
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

BMS 911543
N,N-dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
cas 1271022-90-2
Chemical Formula: C23H28N8O
Exact Mass: 432.23861
UNII-7N03P021J8;
N,N-dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
Bristol-Myers Squibb Company innovator
BMS-911543 is an orally available small molecule targeting a subset of Janus-associated kinase (JAK) with potential antineoplastic activity. JAK2 inhibitor BMS-911543 selectively inhibits JAK2, thereby preventing the JAK/STAT (signal transducer and activator of transcription) signaling cascade, including activation of STAT3. This may lead to an induction of tumor cell apoptosis and a decrease in cellular proliferation. JAK2, often upregulated or mutated in a variety of cancer cells, mediates STAT3 activation and plays a key role in tumor cell proliferation and survival.

The JAK2 selective compound BMS911543 (WO2011028864) is in phase II clinical trials for the treatment of m elofibrosis. BMS91 1543 is shown below.

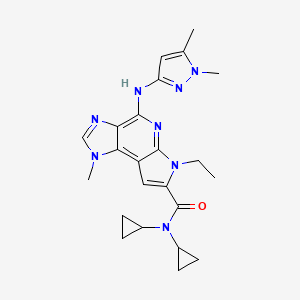
PAPER
ACS Medicinal Chemistry Letters (2015), 6(8), 850-855
Discovery of a Highly Selective JAK2 Inhibitor, BMS-911543, for the Treatment of Myeloproliferative Neoplasms

JAK2 kinase inhibitors are a promising new class of agents for the treatment of myeloproliferative neoplasms and have potential for the treatment of other diseases possessing a deregulated JAK2-STAT pathway. X-ray structure and ADME guided refinement of C-4 heterocycles to address metabolic liability present in dialkylthiazole 1 led to the discovery of a clinical candidate, BMS-911543 (11), with excellent kinome selectivity, in vivo PD activity, and safety profile

MS (ESI) m/z 434.3 (M+H). 1H NMR (CDCl3) δ: 7.96 (s, 1H), 7.65 (s, 1H), 6.83 (s, 1H), 4.67 (q, J = 7.1 Hz, 2H), 4.01 (s, 3H), 3.82 (s, 3H), 2.77 – 2.84 (m, 2H), 2.43 (s, 3H), 1.48 (t, J = 7.2 Hz, 3H), 0.79 – 0.86 (m, 4H), 0.71 – 0.77 (m, 4H).
PAPER
Journal of Organic Chemistry (2015), 80(12), 6001-601
Click to access jo5b00572_si_001.pdf
Ni-Catalyzed C–H Functionalization in the Formation of a Complex Heterocycle: Synthesis of the Potent JAK2 Inhibitor BMS-911543

BMS-911543 is a complex pyrrolopyridine investigated as a potential treatment for myeloproliferative disorders. The development of a short and efficient synthesis of this molecule is described. During the course of our studies, a Ni-mediated C–N bond formation was invented, which enabled the rapid construction of the highly substituted 2-aminopyridine core. The synthesis of this complex, nitrogen-rich heterocycle was accomplished in only eight steps starting from readily available materials.
N,N-Dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide, 1
PATENT
WO 2015031562
These Schemes are illustrative and are not meant to limit the possible techniques one skilled in the art may use to manufacture compounds disclosed herein.
As shown below in Scheme 1, the general preparation of compound 7 is described. Trichloroacetyl pyrrole (Compound 1) is reacted with a halogenating agent to give the C4-bromo pyrrole (Compound 2). Alcoho lysis occurs in the presence of an alcohol and base to generate ester (Compound 3), which can be selectively nitrated through contact with an appropriate nitrating agent (defined as a species that generates N02 ), yielding C5-nitro pyrrole (Compound 4). Compound 4 can be isolated as its free form, or optionally as a salt with an appropriate base. Ethylation with an appropriate alkylating agent generates the N-ethyl pyrrole (Compound 5), which in the presence of an imidazole, base, palladium and an appropriate phosphine ligand, will undergo a coupling process to form Compound 6. Reduction of the nitro-group of Compound 6 in the presence of hydrogen, a metal catalyst and optionally a base will produce Compound 7.
Scheme 1

As shown below in Scheme 2, the preparation of Compound 13 is described. Trichloroacetyl pyrrole is treated with NBS in acetonitrile to produce Compound 8. Treatment with sodium ethoxide in EtOH yields the ethyl ester Compound 9. This may be treated with a range of nitrating systems, in this example, NaNC /SCVPy, to generate nitro-pyrrole Compound 10, which can be isolated directly or as a salt form with an appropriate base, preferably dibenzylamine. Ethylation with ethyl iodide generates Compound 11 which may be isolated, or optionally telescoped directly into the arylation with Compound 32. Arylation proceeds in the presence of palladium, Xantphos, potassium pivylate and Hunig’s base to generate Compound 12. Hydrogenation presence of Pt/C followed by cyclization with NaOEt yields Compound 13.
Scheme 2

Another process of the invention is disclosed in Scheme 3 shown below. Compound 14 is prepared from Compound 3 in the presence of an alkylating agent. Treatment with a suitable diboron reagent produces Compound 15, which can then be coupled with a suitably functionalized imidazole derivative to yield Compound 16. Amino lysis with a suitable nitrogen donor produces Compound 17, which can cyclize under appropriate conditions to produce Compound 7.
Scheme 3
Step 3 Step 4 Step 5

As shown below in Scheme 4, ethylation of Compound 9 with ethyl iodide produces Compound 18. This may be directly reacted with dipinacol-diboron in the presence of Pd(OAc)2 and tricyclohexylphosphin hexafluorophosphate and
tetramethylammonium acetate to yield Compound 19. Subsequent coupling with 5-Br-imidazole derivative yields Compound 20. Treatment with hydroxylamine hydrochloride in the presence of triethylamine yields the Compound 21. Subsequent cyclization with Piv20 in the presence of PRICAT™ and hydrogen yields Compound 13.
Scheme 4
77% isolated over 2-steps%

18
Step 5 Pd(OAc)2
PPh3
78%

As shown below in Scheme 5, Compound 23 may be converted to Compound 26 by two pathways. In one option, Compound 23 can be treated with palladium, ligand and a mild base to prepare Compound 25. Reaction of Compound 25 with a metal hydroxide produces Compound 26.
Alternately, Compound 23 can be treated with palladium and ligand in the presence of a soluble hydroxide base, followed by treatment with the metal counter-ion to prepare Compound 26 directly. Once Compound 26 is formed, it can be coupled to Compound 27 to form compound I.


A solution of Compound 1 in acetonitrile (1238.0 kg, 264.9 kg after correction) was charged into a 5000 L glass-lined reactor at a temperature of 20-30 °C. The mixture was added with stirring over about 2 h and then cooled to 0 °C. NBS (221.8 kg) was charged into the mixture at intervals of 20-30 min at 0-20 °C. The mixture was cooled to 0-5 °C and reacted until the content of Compound 8was < 1.0%. Additional NBS (4.0 kg) was charged into the mixture at 0-20 °C. The mixture was reacted over 3 h until the content of Compound 8 was < 1.0%. Purified water (2650.0 kg) was added over about 1.5 – 2.5 h at 0-20 °C. The mixture was cooled to 0-5 °C and then stirred for about 1 h for crystallization. The mixture was filtered and the filter cake was rinsed with water.
Example 2

While maintaining the temperature at 20-30 °C, anhydrous ethanol (950.0 kg) was charged into a 3000 L glass-lined reactor followed by Compound 8 (342.7 kg). The mixture was cooled to 0-5 °C over about 2 h. Sodium alcoholate solution in ethanol (21%, 36.4 kg) was added dropwise over about 1-1.5 h at 0-5 °C. The reaction mixture was then heated to about 25-30 °C and tested until the content of Compounds 8/9 was < 1.0%. The reaction mixture was concentrated at a temperature < 50 °C until about 1.3-1.4 volume of Compound 8 was left. The concentrated mixture was cooled at 25-30 °C. The mixture was quenched into cooled water (3427.0 kg) over about 2 h. After addition, the mixture was stirred at 0-5 °C over about 2 h for crystallization. The mixture was filtered and the filter cake was rinsed. The solid was dried at 30-40 °C over 40-45 h to afford 234.3 kg of Compound 9 , 99.9% purity and 91.3% yield.
Example 3

9 10
A mixture of NaN03, NaHS04, and Na2S04 in CH3CN is wet-milled to constant particle size of -50 micron. To the slurry of inorganic salts is added S03 -pyridine and Compound 9. The reaction mixture is agitated at 25 °C until 90-95% conversion is achieved. The reaction is quenched with aqueous sodium hydroxide and the spent inorganic salts are removed by filtration. The filtrate is passed through a carbon pad and distilled under constant volume distillation and diluted with water to a target 15
volumes/kg of Compound 9 and a target ratio 1.0:2.0 vol/vol MeCN to water. The resulting solids are deliquored, washed, and dried to afford Compound 10.
Example 4

Toluene (10 L/Kg)
65 °C
Compound 10 (1.0 eq) and TBABr (1.0 eq) were added to a biphasic mixture of toluene (8 L/kg 10) and potassium carbonate (1.5 eq) in water (5 L/kg 10). The batch temperature was held at 25 °C. The resulting triphasic slurry was heated to 60-65 °C and diethylsulfate (1.5 eq, in a solution of toluene 2 L/kg 10) was slowly added over ~ 1 h. The reaction was aged until less than 1 RAP of Compound 10 (10:11) remained. The resulting homogeneous biphasic mixture was cooled to 20 °C and the lean aq. phase was removed. The rich organic phase was washed with water (2×7 L/kg 10) and concentrated to 6 mL/g 10. The concentrated stream was dried via azeotropic, constant volume distillation with toluene until the water content of the stream was <0.1 wt %. The resulting stream was telescoped into the subsequent direct arylation reaction.
Example 5

11 28 12
To the toluene stream of Compound 11, with potassium pivalate (1.5 equiv.) was charged, followed by DIPEA (3 eq.), Compound 28 (3 eq.) and Pd(Xantphos)Cl2 (0.04 eq.). The vessel was evacuated to < 200 torr and backfilled with nitrogen (3 X) followed by heating to 95 °C until residual Compound 11 was less than 1 RAP (11: 12). The reaction mixture was cooled to 25 °C and diluted with ethyl acetate (15 mL/g vs input pyrrole) and aq. N-acetylcysteine (0.2 eq., 5 wt % solution, 1.8 mL/g vs. input pyrrole) and heated to 50 °C for 1 h. The biphasic mixture was cooled to 25 °C. The lower aqueous layer was removed. The ethyl acetate stream was washed with water (2×7 mL/g vs. input pyrrole). The rich organic phase was polish filtered followed by a vessel/polish filter rinse with ethyl acetate (2 mL/g vs. input pyrrole). The rich organic stream was concentrated to 4 mL/g vs. input pyrrole via vacuum distillation, while maintaining the batch temperature above 50 °C. If spontaneous nucleation did not occur, Compound 12 seeds (1 wt %) were charged, followed by aging for 30 min at temperature. MTBE (5 mL/g vs. 11) was charged to the slurry over 1 hour while maintaining the batch temperature above 40 °C, followed by aging at 40 °C for 1 h. The slurry was cooled to 0 °C over 6 h and aged at 0°C for 6 h. The slurry was filtered and washed with
EtO Ac : Toluene : MTBE (1.5: 1.0: 1.5, 2 mL/g vs. input 11 ). The wet cake was dried (50 °C, 100 torr) until LOD was < 1 wt %.
Example 6

Compound 12 (1 eq., limiting reagent (LR)) is dissolved in THF/NMP (20 Vol wrt LR, 9/1 ratio) and submitted to hydrogenation using 10 wt% (wrt LR) Pt/C (5 wt%) at 25 to 40° C for 5-10 h. The reaction containing the corresponding amine is filtered. The rich organic stream is concentrated to Compound 12 Vol (wrt LR) and subjected to 0.1 eq of 21 wt% NaOEt/EtOH for 5 h at 20-25 °C, upon which Compound 13 forms. The stream is cooled to 0-10 °C, and water (5L/Kg, wrt to LR) is added and then filtered to isolate Compound 13. The product is dried at 50 °C under vacuum.
Example 7

in toluene solution
9
18
Compound 18 was prepared by treating the pyrrole with ethyl iodide and pulverized potassium carbonate in DMF at 25-30°C under inert atmosphere. After the reaction was completed, the batch mass was cooled to 15°C to 20°C and quenched by slow addition of water then MTBE. The MTBE layer was separated and washed with water. The MTBE layer was distilled to 4 Vol and solvent swapped with toluene. The toluene stream was then taken into the next step.
Example 8

18 19
Tetra-methyl ammonium acetate in toluene slurry was heated to 75-80°C to get a clear solution. The mass was cooled to below 30°C and pyrrole in toluene and bis (pinacolato) diborane were added. The reactor was inerted by nitrogen purging then the reaction was heated to 75-80°C. A freshly prepared catalyst/ligand complex (0.0 leq of palladium acetate, 0.025eq of tricyclohexyl phosphino hexafluoroborate and 0.2eq of tetra methyl ammonium acetate in toluene) was charged under nitrogen atmosphere at RT and stirred for 2h. The mass was then stirred at 75-80°C under nitrogen atmosphere. After the reaction was completed, the mixture was cooled below 30°C and quenched with aq. sodium bisulphate solution. The organic layer was polish filtered through a Celite bed and the filtrate was washed with water. The solvent swapped to ethanol until the toluene content became less than 0.5 %. The solution was cooled to 0-5°C and water was added for crystallization. The product was then isolated by filtration.
Example 9

Compound 20 was prepared by treating Compound 19 with Compound 34 in the presence of palladium acetate, triphenyl phosphine and potassium carbonate in dimethyl acetamide with the water mixture as the solvent. Dimethyl acetamide, water, potassium carbonate and the two starting materials were charged into the reactor. The mixture was made inert with nitrogen for 30 min and then charged with freshly prepared catalyst mixture (palladium acetate, triphenyl phosphine and potassium carbonate in dimethyl acetamide). The temperature was raised to 78-83 °C then the mass was stirred at this temperature. After the reaction was completed, the reaction mass was cooled to ambient temperature and purified water was added slowly into the mass for product
crystallization. The mass was stirred for a period of 3 h and filtered. The wet cake was washed with purified water and dried in VTD at 50-55 °C under vacuum.
Example 10

Compound 21 was prepared by treating Compound 20 with hydroxylamine hydrochloride and triethyl amine using ethanol as the solvent. Compound 20 was added into ethanol (15 Vol) and the reaction mass was heated to 38-40 °C. Hydroxylamine hydrochloride was charged and stirred for 10 min, then triethyl amine was added slowly at 38-40 °C over a period of lh. The above mass was stirred at 38-40 °C until Compound 20 becomes less than 5.0%, typically in about 15 h. After the reaction was completed, the above reaction mass was cooled to ambient temperature (below 30 °C) and filtered. The wet cake was washed with purified water (4 Vol) and dried under vacuum in VTD at 55-60 °C.
Example 11

Initially Compound 21 was treated with pivalic anhydride using toluene and acetic acid mixture as solvent under inert atmosphere until Compound 21 becomes less than 3.0% with respect to Compound 21, typically in about 30 min. PRICAT Nickel was then added under nitrogen atmosphere. The reaction mass was inerted with nitrogen for three cycle times and then degassed with hydrogen gas for three cycle times. Following this, 3.0 kg/cm2 hydrogen pressure was applied to the reaction mass which was stirred for about 12h. After the reaction was completed, the reaction mixture was filtered through a sparkler filter. The filtrate was distilled and the solvent exchanged with toluene until the ratio of acetic acid & toluene reaches 1 :20. At this time, n-Heptane was charged and cooled to 15°C. Then the product was filtered and the wet cake was dried in VTD at 50-55°C under vacuum.

Compound 30 was prepared by the coupling of Compound 22 with Compound 29, 3 -bromo- 1,5 -dimethyl- lH-pyrazole in the presence of
Tris(dibenzylideneacetone)dipalladium chloroform adduct, t-Brettphos and potassium phosphate in tert-amyl alcohol at 98-103 °C under inert atmosphere. After completion of the reaction (typical level of Int.9 -5% & typical reaction hrs 20 h), the mass was cooled to ambient temperature and t-amyl alcohol (4 Vol) and 20 Vol of water were charged into the reaction mass. The reaction mass was stirred for 15 min. and then phase split. The organic layer was diluted with 10 Vol of MTBE and product was extracted with 20 Vol of 1M methane sulphonic acid. The MSA stream was treated with 15 wt % charcoal to reduce the residual palladium numbers. The filtrate was cooled to below 20 °C and the pH was adjusted to 1.7-1.9 using IN NaOH for product crystallization and then iltered. The wet cake was washed with purified water (3 x 5 Vol), followed by methanol (5 Vol). The cake was vacuum dried for 3 h. then the wet cake and dimethyl sulfoxide (20 Vol) were charged into a reactor. The mass was heated to 120-125 °C to get clear solution then the mass was cooled to ambient temperature and stirred for 2 h, then filtered. The wet cake was washed with methanol (3x 4.0 Vol) and vacuum dried for 2 h. The wet cake was dried in VTD at below 55°C under vacuum.
Example 13

Compound 30 , ethanol (16.5 Vol), water and aq sodium hydroxide solution were charged into a reactor then the mass was heated to 70-75 °C and stirred until Compound 30 becomes less than 1.0%. After the reaction was completed, the mass was diluted with ethanol for complete product precipitation at 65-75 °C. Then the mass was cooled to 50 °C for a period of lh and stirred for lh at 50 °C. The mass was further cooled to 20 °C and stirred for lh at 20 °C and then filtered. The wet cake was washed with 5 Vol of 15% aqueous ethanolic solution followed by THF. The wet cake was dried under vacuum at 70-75 °C till LOD comes to less than 5.0 %, typically in about 40 h.
Example 14

In a vessel 36.5 mmol (-42.6 mL) of Compound 29 solution in 2-methyl-2-butanol was combined with 30.7g (65.1 mmol) tetrabutylammonium hydroxide (55 wt% in water), 8.01g (27.0 mmol) Compound 13 , and 10 mL 2-methyl-2-butanol. The mixture was heated at 70 °C until hydrolysis of Compound 13 was complete (full dissolution, <15 min). The solution was cooled to 60 °C and 1.12g (2.22 mmol) of tBuBippyPhos followed by 384 mg (1.028 mmol) allylpalladium chloride dimer (L:Pd = 1 :1) was added. The mixture was heated to 80 °C and was aged at this temperature for 20h before cooling to 22 °C.
Water was added and the mixture concentrated, a constant volume distillation was then performed to swap to ethanol (40-55 °C, 150 mbar). The resulting solution was passed through a 5 micron filter to remove any particulates. The solution was heated to 55 °C and 8.10 mL (40.52 mmol, 1.5 equiv) 5N NaOH (aq) was added dropwise over a 3 h period. Crystals of Compound 31 began to form, and after aging for an additional lh, the mixture was cooled to 20 °C over 3 h. After an additional 6h of aging, crystals were collected on a frit and the cake was washed with 40 mL of 90: 10 ethanol: water, followed by 48 mL acetone. After drying at 80 °C in a vacu-oven for 16 h, Compound 31 was collected as an off-white solid (8.89g, 85%).
Example 15

Compound 31 was added into dichloromethane (20 Vol) and cooled to 15-20 °C. The reaction mass was charged with DMC in DCM solution (1.4 eq of DMC in 5.0 Vol of DCM). The mixture was stirred until Compound 31 becomes less than 2.0% with respect to the corresponding acid chloride, typically in about lh. After completion of the reaction, Compound 27 (1.4 eq) and N,N-diisopropylethyleneamine (3.0 eq) were charged and the mixture was stirred. After completion of the reaction, the mass was quenched with 12 Vol of water then the layers were separated. The organic layer was washed with water and filtered through a celite bed. The filtrate was concentrated to ~6.0 vol and then the mass was cooled to 35 °C. To the resulting solution was added THF, followed by seeds of product, then stirred for 3 h. The solvent was swapped with THF until
dichloromethane becomes less than 2 wt% (wrt THF). The mass was cooled to -5 to 0 °C over a period of 2 h and stirred for 2 h. The reaction mass was then filtered under a nitrogen atmosphere. The material was slurried with pre-cooled THF (2*2 Vol) and filtered. The wet cake was dried in VTD at 60 °C under vacuum till LOD becomes < 1%, typically in about 20 h.
Example 16

DC , RT
I
To a slurry of Compound 31 (15.00 g, 40.0 mmol) in dichloromethane (300 ml) was added diphenylphosphinic chloride (12.29 g, 51.9 mmol). The mixture was stirred at room temperature for 2 h and Ν,Ν-diisopropylethylamine ( 16.53 g, 127.9 mmol) was then added and stirred for another 30 min. Compound 27 (6.94 g, 51.9 mmol) and 4-dimethylaminopyridine (0.49 g, 4.0 mmol) were subsequently added and stirred for 16 h until the reaction was completed. The reaction mixture was treated with N-acetyl-L-cysteine (3.26 g, 20.0 mmol) and citric acid (10.10 g, 48.0 mmol) in deionized water (180 ml) for 2 h. After phase split, the dichloromethane phase was washed once with 0.42 N NaOH solution (180 ml) and washed twice with deionized water (180 ml each). The final dichloromethane phase was concentrated (to 90 ml) and acetone (30 ml) was added. The solution was cooled to 35 °C and N-2 form seed of Compound 1 ( 150 mg ) was added and aged for 1 h. The resulting slurry was solvent-swapped to acetone (DCM < 10% v/v), and cooled to 0 °C. The solid was filtered and washed with cold acetone and dried to afford 14.69 g (85%) of Compound I (HPLC AP 99.8) as off-white crystals.
Patent
WO 2011028864
http://www.google.com/patents/WO2011028864A1?cl=en
Compounds of general formula I in which the R group is thiazole (as in Ial) and R1 and R2 groups are CF3 or alkyl or cycloalkyl or combine to form a saturated carbocyclic or heterocyclic ring or where R2 group is COORb could be prepared using the general method depicted in Scheme 1. Dichloro intermediate II (prepared using procedure reported in WO200612237) could be combined with a 2,4-dimethoxybenzyl and the resulting secondary amine is capped with suitable protective group (Boc) (III). The second chlorine atom could be converted into the
corresponding amine (IV) through the benzophenone imine intermediate. The amino compound could be halogenated to intermediate V. V could be subjected to transition metal mediated indole ring formation and the resulting indole nitrogen is capped with ethyl iodide to afford VI. Ester hydrolysis followed by amide bond formation and cleavage of protective groups with acid treatment would yield amine VII. Amine VII could be converted into thiourea VIII by first coupling with benzoyl isothiocyanate followed treatment with aqueous base. Formation of thiazole could be achieved by condensation with an a-bromoketone derivative (R^HBrCOR2).


a) 2,4-dimethoxybenzylamine, heat; b) NaHMDS, Boc20; c) (Ph)2=NH; d) HCl; e) NIS; f) Pd2(dba)3, ethyl pyruvate; g) Etl, Cs2C03; h) NaOH (aq); i) dicyclopropylamine HCl, HATU, DIPEA; j) TFA; k) Benzoyl isothiocyanate;
1) NaOH (aq); m) I^CHBrCOR1
Scheme 1
Compounds of general formula Ia2 in which the R1 group is CONRaRa could be made using Scheme 2. Thiourea intermediate (VIII) could be combined with Et02CCHBrCOR1 to afford the thiazole ester (IX). The ester could be hydrolyzed and the acid could be coupled with amine to afford thiazole amide derivative (la)

a) Et02CCHBrCOR1; b) NaOH (aq); c) HNRaRa, HATU, DIPEA
Scheme 2
Similarly, compounds of general formula Ia3 in which the R1 group is CONRaRa could be prepared using the general protocol depicted in Scheme 3.

a) R2CHBrCOC02Me; b) NaOH (aq); c) HNRaRa, HATU, DIPEA
Scheme 3
Compounds of general formula la in which R1 is halogen (CI, Br or I) could be prepared by condensing an a,a’-dihaloketone as depicted in Scheme 4.
a) R2COCH(Hal)2
Scheme 4
Alternatively, thiourea derivative VIII could be converted to room temperature into C-5 un-substituted thiazole XI and then directly halogenated using electrophilic halogen source or through metallation followed by quenching with an electrophilic halogenating agent (Scheme 5).

a) BrCH2COR2; b) Selectfluor or NCS or NBS or NIS or tBuLi followed Selectfluor or NBS or NCS
Scheme 5
Compounds of general formula Ia5 in which R1 is S02Rb could be synthesized using the general synthetic approach shown in Scheme 6


a) Br2-acetic acid; b) EtOH, heat
Scheme 6
Compounds with general formula la in which R1 and R2 combine to form an aromatic or heteroaromatic ring could be prepared using Scheme 7.

X = hal, -S02Me
a) Pd(0) catalyst, NaOtBu, phosphine ligand, heat
Scheme 7
Alternatively, these compounds could be made by first coupling aniline or heteroaniline (XVI) with the isothiocyanate (XV) followed by oxidative cyclization (Scheme 8).

a) 1, 1 ‘-Thiocarbonyldi-2( 1 H)-pyridone; b) NaH; c) NIS
Scheme 8
Compounds of general formula Ibl could be prepared using the general synthetic approach depicted in Scheme 9. Aniline VII could be combined with γ-dithiomethylketone compound XVII, (prepared using the procedure reported at room temperature in Synlett, p 2331 (2008)) under basic condition to afford XVIII.
Stepwise condensation of the Boc-protected hydrazine derivative would give the required pyrazole Ibl.

a) NaH, THF; b) R1N(Boc)NH2, AcOH, 35-40°C; c) HCO2H or TFA, 60°C
Scheme 9
Compounds of general formula Ibl or Ifl and If could also be prepared by coupling C-4 halo derivative (XIX) with an appropriately substituted 2-aminopyrazole derivative (XX) using a transition metal catalyzed reaction (Scheme 10).

a) isoamyl nitrite, CH2I2 or isoamyl nitrite, CH2Br2; b) Pd2(dba)3, Xanphos, Cs2C03
Scheme 10
Compounds of general formula Ib2 in which R2 group is CONRaRa could be synthesized using Scheme 11. Aniline VII could be combined with γ-dithiomethylketone derivative XXII, (prepared using the procedure from
Tetrahedron, p 2631 (2003)) to afford intermediate XXIII. Stepwise condensation of Boc-protected hydrazine derivative would give the required pyrazole aldehyde XXIV. Aldehyde could be oxidized using oxone or sodium hypochlorite to furnish carboxylic acid XXV. Coupling of acid XXV with amine would give pyrazole amide Ib2.

a) NaH, THF, heat; b) R1N(Boc)NH2, AcOH; c) TFA; d) oxone or sodium hypochlorite; e) HNRaRa, HATU, DIPEA
Scheme 11
Compounds of general formula Icl could be prepared using the general protocol as shown in Scheme 12. Aniline VII could be coupled with chloroacetyl chloride and the resulting amide could be treated with thioamide (R2CS H2) to furnish thiazole Icl .

a) chloroacetyl chloride, base; b) R2CSNH2
Scheme 12
00120] Compounds of general formula ldl could be made as per Scheme 13. Previously described isothiocyanate derivative XV could be combined with amidine XXV under dehydrating reaction conditions to give 1,2,4-thiadiazole (ldl).

Scheme 13
Compounds of general formula lei could be prepared using a synthetic approach as shown in Scheme 14. Isothiocyanate XV could be combined with azide XXVI in the presence of phosphine to yield 1,3-oxazole Iel .

Scheme 14
Compounds of general formula lgl could be prepared using a synthetic approach as shown in Scheme 15. Amine VII could be combined with acyl isothiocyanate XXVII. The acylthioureaido could be condensed with hydrazine derivative to yield the 1,2,4-triazol derivative lgl.

igi
Scheme 15
without a methyl

Preparation of 7V,7V-dicyclopropyl-6-ethyl-l-methyl-4-(5-m ethyl- lH-pyrazol-3- ylamino)-l,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
[00437] Prepared using similar protocol as for example 72 from hydrazine.
[00438] MS (ESI) m/z 419.3 (M+H)
[00439] 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 8.70 (br s, 1 H), 7.91 (br s, 1 H), 6.87 (s, 1 H), 6.09 (br s, 1 H), 4.64 (q, 2 H, J= 7.03 Hz), 4.08 (s, 3 H), 2.74 -2.95 (m, 2 H), 2.41 (s, 3 H), 1.51 (t, 3 H, J= 7.15 Hz), 0.81 – 0.95 (m, 4 H), 0.70 -0.81 (m, 4 H)
with an ethyl

7V,iV-dicyclopropyl-6-ethyl-4-(l-ethyl-5-methyl-lH-pyrazol-3-ylamino)-l-methyl- 1,6-dihydroimidazo [4,5-d] pyrrolo [2,3-b] pyridine-7-carboxamide
74A Preparation of fe/t-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate

Diisopropyl azodicarboxylate (2.92 mL, 15.00 mmol) was added in one portion to a solution of tert-butyl l,3-dioxoisoindolin-2-ylcarbamate (2.62 g, 10 mmol, prepared following the procedure described by Nicolas Brosse et al. in Eur. J. Org. Chem. 4757-4764, 2003), triphenylphosphine (3.93 g, 15.00 mmol) and ethanol (0.691 g, 15.00 mmol) in THF (20 mL) at 0 °C and the reaction solution was stirred at room temperature for lh (monitored by TLC until completion). Solvent was evaporated and the residue was purified by flash chromatography on silica gel using an automated ISCO system (80 g column, eluting with 5-35% ethyl acetate / hexanes) to provide tert-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate (2.6 g, 90 % yield) as a white solid which was used as it in the next step
74B Preparation of fe/t-butyl l-ethylhydrazinecarboxylate
Boc
H2N-N
\
Methylhydrazine (1.415 niL, 26.9 mmol) was added to a solution oi tert-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate (example 74A, 5.2 g, 17.91 mmol) in THF (40 mL) at 0 °C and the reaction mixture was stirred at room temperature overnight. A white precipitate formed and was filtered off through a pad of Celite, The filtrate was concentrated in vacuo. The residue was dissolved in ethyl acetate (50 ml) and extracted with IN HC1 (3×30 ml), the acid layer was washed with ethyl acetate (50 ml) and basified to pH 10 by addition of 20% NaOH. The basic solution was then extracted with ethyl acetate (3×50 ml) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated in vacuo to give tert-butyl 1 -ethylhydrazinecarboxylate (2.5 g, 87 % yield) as colorless oil.
XH NMR (400 MHz, CDC13) δ: 3.90 (br. s., 2H), 3.35 (q, J = 7.0 Hz, 2H), 1.42 (s, 9H), 1.07 (t, J = 7.0 Hz, 3H)
74 Preparation of N.N-dicyclopropyl-6-ethyl-4-(l-ethyl-5-methyl-lH-pyrazol-3-ylamino)-l-methyl-l ,6-dihydroimidazor4,5-d1pyrrolor2,3-b1pyridine-7-carboxamide
A mixture of (Z)-N,N-dicyclopropyl-6-ethyl- 1 -methyl-4-( 1 -(methylthio)-3-oxobut-l-enylamino)-l,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide (example 74B, 70 mg, 0.155 mmol) and tert-butyl 1-ethylhydrazinecarboxylate (49.6 mg, 0.309 mmol) in acetic acid (1 mL) wan stirred at 35 °C for 4 h (monitored by LC/MS until no starting material left). Formic acid (1 mL) was added and the reaction mixture stirred at 60 °C for 6 h. The solvent was evaporated and the crude product was purified by flash chromatography on silica gel using an automated ISCO system (12 g column, eluting with 2-10% methanol / dichloromethane). The material was further purified by preparative HPLC to afford N,N-dicyclopropyl-6-ethyl-4-( 1 -ethyl-5-methyl- lH-pyrazol-3-ylamino)- 1 -methyl- 1 ,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide (38 mg, 53.4 % yield) as an off-white solid.
MS (ESI) m/z 447.3 (Μ+Η).
XH NMR (500 MHz, CDC13) δ: 8.08 (s, 1H), 7.61 (s, 1H), 6.93 (s, 1H),
6.84 (s, 1H), 4.66 (q, J = 7.1 Hz, 2H), 4.02 (q, J = 7.2 Hz, 2H), 3.98 (s, 3H), 2.79 – 2.85 (m, 2H), 2.34 (s, 3H), 1.49 (t, J = 7.1 Hz, 3H), 1.41 (t, J = 7.2 Hz, 3H), 0.82 -0.87 (m, 4H), 0.72 – 0.78 (m, 4H).
Patent
JAK2 INHIBITORS AND THEIR USE FOR THE TREATMENT OF MYELOPROLIFERATIVE DISEASES AND CANCER [US8202881]2011-03-102012-06-19
JAK2 inhibitors and their use for the treatment of myeloproliferative diseases and cancer [US8673933]2012-04-302014-03-18
: Purandare AV, McDevitt TM, Wan H, You D, Penhallow B, Han X, Vuppugalla R, Zhang Y, Ruepp SU, Trainor GL, Lombardo L, Pedicord D, Gottardis MM, Ross-Macdonald P, de Silva H, Hosbach J, Emanuel SL, Blat Y, Fitzpatrick E, Taylor TL, McIntyre KW, Michaud E, Mulligan C, Lee FY, Woolfson A, Lasho TL, Pardanani A, Tefferi A, Lorenzi MV. Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2. Leukemia. 2012 Feb;26(2):280-8. doi: 10.1038/leu.2011.292. Epub 2011 Oct 21. PubMed PMID: 22015772.
Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2http://www.nature.com/leu/journal/vaop/ncurrent/full/leu2011292a.html
GRAPHS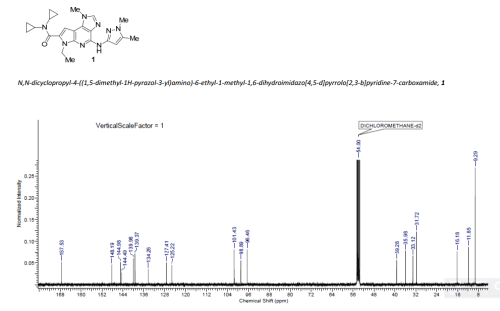
Click to access jo5b00572_si_001.pdf

//////BMS 911543, phase 2, bms,

