivir INN /zəˈnæmɨvɪər/ is a neuraminidase inhibitor used in the treatment and prophylaxis of influenza caused by influenza A virus andinfluenza B virus. Zanamivir was the first neuraminidase inhibitor commercially developed. It is currently marketed by GlaxoSmithKline under the trade name Relenza as a powder for oral inhalation.
The drug is approved for use for the prevention and treatment of influenza in those over the age of 7 in the United States, Canada, European Union, and many other countries. It is not recommended for people with respiratory problems and ailments.
| United States | 6294572 | APPROVED 1994-12-15 | EXPIRY 2014-12-15 |
| United States | 5360817 | 1993-07-26 | 2013-07-26 |
| Canada | 2291994 | 2003-10-14 | 2011-04-24 |
| Canada | 2081356 | 2000-02-22 | 2011-04-24 |
| Patent No | PatentExpiry | use code |
|---|---|---|
| 5360817 | Jul 26, 2013 | |
| 5648379 | Jul 15, 2014 | U-274 |
| 5648379 | Jul 15, 2014 | U-721 |
| 5648379 | Jul 15, 2014 | U-722 |
| 6294572 | Dec 15, 2014 |
Zanamivir was discovered in 1989 by scientists led by Peter Malcolm Colman and Joseph Varghese at the CSIRO, in collaboration with theVictorian College of Pharmacy, Monash University, and scientists at Glaxo, UK. Zanamivir was the first of the neuraminidase inhibitors. The discovery was initially funded by the Australian biotechnology company Biota and was part of Biota’s ongoing program to develop antiviral agents throughrational drug design. Its strategy relied on the availability of the structure of influenza neuraminidase, by X-ray crystallography. It was also known, as far back as 1974, that 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA), a sialic acid analogue, is an inhibitor of neuraminidase. Sialic acid (N-acetyl neuraminic acid, NANA), the substrate of neuraminidase, is itself a mild inhibitor of the enzyme, but the dehydrated derivative DANA, a transition-state analogue, is a better inhibitor.
Computational chemistry techniques were used to probe the active site of the enzyme, in an attempt to design derivatives of DANA that would bind tightly to the amino acid residues of the catalytic site, and so would be potent and specific inhibitors of the enzyme. The GRID software by Molecular Discovery was used to determine energetically favourable interactions between various functional groups and residues in the catalytic site canyon. This investigation showed that there is a negatively charged zone in the neuraminidase active site that aligns with the C4hydroxyl group of DANA. This hydroxyl is, therefore, replaced with a positively charged amino group; the 4-amino DANA was shown to be 100 times better as an inhibitor than DANA, owing to the formation of a salt bridge with a conserved glutamic acid (119) in the active site. It was also noticed that Glu 119 is at the bottom of a conserved pocket in the active site, just big enough to accommodate a more basic functional positively charged group, such as a guanidino group, which was also larger than the amino group. Zanamivir, a transition-state analogue inhibitor of neuraminidase, was the result.
As Biota was a small company, it did not have the resources to bring zanamivir to market by itself. In 1990, zanamivir patent rights were licensed to Glaxo, now GlaxoSmithKline (GSK). In 1999, the product was approved for marketing in the US and subsequently has been registered by GSK in a total of 70 countries (GlaxoSmithKline News release, 2006). Zanamivir is delivered via Glaxo’s proprietary Diskhaler inhalation device. The license agreement entitled Biota to receive a 7% royalty on Glaxo’s sales of zanamivir.
|
Chemical name: |
5- Acetamido- 2, 6- anhydro- 3, 4, 5- trideoxy- 4- guanidino- D- glycero- D- galacto- non- 2- enonic acid |
| Synonyms: | Zanamivir, GG167, 4-guanidino-Neu5Ac2en and 2,3- Didehydro- 2, 4- dideoxy- 4- guanidino- N- acetyl- D- neuraminic acid(2R,3R,4S)-4-guanidino-3-(prop-1-en-2-ylamino)-2-((1R,2R)-1,2,3-trihydroxypropyl)-3,4-dihydro-2H-pyran-6-carboxylic acid |
| Empirical formula: |
C12H20N4O7 |
| Structural formula: | |
| Molecular weight: | 332.31g |
| Beilstein number: | 7083099 |
| Normal State: | Powder |
| Colour: | White to ‘off white’ |
| Melting point: | 325oC |
| Optical rotary power: | Type [�]Conc: 0.9g/100mlSolvent: H2OOptical rotary power: 41 degWavelength: 589nmTemp: 20oC |
| CAS number: | 139110-80-8 |
| Solubility: | 18mg/mL in water at 20oC |
Zanamivir is used for the treatment of infections caused by influenza A virus and influenza B virus. There is low to moderate evidence that it decreases the risk of one’s getting influenza by 1% to 12% in those exposed. In otherwise-healthy individuals, benefits overall appear to be small.It is unclear whether it affects the risk of one’s need to be hospitalized or the risk of death. An independent analysis of its effects by the Cochrane collaboration was awaiting release of trial data as of 2012. The evidence for a benefit in preventing influenza is weak in children with concerns of publication bias in the literature. As of 2009 no influenza has shown any signs of resistance. Since then genes expressing resistance to were found in patients infected with Influenza A H7N9 and who were treated with corticosteroids.
 ZANAMIVIR
ZANAMIVIR
Mass

| 1H NMR |
| Hydrogen | Chemical shift /ppm |
| (1H, d, 3-H) | 5.53 |
| (2H, 2dd, 4- and 6-H) | 4.50 – 4.38 |
| (1H, dd, 5-H) | 4.21 |
| (2H, dd+ddd, 9-Ha and 8-H) | 4.00-3.88 |
| (2H, 2dd, 9-Hb and 7-H) | 3.70-3.62 |
| (3H, s, Ac) | 2.05 |
|
|
| 13C NMR |
| Carbon | Shift /ppm |
| (C=O, Ac) | 177.3 |
| (C-1) | 172.1 |
| (guanidino) | 159.9 |
| (C-2) | 152.1 |
| (C-3) | 106.8 |
| (C-6) | 78.3 |
| (C-8) | 72.6 |
| (C-7) | 71.0 |
| (C-9) | 65.9 |
| (C-4) | 54.0 |
| (C-5) | 50.6 |
| (Me) | 24.8 |
ref 12
IR spectra:
| The following peaks are present in the IR spectra of Relenza: 3332cm-1, 1676cm-1, 1600cm-1, 1560cm-1, 1394cm-1, 1322cm-1 and 1281cm-1. |
UV spectra
| The maximum peak is 235nm giving E = 199 dm-3 mol-1cm-1 |
ref 13for above

Although zanamivir was the first neuraminidase inhibitor to the market, it had only a few months lead over the second entrant, oseltamivir (Tamiflu), with an oral tablet formulation.
According to the CDC, Tamiflu, zanamivir’s main competitor, is not as effective at treating the influenza viruses as zanamivir, especially in H1N1 seasonal flu. In fact, tests showed 99.6% of the tested strains of seasonal H1N1 flu and 0.5% of 2009 pandemic flu were resistant to Tamiflu, while no flu samples, seasonal or pandemic, showed any resistance to zanamivir.
When first marketed in the US in 1999/2000, zanamivir captured only 25% of the influenza antiviral market, despite a huge promotional campaign. By the end of that season, Tamiflu was outselling zanamivir 3:1. During that season, zanamivir experienced worldwide safety warnings involving the risk of bronchospasm and death. Glaxo then reduced the marketing of zanamivir, and Tamiflu’s dominance increased. More than US$20 million worth of zanamivir sold by Glaxo in the first US season was returned to the company in the next two seasons because zanamivir’s sales to patients were far less than expected.
Biota commenced legal proceedings in 2004 alleging Glaxo’s reduced marketing of zanamivir to be a breach of contract. Biota claimed approximately A$700m from Glaxo. After Biota spent four years trying to progress its case, and incurring A$50m in legal costs, the company abandoned the claim in July 2008, recovering only A$20 million, including legal costs following settlement at mediation. Biota had refused an earlier tactical offer from Glaxo of A$75 million plus legal costs.
In August 2006, Germany announced it would buy 1.7 million doses of zanamivir, as part of its preparation strategy against bird flu. “Germany’s purchase shows that countries are starting to take a balanced view of influenza preparedness,” says Simon Tucker, head of research at Melbourne-based Biota, where zanamivir was originally developed.
In April 2009, many cases of swine flu (H1N1-type virus) were reported in US and Mexico. Zanamivir is one of only two drugs prescribed to treat it. A study published in June 2009 emphasized the urgent need for augmentation of oseltamivir (Tamiflu) stockpiles, with additional antiviral drugs including zanamivir, based on an evaluation of the performance of these drugs in the scenario that the 2009 H1N1 swine flu neuraminidase (NA) were to acquire the Tamiflu-resistance (His274Tyr) mutation, which is currently widespread in 99.6% of all tested seasonal H1N1 strains.n January 2011, GSK announced that it would commence phase III trials for intravenous zanamivir in a study that will span 20 countries in the Northern and Southern Hemispheres.
Recently, the reported oseltamivir-resistance H5N1 virus neuraminidase still retaining susceptibility to zanamivir indicates that the structure of zanamivir has some advantages over oseltamivir in binding to the active pocket of H5N1 neuraminidase.
As a proven anti-influenza drug target, neuraminidase continues to be attractive for the development of new inhibitors. The crystal structure of H5N1 avian influenza neuraminidase (PDB code: 2HTY) provides the three-dimensional structural information and opportunity for finding new inhibitors in this regard, because the existing inhibitors, such as oseltamivir and zanamivir, were developed based on different structures of neuraminidase, such as subtypes N9 and N2, and type B genus of influenza virus.
ZANAMIVIR
Chemistry

- Scheigetz, J.; Zamboni, R.; Bernstein, M. A.;Roy, B. (December 1995). “A syntheses of 4-a-guanidino-2-deoxy-2,3-didehydro n-acetylneuraminic acid”. Organic Letters 27 (6): 637–644.doi:10.1021/ol901511x. Retrieved 2010-11-14.
Zanamivir synthetic process in the world
Together with oseltamivir, zanamivir is the only medicine which can prevent influenza on humans caused by H5N1 and H1N1 virus. Vietnam prepared oseltamivir (Tamiflu) medicine. But there was no zanamivir – the first influenza medicine belonging N1 kind, discovered and commercialized before oseltamivir. The scientific name of zanamivir is acid 5-acetamido-4-guanidino-6-(1,2,3-trihydroxy-propyl)-5,6-dihydro-4H-pyran-2-carboxylic. The discovery of zanamivir opens research possibilities for new medicines which have the same effect on enzyme neuraminidase inhibitor to prevent and treat influenza.
Acid sialic is an input to synthetize zanamivir. The name acid sialic (Neu5Ac2en) is used to indicate derivation at O- and N- positions of acid neuraminic, just for acid N-axetylneuraminic. Acid sialic of carbohydrate groups is on animal cells and microorganism, especially in glycoprotein and gangliosid. The commercial acid sialic is extracted from whey of the cheese and milk process as well as egg yolk, and costs about 5,000 USD per kilo.
In 1994, zanamivir was first synthesized and made public by Von Itzstein and other scientists from the Department of Pharmaceutical Chemistry under Monash University (Australia). Then, Chandler and co-workers of Glaxo company (GSK, Britain) acquired results, improved reaction steps and made them public in 1995. Accordingly, this method produced 8.3% of general output. The synthetic process is described in Figure 1.

Figure 1: Zanamivir synthetic process according to Chandler
Up to now, the research of Chandler has been the only publication about zanamivir synthetic method, the output of which is greater than milligrams, and it reproduces details about reaction conditions and physiochemical properties of the requisite substances.
Recently, a research group of Yao (China) proposed a new approach to synthetize into intermediate compound 5. Researchers started from another material – D-glucono-δ-lactone, which is cheaper than acid sialic. However, the synthetic process is longer and much complicated, including 24 steps, with lower productivity (0.2%).
Researching on synthesizing Zanamivir from Acid sialic by Institute of Chemistry
Synthetizing methyl N-acetylneuraminate (2) and O-pentaacetoxy (3) from acid sialic
Scientists from the Institute of Chemistry used acid sialic (axit N-acetylneuraminic) 98% from China as the input for the zanamivir synthetic process. They decided to use the method of Warner, using ion exchange resin Dowex-H, with the role of catalyst. Reaction was performed in the room in 10 hours. The output was metyl (2) este product of acid N-acetylneuraminic with a productivity of 99%.
Then, to synthetize O-pentaacetoxy (3), scientists applied axetyl effective chemistry method recently published, using BF3.OEt2catalysis at 00C. Productivity in this case exceeded 95%.

Figure 2: The diagram of O-pentaacetoxy 3 derivative making
The use of catalysts which were ion exchange resin Dowex-H (for este chemical reaction) and BF3.OEt2 (for axetyl chemical reaction) had more advantages than the method by scientists from Glaxo.
Synthesizing intermediate compound – oxazoline (4) key from O-pentaacetoxy (3)

Figure 3: Diagram to synthesize oxazoline (4) from O-pentaacetoxy (3) according to a and b methods
Firstly, scientists conducted a survey on oxazoline (4) synthetic process according to Chandler’s process. O-pentaacetoxy (3) compound was separated from two types of OAc and formed oxazoline round thanks to the effect of strong acid Lewis, which was TMSOTf at 520C in 2.5 hour. The productivity of this reaction achieved 40%. The pilot instead of TMSOTf by BF3.OEt2 catalysis in dichloromethane at room temperature at night, the productivity of the reaction to form oxazoline round from penta-acetoxy (5) was similar to the method using TMSOTf (42%). To increase productivity, scientists made a survey on one-pot method, directly from metyl este (2) to oxazoline (4), without passing O-pentaacetoxy (3), gave the highest productivity (73,3%) and was the most economic effectiveness.
Synthesizing zanamivir from oxazoline (4) intermediate compound
The next, scientists successfully conducted reactions from oxazoline (4) intermediate compound to Zanamivir (9) final product (Figure 1). Zanamivir product had IR and NMR data which were compatible with their structure.
Therefore, scientists from the Institute of Chemistry under Vietnam Academy of Science and Technology built a stable process, including seven major steps, synthesizing from acid sialic with the general productivity of 6.6% (the productivity made public in the world was 8.3%). Especially, in the first period, from acid sialic to oxazolin (4) was optimized and gave a general productivity of 74%, higher than the productivity made public by (61.7%). However, the productivity gained in the later period is still low. Now, synthesizing zanamivir influenza medicine still continues to be researched.
……………………
Beau and coworkers assembled the core dihydropyran framework of zanamivir congeners via a combination of PBM reaction and Iron(III)-promoted deprotection-cyclization sequence. A stereochemically-defined α-hydroxyaldehyde 2, diallylamine and a dimethylketal-protected boronic acid 1 is coupled to form the acyclic, stereochemically-defined amino-alcohol 3, which then undergoes an Iron(III)-promoted cyclization to form a bicyclic dihydropyran 4. Selective opening of the oxazoline portion of the dihydropyran intermediate 4 with water or timethylsilyl azide then furnish downstream products that have structures resembling the Zanamivir family members.

| Reaction scheme part 1: |
| The commercially available N-acetyl-neuraminic acid 1 is the starting reagent for the most direct approach to the synthesis of 4-guanidino-Neu5Ac2en (Relenza). In reaction scheme 1 the steps for the conversion of N-acetyl-neuraminic acid 1 to its 4-amino analogue is shown. Step 1 is the addition of methanolic HCl (MeOH and HCl gas), which produces the methyl ester of 1, followed by acetic anhydride in pyridine with 4-(dimethylamino)pyridine catalysis, which produces the penta-acetoxy compound, 2. In step 2, 2 is converted into the oxazoline 3 at high yield using trimethylsilyl trifluoromethanesulfonate (TMSOTf) in ethyl acetate at 52oC. In step 3, the azido compound, 4, is produced by the reaction of 3 with trimethylsilyl azide in tert-butyl alcohol at 80oC. In step 4 catalytic sodium methoxide in methanol was used to remove the acetate protecting groups from 4 to give triol 5. The 4-amino analogue, 6 was made in step 5, by hydrolysis using triethylamine in water, hydrogenolysis with a Lindlar catalyst and finally the addition of Dowex 2 * 8 resin. The triethylamine salt of the 6 was made during hydrogenolysis and the purpose of the Dowex 2 * 8 resin was to desalt this intermediate. The chemical names of the compounds are: |
| 1: N-acetyl-neuraminic acid |
| 2: 5- Acetamido- 3,5- dideoxy- D- glycero- �- D- galacto- 2- nonulo- pyranosonic acid methyl ester |
| 3: Methyl (3aR, 4R, 7aR)- 2- Methyl- 4- [(1’S, 2’R)- 1′, 2′, 3′ – triacet- oxypropyl]- 3a, 7a- dihydro- 4H- pyrano [3, 4-d] oxazole- 6- carboxlate. |
| 4: 5- Acetamido- 7, 8, 9- tri- O- acetyl- 2, 6- anhydro- 4- azido- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid methyl ester. |
| 5: 5- Acetamido- 2, 6- anhydro- 4- azido- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid methyl ester. |
| 6: 5- Acetamido- 4- amino- 2, 6- anhydro- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid. |

Part one of reaction scheme
| Synthesis of reactant necessary for part 2 of reaction: |
| Aminoiminomethane-sulfonic acid (AIMSA), 7, which is necessary for the conversion of compound 6 into Relenza, 9, is synthesised in Reaction scheme 2. The oxidizing solution necessary for the reaction is prepared by the addition of peracetic acid to 30% hydrogen peroxide and then conc. sulfuric acid. This is followed by acetic anhydride and, once the reaction has completed, methanol. Thiourea is dissolved in methanol and added slowly to the oxidizing solution.to produce compound 7. Note that any crystals that form are removed and that the reaction needs to be carried out under cooled conditions. See the reference source for more experimental details. |

Synthesis of AIMSA
| Reaction scheme part 2: |
| Reaction scheme 3 shows the conversion of compound 6 into Relenza For route A, 3 mol equivalent of AIMSA, 7, and 3 mol equivalent of potassium carbonate are added in a portionwise manner to compound 6 over an eight hour period. A yield of about 48% of the crystalline product 8 should be obtained for this method. An alternative route is to treat compound 6 with 1.1 mol equivalent of cyanogen bromide in the presence of sodium acetate in methanol. Route B step 1 gives compound 9, which can be converted into the final product 8 by treating it with ammonium hydroxide and ammonium formate at 85oC. A 36% yield of the purified product can be obtained after purification with ion-exchange chromatography and crystallisation. The chemical names of the compounds in this scheme are: |
| 8. 5- Acetamido- 2, 6- anhydro- 3, 4, 5- trideoxy- 4- guanidino- D- glycero- D- galacto- non- 2- enonic acid. (Relenza) |
| 9. 5- Acetylamino- 2, 6- anhydro- 4- cyanoamino- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid |

Part 2 of reaction scheme
ref are 13 and 14
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
SYNTHESIS FROM PATENT EP2276479A2
ZANAMIVIR AND BOC PROTECTED ZANAMIVIR
The synthesis of zanamivir is shown in Scheme 1. The starting material used for zanamivir synthesis is sialic acid 1, which was converted to the methyl ester 2, in presence of Dowex H+ as described in detail in reference 104. The hydroxyl groups of 2 are protected with acetyl groups to give compound 3, which was then converted to the oxazoline derivative 4 in the presence of trimethyltrifluoromethanesulfonate as described in detail in reference 105. Azide 5 was synthesized from 4 in presence of azidotrimethylsilane as described in detail in reference 105. The azide is reduced to the corresponding amine 6 by using Lindlar’s catalyst, and the amine is in turn converted to the guanidine derivative 7 as described in detail in reference 106. The final step involves the deprotection of the methyl ester and acetyl groups in the presence of methanolic sodium hydroxide to give Boc-protected zanamivir 8 as described in detail in reference 106. 8, 1H NMR (CD3OD) δ (ppm) 5.6 (d, J = 2.0 Hz, IH), 5.01 (dd, J = 9.6, 2.1 Hz, IH), 4.25 (dd, J = 10.8, 1.1 Hz, IH), 4.18 (dd, J = 10.6, 9.6 Hz, IH), 3.89 (ddd, J = 9.4, 6.2, 2.7 Hz, IH), 3.84 (dd, J = 11.3, 2.8 Hz, IH), 3.67 (dd, J = 11.3, 5.8 Hz, IH), 3.57(d, J = 9.3 Hz, IH), 1.9 (s, 3H), 1.55 (s, 9H), 1.50 (s, 9H); ESI-MS: 533 (M+H)+.
Scheme 1



a) Dowex H Methanol b) Aceticanhydride DMAP pyridine c) trimethylsilyl tπfluorαmethane sulfonate ethylacetate d) azidotrimethylsilane butanol e) Lindlar’s catalyst ethanol f) N N’-bis-tert-butoxycarbonyMH-pyrazole-i carboxamidine tetrahydrofuran g) sodium hydroxide methanol
104. Martin, R., K.L. Witte, and C-H. Wong, The synthesis and enzymatic incorporation of sialic acid derivatives for use as tools to study the structure, activity, and inhibition of glycoproteins and other glycoconjugates. Bioorganic & Medicinal Chemistry, 1998. 6(8): p. 1283-1292.
105. Malcolm Chandler, M.J.B., Richard Conroy, Brian Lamount, Bina Patel, Vipulkumar K. Patel, Ian P. Steeples, Richard Storer, Naill G. Weir, Michael
Wrightm Christopher Williamson, Synthesis of the potent influenza neuraminidase inhibitor 4-guanidino Neu5Ac2en. X-Ray molecular structure of S-acetamido^-amino^^-anhydro-S^^-trideoxy-D-erythro-L-gluco- nononic acid. J. Chem. Soc, Perkin Trans. 1, 1995: p. 1173 – 1180.
106. Masuda, T., et al., Synthesis and anti-influenza evaluation of polyvalent sialidase inhibitors bearing 4-guanidino-Neu5Ac2en derivatives. Chem Pharm Bull (Tokyo), 2003. 51(12): p. 1386-98
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
The active component of RELENZA is zanamivir. The chemical name of zanamivir is 5- (acetylamino)-4-[(aminoiminomethyl)-amino]-2,6-anhydro-3,4,5-trideoxy-D-glycero-D-galacto non-2-enonic acid. It has a molecular formula of C12H20N4O7 and a molecular weight of 332.3. It has the following structural formula:
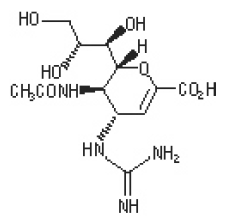 |
Zanamivir is a white to off-white powder for oral inhalation with a solubility of approximately 18 mg/mL in water at 20°C.
RELENZA is for administration to the respiratory tract by oral inhalation only. Each RELENZA ROTADISK contains 4 regularly spaced double-foil blisters with each blister containing a powder mixture of 5 mg of zanamivir and 20 mg of lactose (which contains milk proteins). The contents of each blister are inhaled using a specially designed breath-activated plastic device for inhaling powder called the DISKHALER. After a RELENZA ROTADISK is loaded into the DISKHALER, a blister that contains medication is pierced and the zanamivir is dispersed into the air stream created when the patient inhales through the mouthpiece. The amount of drug delivered to the respiratory tract will depend on patient factors such as inspiratory flow. Under standardized in vitro testing, RELENZA ROTADISK delivers 4 mg of zanamivir from the DISKHALER device when tested at a pressure drop of 3 kPa (corresponding to a flow rate of about 62 to 65 L/min) for 3 seconds.
CLIP
On Zanamivir
Total Synthesis of Anti-Influenza Agents Zanamivir and Zanaphosphor via Asymmetric Aza-Henry Reaction

The potent anti-influenza agents, zanamivir and its phosphonate congener, are synthesized by using a nitro group as the latent amino group at C4 for asymmetric aza-Henry reaction with a chiral sulfinylimine, which is derived from inexpensive d-glucono-δ-lactone to establish the essential nitrogen-containing substituent at C5. This method provides an efficient way to construct the densely substituted dihydropyran core of zanamivir and zanaphosphor without using the hazardous azide reagent.
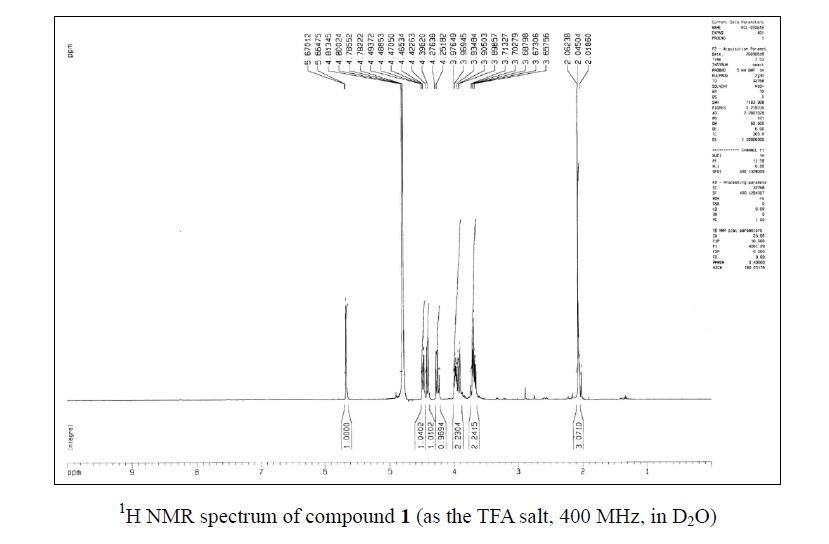
Zanamivir as the TFA salt (40 mg, 90 %). C14H21F3N4O9; colorless solid, mp 260262 oC;
1H NMR (400 MHz, D2O) δ 5.67 (1 H, d, J = 2.1 Hz), 4.48 (1 H, dd, J = 9.3, 2.1 Hz), 4.41 (1H, d, J = 10.6 Hz), 4.26 (1 H, dd, J = 10.6, 9.3 Hz), 3.98–3.90 (2 H, m), 3.71–3.66 (2 H, m),2.06 (3 H, s);
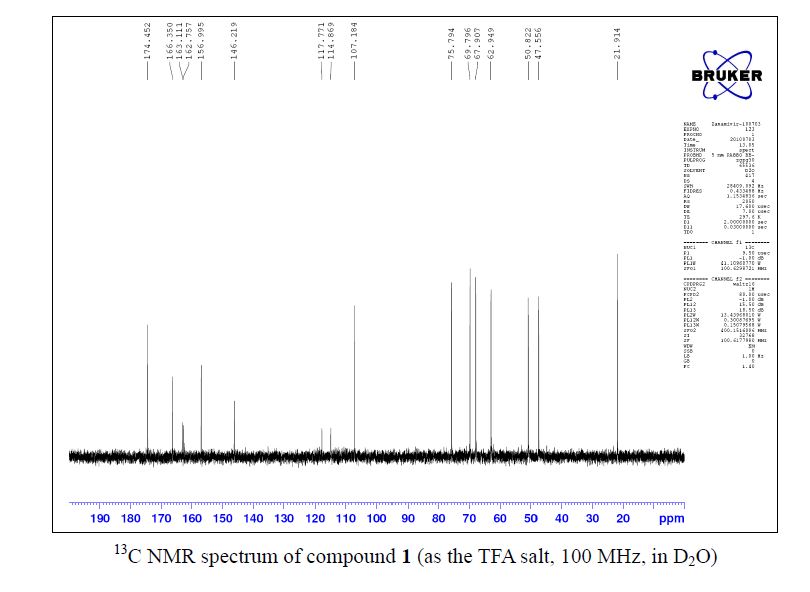
13C NMR (100 MHz, D2O) δ 174.5, 166.4, 162.9 (CO2 of TFA, q, J = 35.4 Hz ),157.0, 146.2, 116.3 (CF3 of TFA, q, J = 290.2 Hz ), 107.2, 75.8, 69.8, 67.9, 62.9, 50.8, 47.6,21.9;
ESI–HRMS calcd for C12H20N4O7Na: 355.1230, found: m/z 355.1288 [M + Na]+.
Introduction
Relenza (Zanamivir for oral inhalation) is the first in a new generation of influenza virus-specific drugs known as neuraminidase inhibitors, which work by interferring with the life cycles of influenza viruses A and B. It prevents the virus spreading infection to other cells by blocking the neuraminidase enzyme present on the surface of the virus. Relenza is available as a powder that is administered by inhalation of 2 blisters from the rotadisk inside the diskhaler (Fig. 1) twice daily for five days. This means that 20mg of Relenza is delivered to the principal site of viral replication each day.The main method for preventing influenza since the 1960s is by vaccination and although this and anti-viral drugs such as amantadine and its analogue rimantadine have long been available (since 1976 and 1993 respectively), they are only of limited use because of the constant mutation of the virus. This chameleon-like nature also means that the virus can become unrecognizable to the human immune system and thus repeatedly infect millions of people year after year.

Fig 1: The diskhaler used to administer Relenza. Each blister in the Rotadisk contains 5mg of the drug
Why there is a need for a more effective influenza treatment: At present influenza is basically an uncontrolled disease and an effective method is needed for both the prevention and treatment of it. In the 20th century there were some major pandemics such as the 1918-1919 Spanish ‘flu which killed 20 million people world wide, the 1957 Asian ‘flu, the 1968 Hong Kong ‘flu and the 1977 Russian ‘flu12 These viruses also affect different animals, especially domesticated chickens and turkeys and in Hong Kong in 1997 a virulent bird flu virus, started infecting and killing people for the first time ever. Of the 18 people affected 6 died, although there was no evidence that the virus was able to spread between people. Given the antigenic properties of the influenza virus, in the future the virus may be passed from person to person, and because human immune systems are not prepared for avian viruses the effects on the population could be grave. It would not be possible to prepare vaccines in time and anti-viral drugs are not always adequate.
Advantages of Relenza over previous treatments:
Relenza has a number of advantages over the existing treatments for influenza. It does not cause significant side effects and the development of zanamivir-resistant viruses is not expected to occur readily in patients. This is because selection of drug-resistant mutants characterized by changes in neuraminidase requires prolonged passage in tissue culture and may be a biological cripple. If started within two days of the onset of influenza symptoms and if a fever is present, the duration of illness is decreased by an average of 1.5 days. It appears to decrease the severity of flu symptoms for the remainder of the illness, as well as decreasing the number of complications from the flu. It is also possible that Relenza could be used as a method of ‘flu prevention although it has not yet been approved for this use.
Comparison of the symptoms of the ‘flu with that of a common cold:
| People infected by an influenza virus suffer a lot more than those with a cold. As you can see from the table below, some of the symptoms are similar, but with a cold they are less severe.Influenza also becomes more serious when it leads to secondary bacterial pneumonia or primary influenza viral pneumonia or when it exacerbates underlying medical conditions such as pulmonary or cardiac disease. In children, the symptoms are similar to those observed in adults, however children often have higher fevers and younger ones may develop gastrointestinal manifestations. It should be noted that Relenza is not effective on people with colds or other viral illnesses. |
| Influenza | Cold |
| Sore throat | Mild sore throat |
| High fever and chills | Low-grade fever |
| Non-productive cough | Cough |
| Severe muscle aches | Congestion |
| Headache | |
| Intense fatigue. |
The effect of Relenza on patients with respiratory diseases:Relenza is not generally recommended for the treatment of patients with respiratory dieseases such as asthma or chronic obstructive pulmonary disease (COPD) and has carried an approval since its approval in July 1999. Some patients with underlying airway diseases have experienced serious adverse events following treatment, with some fatal outcomes although causality has been difficult to establish. It has been recommended that patients with asthma have a fast-acting bronchodilator inhaler available and use it about 15 minutes before taking Relenza
Successfulness of Relenza:The sialidase inhibitory activities (determined by methods described in reference 7) of Relenza compared to the more recent neuraminidase inhibitor Oseltamivir are shown in the table below9.IC50 is the concentration that reduces enzyme activity by 50%.
| Compound | Influenza A IC50 (�M) | Influenza B IC50 (�M) |
| Relenza | 0.005 | 0.004 |
| Oseltamivir | 0.002 | 0.032 |
The results demonstrate that both compounds are good inhibitors of influenza A and B, with Oseltamivir being more selective towards Influenza A and Relenza showing a better overall performance. In phase I and II tests reported by the Lancet5, no important adverse effects were found in healthy patients or those reported to have mild to moderate asthma following an inhaled administration of 40mg/day of Relenza. There was a significant improvement of the symptoms of people taking Relenza compared to those taking the placebo.
1940s: Discovery that the influenza virus’s enzyme was destroying receptors on red blood cellsF.This was discovered by George Hirst, who noticed that when red blood cells were mixed with fluids from influenza infected chicken embryos in cold conditions the cells were very heavily agglutinated by the virus. These red cells dispersed when warmed up and could not be re-agglutinated in the cold with fresh virus. This led him to the conclusion that the influenza virus’s enzyme was destroying receptors on red blood cells.
The finding of sialidase (also known as neuraminidase):Alfred Gottschalk heard of Hirst’s experiment and interpretation of results, and this led him to believe that there was a “split product”. He discovered sialic or neuraminic acid (Fig 2), a type of sugar, and the enzyme on the virus was called neuraminidase (or sialidase). At this time it was thought that it was the neuraminidase which was responsible for the observations made by Hirst, but it was later shown by Robin Valentine, W. Graeme Laver, Norbert Bischofberger and Robert G. Webster that the hemagglutinin (receptor-binding) and neuraminidase (receptor-destroying) activities of the virus resided in two quite different spikes on the surface of the virus.

Fig 2: Sialic Acid
Discovery of how new pandemic strains of ‘flu A occured.
Ed Kilbourne, W. Graeme Laver, Norbert Bischofberger and Robert G. Webster realised that hybrid viruses could be formed by infecting cells simultaneously with two different Type A flu viruses. This was because the RNA pieces coding the various virus proteins reassorted, some of the viruses contained the hemagglutinin from one parent and the neuraminidase from the other. This “mating” of two parent viruses to give a hybrid virus explained how new pandemic strains of ‘flu A occurred, and led to a very good way of producing influenza viruses with any desired combination of hemagglutinin and neuraminidase spikes. This helped towards finding a way of producing pure neuraminidase which was later essential for crystal growth and drug design experiments.
The crystallization of neuraminidase:
Laver, Bischofberger and Webster isolated one type of influenza virus by sucking off the allantoic fluid surrounding the embryo of infected chicken eggs and purifying this. The virus particles were incubated with an enzyme capable of digesting proteins. This enzyme was selected to split the “heads” of the neuraminidase spikes off the virus particle without destroying them and to leave behind or destroy the hemagglutinin spike. The neuraminidase “heads” obtained were concentrated using high-speed centrifugation. The tiny pellet of neuraminidase heads examined had a crystalline appearance, and X-ray diffraction analysis of larger crystals showed that they were made of protein.
Neu5Ac2en (DANA) was shown to inhibit influenza neuraminidase:
Different variants of ‘flu neuraminidase were known to exist, each containing an amino acid sequence that varies between types of neuraminidase apart from one small sequence.It was seen that the conserved amino acids came together when the neuraminidase polypeptide folded up to form the active enzyme. This formed a well conserved cavity which was the active catalytic site of the neuraminidase enzyme. It became apparent that a plug-drug could be made to exactly fit into the active site and inhibit the neuraminidase activity from other influenza viruses. A synthetic analog of sialic acid called Neu5Ac2en (DANA) (Fig 3) was shown to inhibit the influenza virus neuraminidase, but not sufficiently enough to be used treatment for the ‘flu in humans.
Fig 3: Neu5Ac2en (DANA)

Fig 3: Neu5Ac2en (DANA)
The plug drug.Mark von Itzstein and colleagues discovered that replacing the OH at the 4 position of sialic acid with a positively charged amino group made a better inhibitor than sialic acid or its analogue, DANA. Replacing the OH at the 4 position of sialic acid with a guanidino group led to a potent inhibitor of ‘flu neuraminidase. This compound was given the names GG167 and Zanamivir and is now more commonly known as Relenza. Peter Colman soaked the substrate for sialic acid in neuraminidase crystals and used X-ray crystallography to determine the three-dimensional structure of the crystals. The strong binding of Relenza by ‘flu neuraminidase which was seen is due to the positively charged guanidino group being anchored by the negatively charged glutamic acids. More details about this are provided in the immunology section.
Immunology

|
Fig 4: The influenza viruses as seen under the electron microscope. Neuraminidase and haemagglutin spikes are visible. |
||
Structure of the flu virus:Influenza (Fig 4) is an RNA virus which may exist as any shape from round balls to long, spaghetti-like filaments. The genome of this virus is associated with five different viral proteins and is surrounded by a lipid membrane, which means that influenza belongs to the “enveloped” group of viruses. Eight separate pieces of ribonucleic acid (RNA) make up the influenza virus genome and each piece of RNA specifies the amino acid sequence of one and sometimes two of the virus’s proteins. The segmented nature of the RNA allows differenet flu viruses to easily “mate” with each other to form hybrid progeny viruses with bits of RNA from each parent virus.Two glycoprotein molecules, known as hemagglutinin (HA) and neuraminidase (NA) (Fig 5) are stuck onto the lipid envelope of the virus and both play a crucial role in the infection of the epithelial cells of the upper respiratory tract. HA is a rod-shaped triangular molecule.and NA exists as a mushroom shaped spike with a box-like head on top of a long stalk, containing a hydrophobic region by which it is embedded in the viral membrane.. |
||
|
Fig 5: The Neuraminidase enzyme |
||
| The enzyme Neuraminidase, also known as sialidase, is a tetramer with C-4 symmetry and an approximate molecular weight of 250 000. It contains a symmetrical folding pattern of six four-stranded antiparallel �-sheets arranged like propeller blades. Nine types of neuraminidase have been identified for influenza A and only one subtype for influenza B, and only 30% of the overall amino acid sequence is conserved between all known types of neuraminidase8 – these are the amino acids which line and surround the walls of the binding pocket. If they mutate, the enzyme is inactivated, so the virus could not mutate to escape from a drug which interfered with this site. So neuraminidase offers an attractive site for therapeutic intervention in influenza infections. | ||
|
|
||
How the influenza virus works:The influenza virus (like all viruses) can only replicate after invading selected living cells and growing inside them. It makes thousands of new virus particles from the cellular machinery and then goes on to infect other cells.. Hemagglutinin allows the virus to infect the epithelial cells of the upper respiratory tract by attaching it to cells through receptors on the cell containing sialic acid, it fuses the cell membrane with the membrane of the virus, allowing the RNA of the virus to get inside the cell and thus instruct the cell to make thousands of new virus particles. After this viral replication, the progeny virions must be released from the cell to repeat the cell cycle of infection.Neuraminidase removes the sialic acid receptors from the host cell and other newly made virus particles by cleavage of -glycosidic bonds. This enables the virus to escape from the cell in which it grew and spread in the body to infect other cells. The action of NA may also facilitate viral mobility through the mucus of the respiratory tract. 
|
| Fig. 6: The life cycle of the influenza virus. Click once on this image to see a larger version | ||
|
The life cycle of the influenza virusG begins with the individual virus entering the cell lining of the respiratory tract (letter a in Fig. 6), and the cell being induced to take up the virus because hemagglutinin on the virus binds to the sialic acid (b and c in Fig 6). The virus then dispatches its genetic material (made up of RNA) and its internal proteins to the nucleus of the cell (e and f). Messenger RNA is produced when some of the internal proteins duplicate the RNA (f). This messenger RNA is used by the cell as a template for making viral proteins (g and h) and genes which become new viral particles and leave the cell covered in sialic acid. This sialic acid needs to be removed so that the hemagglutinin molecules on one particle don’t attach to the sialic acid on other ones, thus causing the new viruses to clump together and stick to the cell. The sialic acid is removed from the surface of the new viral particle by neuraminidase (j) and the new viral particles are able to travel and invade other cells (k). |
||
| How Relenza works:
Relenza adopts a position within the active site of the enzyme and copies the geometry of the sialoside hydrolysis transition state9. It can achieve very good binding through appropriate presentation of its four pendent substituents and contains a hydrogen bonding glycerol sidechain. The guanidino group in Relenza is believed to form salt bridges with Glu 119 in the neuraminidase active site and add a strong charge interaction with Glu 2278. Two hydroxyl groups of the 6-glycerol side chain are hydrogen bonded to Glu276 and the 4-hydroxyl is oriented towards Glu119. The NH group of the 5-N acetyl side chain interacts with a bound water molecule on the floor of the active site. The carbonyl oxygen of the same side chain is hydrogen bonded to Arg152 and the methyl group enters a hydrophobic pocket lined by Ile222 and Trp178. The glycosidic oxygen projects into bulk solvent.
|

|
Fig 7. Relenza bound to neuraminidase |
||
|
The binding involved in Fig 7 is shown more clearly in Fig 8 below. Neuraminidase can no longer remove the sialic acid receptors from the host cell and newly made virus particles because of this binding. Therefore the virsuse ‘clump’ together or to the host cell and cannot go on to effect new cells. |
||
 |
||
|
Fig 8: Depiction of interaction of Relenza (GG 167) in the neuraminidase binding site6 |
||
References
| 1): K. J. Lui and A. P. Kendal, Am. J. Public Health, 1987, 77, 712 |
| 2): Scheiget, Zambonis, Bernstein and Roy, Org. Prep. Proced. Int., 1995, 27, 637- 644 |
| 3): Glaxo Wellcome Inc. Relenza� (zanamivir for inhalation) [package insert]. Research Triangle Park, NC: Glaxo Wellcome, Inc., 1999 |
| 4): N Seppa, Scientific American, July 10th 1999, Volume 156 |
| 5): L. Gubareva, Lancet, March 4th 2000, 355: 827-35 |
| 6): J. Medicinal Chemistry. 1999, 42, 2332-2343 |
| 7):P Smith, S Sollis, P Howes, P Cherry, I Starkey, K Cobley, H Weston, J Scicinski, A Merritt, A Whittington, P Wyatt, N Taylor, D Green, R Bethall, S Madar, R Fenton, P Morley, T Pateman, A Beresford. A. J. Med. Chem, 41, 1998, 787-797 |
| 8): C Kim, W Lew, M Williams, H Liu, L Zhang, S Swaminathan, N Bischofberger, M Chen, D Mendel, C Tai, G Laver, R Stevens, J Am Chem Soc, 1997, 119, 681-690 |
| 9): P Smith, J Robinson, D Evans, S Sollis, P Howes, N Trivedi and R Bethell, Bioorganic and Medicinal Chemistry Letters 9, 1999, 601-604 |
| 10): A. J. Hay, A. J. Wolstenholme, J. J. Skehel and M. H. Smith. EMBO J,. 1985, 4, 3021: L. J. Holsinger and R. A. Lamb, Cell, 1992, 69, 517 |
| 11): J. C. Stoof, J. Booij, B. Drukarch and E. C. Wolters, Eur. J. Pharmacol., 1992, 213, 439 |
| 12): W. Graeme Laver, Norbert Bischofberger, and Robert G. Webster, Perspectives in Biology and Medicine 43.2 (2000) 173-192. This can be seen by visitinghttp://www.press.jhu.edu/journals/perspectives_in_biology_and_medicine/v043/43.2laver.html nmr |
| 13): M. Chandler, M. J. Bamford, R. Conroy, B. Lamont, B. Patel, V. K. Patel, I. P. Steeples, R. Storer, N. G. Weir, M. Wright, C. Williamson, J. Chem. Soc. Perkin Trans. 1, 1995, 1173- 1180 nmr synth |
| 14): A. E. Miller, J. J. Bischoff, Synthesis, 1986, 777- 779 |
| 15): G. D. Allena, S. T. Brookesa, A. Barrow, b, J. A. Dunnc and C. M. Grossec, Journal of Chromatography B: 1999, 732, 383-393 |
Zanamivir
139110-80-8
APPROVED 26-7-96……. GSK NDA 021036
A guanido-neuraminic acid that is used to inhibit neuraminidase.
READ AT


Reblogged this on DRUG REGULATORY AFFAIRS INTERNATIONAL.
LikeLike