

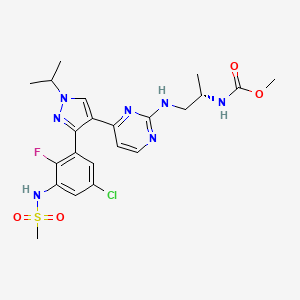
ENCORAFENIB, エンコラフェニブ
UNII:8L7891MRB6
Formula:C22H27ClFN7O4S, Average: 540.01
1269440-17-6
- BRAFTOVI
- NVP-LGX818
- NVP-LGX-818-NXA
- NVP-LGX818-NXA
- ENCORAFENIB [USAN]
- ENCORAFENIB [WHO-DD]
- ENCORAFENIB
- ENCORAFENIB [INN]
- METHYL N-((2S)-1-((4-(3-(5-CHLORO-2-FLUORO-3-(METHANESULFONAMIDO)PHENYL)(-1-(PROPAN-2-YL)-1H-PYRAZOL-4-YL(PYRIMIDIN-2-YL)AMINO)PROPAN-2-YL)CARBAMATE
- CARBAMIC ACID, N-((1S)-2-((4-(3-(5-CHLORO-2-FLUORO-3-((METHYLSULFONYL)AMINO)PHENYL)-1-(1-METHYLETHYL)-1H-PYRAZOL-4-YL)-2-PYRIMIDINYL)AMINO)-1-METHYLETHYL)-, METHYL ESTER
- LGX818
- LGX-818
Encorafenib, also known as BRAFTOVI, is a kinase inhibitor. Encorafenib inhibits BRAF gene, which encodes for B-raf protein, which is a proto-oncogene involved in various genetic mutations Label. This protein plays a role in regulating the MAP kinase/ERK signaling pathway, which impacts cell division, differentiation, and secretion. Mutations in this gene, most frequently the V600E mutation, are the most commonly identified cancer-causing mutations in melanoma, and have been isolated in various other cancers as well, including non-Hodgkin lymphoma, colorectal cancer, thyroid carcinoma, non-small cell lung carcinoma, hairy cell leukemia and adenocarcinoma of the lung 6.
On June 27, 2018, the Food and Drug Administration approved encorafenib and Binimetinib(BRAFTOVI and MEKTOVI, Array BioPharma Inc.) in combination for patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test Label.
Array Biopharma (a wholly owned subsidiary of Pfizer ), under license from Novartis , and licensees Pierre Fabre and Ono Pharmaceutical have developed and launched the B-Raf kinase inhibitor encorafenib . In January 2020, the US FDA’s Orange Book was seen to list encorafenib patents such as US8946250 , US8501758 , US9314464 and US9763941 , expiring in the range of 2029-2032. At that time Orange Book also reported that encorafenib as having NCE exclusivity expiring on July 27, 2023.
Encorafenib (trade name Braftovi) is a drug for the treatment of certain melanomas. It is a small molecule BRAF inhibitor [1] that targets key enzymes in the MAPK signaling pathway. This pathway occurs in many different cancers including melanoma and colorectal cancers.[2] The substance was being developed by Novartis and then by Array BioPharma. In June 2018, it was approved by the FDA in combination with binimetinib for the treatment of patients with unresectable or metastatic BRAF V600E or V600K mutation-positive melanoma.[3][4]
The most common (≥25%) adverse reactions in patients receiving the drug combination were fatigue, nausea, diarrhea, vomiting, abdominal pain, and arthralgia.[3]
Indication
Used in combination with Binimetinib in metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test 5.
Associated Conditions
Pharmacodynamics
Encorafenib has shown improved efficacy in the treatment of metastatic melanoma 3.
Encorafenib, a selective BRAF inhibitor (BRAFi), has a pharmacologic profile that is distinct from that of other clinically active BRAFis 7.
Once-daily dosing of single-agent encorafenib has a distinct tolerability profile and shows varying antitumor activity across BRAFi-pretreated and BRAFi-naïve patients with advanced/metastatic stage melanoma 7.
Mechanism of action
Encorafenib is a kinase inhibitor that specifically targets BRAF V600E, as well as wild-type BRAF and CRAF while tested with in vitro cell-free assays with IC50 values of 0.35, 0.47, and 0.3 nM, respectively. Mutations in the BRAF gene, including BRAF V600E, result in activated BRAF kinases that mahy stimulate tumor cell growth. Encorafenib is able to bind to other kinases in vitro including JNK1, JNK2, JNK3, LIMK1, LIMK2, MEK4, and STK36 and significantly reduce ligand binding to these kinases at clinically achievable concentrations (≤ 0.9 μM) Label.
In efficacy studies, encorafenib inhibited the in vitro cell growth of tumor cell lines that express BRAF V600 E, D, and K mutations. In mice implanted with tumor cells expressing the BRAF V600E mutation, encorafenib induced tumor regressions associated with RAF/MEK/ERK pathway suppression Label.
Encorafenib and binimetinib target two different kinases in the RAS/RAF/MEK/ERK pathway. Compared with either drug alone, co-administration of encorafenib and binimetinib result in greater anti-proliferative activity in vitro in BRAF mutation-positive cell lines and greater anti-tumor activity with respect to tumor growth inhibition in BRAF V600E mutant human melanoma xenograft studies in mice. In addition to the above, the combination of encorafenib and binimetinib acted to delay the emergence of resistance in BRAF V600E mutant human melanoma xenografts in mice compared with the administration of either drug alone Label.

Pharmacology
Encorafenib acts as an ATP-competitive RAF kinase inhibitor, decreasing ERK phosphorylation and down-regulation of CyclinD1.[5]This arrests the cell cycle in G1 phase, inducing senescence without apoptosis.[5] Therefore it is only effective in melanomas with a BRAF mutation, which make up 50% of all melanomas.[6] The plasma elimination half-life of encorafenib is approximately 6 hours, occurring mainly through metabolism via cytochrome P450 enzymes.[7]
Clinical trials
Several clinical trials of LGX818, either alone or in combinations with the MEK inhibitor MEK162,[8] are being run. As a result of a successful Phase Ib/II trials, Phase III trials are currently being initiated.[9]
History
Approval of encorafenib in the United States was based on a randomized, active-controlled, open-label, multicenter trial (COLUMBUS; NCT01909453) in 577 patients with BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma.[3] Patients were randomized (1:1:1) to receive binimetinib 45 mg twice daily plus encorafenib 450 mg once daily, encorafenib 300 mg once daily, or vemurafenib 960 mg twice daily.[3] Treatment continued until disease progression or unacceptable toxicity.[3]
The major efficacy measure was progression-free survival (PFS) using RECIST 1.1 response criteria and assessed by blinded independent central review.[3] The median PFS was 14.9 months for patients receiving binimetinib plus encorafenib, and 7.3 months for the vemurafenib monotherapy arm (hazard ratio 0.54, 95% CI: 0.41, 0.71, p<0.0001).[3] The trial was conducted at 162 sites in Europe, North America and various countries around the world.[4]
SYN


PATENT
WO2010010154 , expiry , EU states, 2029, US in 2030 with US154 extension.
WO 2011025927
WO 2016089208
Patent
WO-2020011141
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020011141&tab=FULLTEXT&_cid=P20-K5QFFQ-43376-1
Novel deuterated analogs of diarylpyrazole compounds, particularly encorafenib are B-RAF and C-RAF kinase inhibitors, useful for treating proliferative diseases such as melanoma and colorectal cancer. Family members of the product case, WO2010010154 , expire in most of the EU states until 2029 and will expire in the US in 2030 with US154 extension. In January 2020, the US FDA’s Orange Book was seen to list encorafenib patents such as US8946250 , US8501758 , US9314464 and US9763941 , expiring in the range of 2029-2032. At that time Orange Book also reported that encorafenib as having NCE exclusivity expiring on July 27, 2023.

The mitogen-activated protein kinase (MAPK) pathway mediates the activity of many effector molecules that coordinately control cell proliferation, survival, differentiation, and migration. Cells are bound by plasma factors such as growth factors, cytokines, or hormones to plasma membrane-associated Ras and GTP and thereby activated to recruit Raf. This interaction induces Raf’s kinase activity, resulting in direct phosphorylation of MAPK / ERK (MEK), which in turn phosphorylates extracellular signal-related kinase (ERK). Activated ERK phosphorylates a range of effector molecules, such as kinases, phosphatases, transcription factors, and cytoskeleton proteins. Therefore, the Ras-Raf-MEK-ERK signaling pathway transmits signals from cell surface receptors to the nucleus and is essential for cell proliferation and survival.
[0003]
According to Raf’s ability to interact with upstream regulator Ras, Raf has three different isoforms, namely A-Raf, B-Raf, and C-Raf. An activating mutation of one of the Ras genes can be observed in about 20% of all tumors, and the Ras-Raf-MEK-ERK pathway is activated in about 30% of all tumors. Activation mutations in the B-Raf kinase domain occur in approximately 70% of melanoma, 40% of papillary cancer, 30% of low-grade ovarian cancer, and 10% of colorectal cancer. Most B-Raf mutations are found in the kinase domain, with a single substitution (V600E) accounting for 80%. The mutated B-Raf protein activates the Raf-MEK-ERK pathway by increasing kinase activity against MEK or by activating C-Raf. B-Raf inhibitors inhibit cells involved in B-Raf kinase by blocking the signal cascade in these cancer cells and eventually inducing cell arrest and / or death.
[0004]
Encorafenib (aka LGX-818, chemical name is (S)-(1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1-iso Propyl-1H-pyrazol-4-yl) pyrimidin-2-yl) amino) prop-2-yl) methyl carbamate, which has the following structural formula) is a new oral BRAF jointly developed by Novartis and Array Pharmaceuticals Inhibitors can inhibit the activation of the MAPK pathway caused by B-Raf kinase mutations (such as V600 mutations, that is, glutamate mutations at amino acid 600). Encorafenib alone or in combination with MEK inhibitor Binimetinib is used to treat patients with advanced BRAF v600 mutant melanoma. On June 27, 2018, the FDA approved Encorafenib (commercial name BRAFTOVI) capsules in combination with Binimetinib (commercial name: MEKTOVI) tablets for treating melanoma patients with BRAF V600E or BRAFV 600K mutations.
It is known that poor absorption, distribution, metabolism, and / or excretion (ADME) properties are the main cause of the failure of many candidate drug clinical trials. Many drugs currently on the market also limit their scope of application due to poor ADME properties. The rapid metabolism of drugs will cause many drugs that could be highly effective in treating diseases to be difficult to make because they are metabolized from the body too quickly. Although frequent or high-dose medication may solve the problem of rapid drug removal, this method will bring problems such as poor patient compliance, side effects caused by high-dose medication, and rising treatment costs. In addition, rapidly metabolizing drugs may also expose patients to adverse toxic or reactive metabolites.
[0007]
Although Encoratenib as a BRAF inhibitor can effectively treat BRAF V600 mutant melanoma, there are still serious clinical unmet needs in this field, and the Encoratenib compound is a class II BCS with poor water solubility at weakly acidic and neutral pH Compounds have poor oral availability, so finding new compounds that have a therapeutic effect on BRAF kinase mutations, have good oral bioavailability, and have pharmaceutical properties is still a challenging task. Therefore, there remains a need in the art to develop compounds that have selective inhibitory activity and / or better pharmacodynamics / pharmacokinetics for use as BRAF inhibitors, and the present invention provides such compounds.
PAPER
European journal of cancer (Oxford, England : 1990) (2018), 88, 67-76.
References
- ^ Koelblinger P, Thuerigen O, Dummer R (March 2018). “Development of encorafenib for BRAF-mutated advanced melanoma”. Current Opinion in Oncology. 30 (2): 125–133. doi:10.1097/CCO.0000000000000426. PMC 5815646. PMID 29356698.
- ^ Burotto M, Chiou VL, Lee JM, Kohn EC (November 2014). “The MAPK pathway across different malignancies: a new perspective”. Cancer. 120 (22): 3446–56. doi:10.1002/cncr.28864. PMC 4221543. PMID 24948110.
- ^ Jump up to:a b c d e f g “FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations”. U.S. Food and Drug Administration (FDA)(Press release). 27 June 2018. Archived from the original on 18 December 2019. Retrieved 28 June 2018.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Drug Trial Snapshot: Braftovi”. U.S. Food and Drug Administration (FDA). 16 July 2018. Archived from the original on 19 December 2019. Retrieved 18 December 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, Piao Y, Liu J, Cheng W, Bi X, Gong P, Song Z, Meng S (January 2016). “Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells”. Cancer Letters. 370 (2): 332–44. doi:10.1016/j.canlet.2015.11.015. PMID 26586345.
- ^ Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. (July 2012). “A landscape of driver mutations in melanoma”. Cell. 150 (2): 251–63. doi:10.1016/j.cell.2012.06.024. PMC 3600117. PMID 22817889.
- ^ Koelblinger P, Thuerigen O, Dummer R (March 2018). “Development of encorafenib for BRAF-mutated advanced melanoma”. Current Opinion in Oncology. 30 (2): 125–133. doi:10.1097/CCO.0000000000000426. PMC 5815646. PMID 29356698.
- ^ “18 Studies found for: LGX818”. Clinicaltrials.gove.
- ^ Clinical trial number NCT01909453 for “Study Comparing Combination of LGX818 Plus MEK162 and LGX818 Monotherapy Versus Vemurafenib in BRAF Mutant Melanoma (COLUMBUS)” at ClinicalTrials.gov
External links
- “Encorafenib”. Drug Information Portal. U.S. National Library of Medicine.
- Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, Piao Y, Liu J, Cheng W, Bi X, Gong P, Song Z, Meng S: Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016 Jan 28;370(2):332-44. doi: 10.1016/j.canlet.2015.11.015. Epub 2015 Nov 14. [PubMed:26586345]
- Koelblinger P, Thuerigen O, Dummer R: Development of encorafenib for BRAF-mutated advanced melanoma. Curr Opin Oncol. 2018 Mar;30(2):125-133. doi: 10.1097/CCO.0000000000000426. [PubMed:29356698]
- Moschos SJ, Pinnamaneni R: Targeted therapies in melanoma. Surg Oncol Clin N Am. 2015 Apr;24(2):347-58. doi: 10.1016/j.soc.2014.12.011. Epub 2015 Jan 24. [PubMed:25769717]
- Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R, Chiarion-Sileni V, Dutriaux C, de Groot JWB, Yamazaki N, Loquai C, Moutouh-de Parseval LA, Pickard MD, Sandor V, Robert C, Flaherty KT: Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018 May;19(5):603-615. doi: 10.1016/S1470-2045(18)30142-6. Epub 2018 Mar 21. [PubMed:29573941]
- FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations [Link]
- BRAF B-Raf proto-oncogene, serine/threonine kinase [ Homo sapiens (human) ] [Link]
- Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma [Link]
- Encorafenib FDA label [File]
- Encorafenib review [File]
///////////ENCORAFENIB, 1269440-17-6, BRAFTOVI, NVP-LGX818, LGX818, LGX 818, エンコラフェニブ ,
COC(=O)N[C@@H](C)CNc1nccc(n1)c2cn(nc2c3cc(Cl)cc(NS(=O)(=O)C)c3F)C(C)C
patent
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020011141&tab=FULLTEXT&_cid=P20-K5QFFQ-43376-1
Method for preparing compounds of the invention
[0165]
The compounds of the invention, including their salts, can be prepared using known organic synthesis techniques, and can be synthesized according to any of a number of possible synthetic routes, such as those in the schemes below. The reaction for preparing the compound of the present invention can be performed in a suitable solvent, and a person skilled in the art of organic synthesis can easily select a solvent. Suitable solvents may be substantially non-reactive with the starting materials (reactants), intermediates, or products at the temperature at which the reaction is performed (e.g., a temperature ranging from the solvent freezing temperature to the solvent boiling point temperature). A given reaction may be performed in one solvent or a mixture of more than one solvent. The skilled person can select a solvent for a specific reaction step depending on the specific reaction step.
[0166]
The preparation of the compounds of the invention may involve the protection and removal of different chemical groups. Those skilled in the art can easily determine whether protection and removal of protection are needed and the choice of an appropriate protecting group. The chemical nature of the protecting group can be found, for example, in Wuts and Greene, Protective Groups in Organic Synthesis, 4th Edition, John Wiley & Sons: New Jersey, (2006), which is incorporated herein by reference in its entirety.
[0167]
The compound of the present invention can be prepared into a single stereo by reacting a racemic mixture of the compound with an optically active resolving agent to form a pair of diastereomeric compounds, separating the diastereomers, and recovering the optically pure enantiomer isomer. Enantiomeric resolution can be performed using diastereomeric derivatives of the compounds of the present invention, with preferentially dissociable complexes (e.g., crystalline diastereomeric salts). Diastereomers have significantly different physical properties (eg, melting points, boiling points, solubility, reactivity, etc.) and can be easily separated by the advantages of these dissimilarities. Diastereomers can be separated by chromatography, preferably by separation / resolution techniques based on differences in solubility. The optically pure enantiomer is then recovered, along with the resolving reagent, by any practical means that does not allow racemization. A more detailed description of techniques suitable for resolution of stereoisomers of compounds starting from racemic mixtures can be found in Jean Jacques, Andre Collet, Samue1H. Wilen, “Enantiomers, Racemates and Resolution” (“Enantiomers, Racemates and Resolutions “), John Wiley And Sons, Inc., 1981.
[0168]
The reaction can be monitored according to any suitable method known in the art. For example, it may be by spectroscopic means such as nuclear magnetic resonance (NMR) spectroscopy (e.g. 1 H or 13 C), infrared (IR) spectroscopy, spectrophotometry (e.g. UV-visible light), mass spectrometry (MS)) or by chromatography Methods such as high performance liquid chromatography (HPLC) or thin layer chromatography (TLC) to monitor product formation.
[0169]
The compound of formula (I) of the present invention can be prepared by the following reaction scheme 1:
[0170]
Reaction Flowchart 1
[0171]

WO2020011141 / pic / XxJADXdTFKEoDNpTEyy19bUgmH96fty917ouhkO5VZ8DxAcnBrNNXgNmrPfLZTkbnfDDV8tm_ImJg2inA4pPj9gRdLA4C4Y4C4Y4C4Y4C4R4A4
[0172]
Wherein Y 1 , Y 2 , Y 3 , Y 4 , Y 5 , R 1 , R 2 , R 3 , R 4 , X 1 , X 2 , X 3 , X 4 and X 5 are as defined in the present invention. The compound of formula (I) can be obtained by using a compound of formula (I-1) and a sulfonating agent X 5 SO 2 Cl at a suitable base (for example, pyridine, triethylamine, 4- (N, N-dimethylamino) pyridine, etc.) Reaction with a suitable solvent (e.g., dichloromethane, THF, etc.). The reaction is performed at a temperature ranging from about 0 ° C to about 1000 ° C, and may take up to about 20 hours to complete. The reaction mixture is optionally further reacted to remove any protecting groups.
[0173]
The compound of formula (I-1) can be prepared by the following reaction scheme 2:
[0174]
Reaction Flowchart 2
[0175]
WO2020011141 / pic / 0j7t4gaakD7jifc_-mXUo7X65c8la3xpUvQQUfnz6tLaRlcSBbtBx_ehky4qNV0PICK_GRydD0JIoErMNKGqXAa-Pdt7Mtw-IlvJllyprtNJlkwQFY2QFKYFQFY2F2F2F-A
[0176]
Wherein Y 1 , Y 2 , Y 3 , Y 4 , Y 5 , R 1 , R 2 , R 3 , R 4 , X 1 , X 2 , X 3 , X 4 and X 5 are as defined in the present invention, M is a leaving group (for example, iodine, bromine, chlorine, trifluoromethanesulfonyloxy, etc.), each Z may be, for example, hydrogen, methyl, etc., or two Z groups may be connected to form a boric acid ester. Both P groups can be H, or two P groups taken together represent a suitable nitrogen protecting group (eg, one P can be hydrogen and the other can be Boc). The compound of formula (I-2) can be obtained by using a compound of formula (I-4) and a compound of formula (I-3) in a suitable transition metal catalyst (for example, Pd (PPh 3 ) 4 or PdCl 2(dppf)), a suitable solvent (for example, DME, dioxane, toluene, ethanol, etc.) and a suitable base (for example, anhydrous potassium carbonate or sodium carbonate, etc.) are reacted. The reaction is carried out at a temperature ranging from about 20 ° C to 120 ° C, and may take about 2 hours to complete. Compounds of formula (I) can be synthesized by leaving the protecting group P from compounds of formula (I-2) (eg, by treatment with a strong acid such as hydrogen chloride in the presence of DME and dioxane).
[0177]
Compounds of formula (I-4) can be prepared by the following reaction scheme 3:
[0178]
Reaction Flowchart 3
[0179]
WO2020011141 / pic / H1aXUHL0cjl3M_4rpEpbJjUXM5MVl8eWmRAYSGnBPikn5V42NDHXIWwphroHiMSaKEOQI2xHvuG9rOZ0TmtIGAgEd55PYww1WwLNWYpYGOjx5MePjrwW1
[0180]
Wherein Y 1 , Y 2 , R 1 , R 2 , R 3 , R 4 , X 1 , X 2 , X 3 and X 4 are as defined in the present invention, and M is a leaving group (for example, iodine, Bromine, chlorine, trifluoromethanesulfonyloxy, etc.), and V is a leaving group (eg, iodine, bromine, chlorine, trifluoromethanesulfonyloxy, etc.). Compounds of formula (I-4) can be prepared by reacting an amine compound of formula (I-5) and a compound of formula (I-6). The reaction is performed in a suitable solvent (for example, DMSO, NMP, dioxane, or isopropanol) in the presence of a suitable base (for example, sodium carbonate or potassium carbonate, etc.) at a temperature ranging from about 25 ° C to about 120 ° C.
[0182]
The present invention will be further described below with reference to specific embodiments. It should be understood that these examples are only used to illustrate the present invention and not to limit the scope of the present invention. The experimental methods without specific conditions in the following examples are generally based on conventional conditions or conditions recommended by the manufacturer. Unless stated otherwise, parts and percentages are parts by weight and percent by weight.
[0183]
The abbreviations used in this article have the following meanings:
[0184]
[TABLE 0001]
| APCI |
Atmospheric pressure chemical dissociation |
| HPLC |
High performance liquid chromatography |
| TLC |
TLC |
| h |
hour |
| DMF |
N, N-dimethylformamide |
| K 2 CO 3 |
Potassium carbonate |
| DCM |
Dichloromethane |
| THF |
Tetrahydrofuran |
| CH 3 MgBr |
Methyl magnesium bromide |
| PTSA |
p-Toluenesulfonic acid |
| TFA |
Trifluoroacetate |
| NMP |
N-methylpyrrolidone |
| Diguanidinium carbonate |
Guanidine carbonate |
| MTBE |
Methyl tert-butyl ether |
| POCl 3 |
Phosphorus oxychloride |
| DMSO |
Dimethyl sulfoxide |
| Pd (dppf) Cl 2 |
[1,1′-Bis (diphenylphosphino) ferrocene] Palladium dichloride |
| Dioxane |
Dioxane |
| TsCl |
4-toluenesulfonyl chloride |
| Boc |
Tert-butoxy carbon |
| DIPEA |
N, N-diisopropylethylamine |
| CDCl 3 |
Deuterated chloroform |
| TEA |
Triethylamine |
| DMAP |
4-dimethylaminopyridine |
| Na 2 CO 3 |
Sodium carbonate |
| HCl |
hydrochloric acid |
[0185]
[表 0002]
| MsCl |
Methanesulfonyl chloride |
| Tol |
Toluene |
[0186]
Preparation of intermediate A 2-chloro-4- (3-iodo-1- (prop-2-yl-d 7) -1H-pyrazol-4-yl) pyrimidine.
[0188]
Use the following route for synthesis:
[0187]
WO2020011141 / pic / FNMs_XnbU3RObeg6K-VT91xnEa9pD4CszLQIShhoBrnGwf4vFDH7dAkcn-3inZ_bWfKR2ST5u0v_zJNop7mFw4GGCQQ-n-KUOLKt_hScUwRV00GBR1
[0188]
Use the following route for synthesis:
[0189]
WO2020011141 / pic / X5sd0-Eb1TIYnP9Ih5i8tod2iaKSm99ccdy8emg0txiLBrTHdVUkygjUPWlzRjkQFaUW8mpEfWyY68vXxmmbEdx1Q3ZQZFZ1ZYZFZ5ZFJ2
[0190]
Step 1 Synthesis of Compound A-2
[0191]
Compound 1 (5.0 g, 73.4 mmol) was added to a 47% solution of hydrobromic acid (20 ml). The reaction solution was reacted at 80 ° C for 3 hours, and distilled under normal pressure. The 60-70 ° C fraction was collected to obtain a colorless liquid. 6.2g, yield 65%.
[0192]
Step 2 Synthesis of Compound A-4
[0193]
Compound A-3 (3.0 g, 27.8 mmol) was added to a DMF (20 ml) solution, the solution was lowered to 0 ° C, K 2 CO 3 (4.6 g, 33.3 mmol, 10 ml) was added, and the mixture was stirred at low temperature for 0.5 h. Then compound A-2 (4.3g, 33.3mmol) was slowly added dropwise. After the dropwise addition, the temperature was raised to 90 ° C for 10 hours. The reaction solution was extracted with DCM (50ml × 3). The organic phases were combined and dried over anhydrous sodium sulfate. The concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 1: 1) to obtain 3.1 g of a white solid in a yield of 72%. LC-MS (APCI): m / z = 158.21.06 (M + 1) + .
[0194]
Step 3 Synthesis of Compound A-5
[0195]
Under nitrogen protection, compound A-4 (3.0 g, 19.1 mmol) was added to a solution of anhydrous THF (40 ml), and the temperature was lowered to -5 ° C, and CH 3 MgBr (19.1 ml, 57.3 mmol, 3 ml / L) was added dropwise . Anhydrous THF solution. After the dropwise addition was completed, the temperature was gradually raised to reflux for 4 h. The reaction was quenched with saturated ammonium chloride, then the pH was adjusted to neutral with dilute hydrochloric acid, and the mixture was extracted with ethyl acetate (50 ml × 3). The phases were dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 2: 3) to obtain 2.0 g of a yellow solid with a yield of 61%. LC-MS (APCI): m / z = 175.21.06 (M + 1) + .
[0196]
Step 4 Synthesis of Compound A-6
[0197]
Compound A-5 (2.0g, 11.5mmol), PTSA (4.2g, 23.0mmol) were added to the acetonitrile (15ml) solution, and after dropping to 0 ° C, sodium nitrite (1.43g, 20.7mmol) and Aqueous solution (5 ml) of potassium iodide (3.82 g, 23.0 mmol). The reaction solution was reacted at room temperature for 3 hours, and extracted with ethyl acetate (30 ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and spin-dried to obtain 2.5 g of an orange solid with a yield of 75%.
[0198]
Step 5 Synthesis of Compound A-8
[0199]
Under nitrogen protection, compound A-6 (2.0 g, 7.01 mmol) was added to a DMF (15 ml) solution, and the temperature was raised to 120 ° C. Then compound A-7 (1.9 g, 10.5 mmol, 10 ml) was added at 120 ° C. The reaction was stirred for 0.5h. Dichloromethane (30ml × 3) was extracted. The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was separated by column. ) To obtain 1.9 g of the product in a yield of 80%. LC-MS (APCI): m / z = 341.06 (M + 1) + .
[0200]
Step 6 Synthesis of Compound A-9
[0201]
Under nitrogen protection, compound A-8 (1.9 g, 5.6 mmol) and guanidine carbonate (1.6 g, 12.8 mmol) were sequentially added to the NMP (20 ml) solution. At the same time, a water separation device was set up to raise the solution to 130 ° C. The reaction was stirred at 130 ° C for 10 hours. After the reaction was completed, dichloromethane (30ml × 3) was extracted, the organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was separated by column (eluent: petroleum ether / ethyl acetate (v / v) = 2: 3), 1.5 g of product was obtained with a yield of 81%. LC-MS (APCI): m / z = 336.86 (M + 1) + .
[0202]
Step 7 Synthesis of Compound A-10
[0203]
Compound A-9 (1.5 g, 4.5 mmol) was added to the TFA (15 ml) solution. After reducing to 0 ° C, sodium nitrite (0.93 g, 13.4 mmol) was added as a solid. The reaction solution was reacted at room temperature for 1 h. Extract with ethyl acetate (30ml × 3), combine the organic phases, dry over anhydrous sodium sulfate, spin dry the oil with MTBE (10ml), and filter to obtain 1.3g of white solid, 87% yield, LC-MS (APCI) : m / z = 338.15 (M + 1) + .
[0204]
Step 8 Synthesis of intermediate compound A
[0205]
Compound A-10 (1.3 g, 3.86 mmol) was added to a solution of POCl 3 (15 ml), and the temperature was raised to 110 ° C., and the reaction was refluxed at this temperature for 10 h. After the reaction was completed, the reaction solution was spin-dried and dichloromethane (30 ml × 2) Extraction, combined organic phases, dried over anhydrous sodium sulfate, and column separation of the concentrated solution (eluent: petroleum ether / ethyl acetate (v / v) = 4: 1), 1.0 g of product was obtained, yield 73% . LC-MS (APCI): m / z = 356.32 (M + 1) + .
[0206]
Preparation of intermediate B (S)-(methyl-d 3) (1-aminoprop-2-yl) aminocarbonate.
[0208]
Use the following route for synthesis:
[0207]
WO2020011141 / pic / -0strXxact6b2WUIRF3g-qYghbCelI38aof_aRxWyEeaR72see_zBNkAfrwxU-jzi8mdXg4_x4dVwb8bvcLmC0ELLoGLnitco1K2i6cFdUmLPY-LVCRcRcRiOsrQrCsIrOc
[0208]
Use the following route for synthesis:
[0209]
WO2020011141 / pic / luvqF_emaX_eXgTd5ug-arAL8ywwxiu1gGgclql8FZMllvX_6O0eC2cCrB0EEspypcf5ZTRPbOib3MqPf8rPV8752UgYWY2ZwOYZY
[0210]
Step 1 Synthesis of Compound B-2
[0211]
Compound B-1 (1.3 g, 4.5 mmol) was dissolved in a toluene (15 ml) solution, the temperature was lowered to 0 ° C, and CD 3 OD (0.5 g, 15 mmol) and triethylamine (1.7 g, 17 mmol) in toluene were added dropwise . (10ml) solution, reacted at room temperature for 2h after the dropwise addition, washed three times with ice water, dried over anhydrous sodium sulfate, filtered to obtain a toluene solution of compound B-2, and directly used in the next step.
[0212]
Step 2 Synthesis of Compound B-4
[0213]
At 0 ° C, the hydrochloride (0.5 g, 2.4 mmol) and triethylamine (0.73 g, 7.2 mmol) of compound B-3 were added to a solution of dichloromethane (10 ml) in this order, and one step of compound B was added dropwise. -2 toluene solution, reacted for 5 hours at room temperature after the addition, quenched by adding water (10ml), extracted with dichloromethane (20ml × 3), combined organic phases, dried over anhydrous sodium sulfate, and concentrated the column for separation (elution Agent: petroleum ether / ethyl acetate (v / v) = 4: 1), 0.45 g of white solid product was obtained with a yield of 80%.
[0214]
Step 3 Synthesis of intermediate compound B
[0215]
At 0 ° C, a solution of 4M hydrochloric acid in dioxane (4ml) was slowly added to a solution of compound B-4 (0.45g, 1.9mmol) in dichloromethane (10ml), and the reaction was continued at room temperature for 6h. After the reaction was completed, the solution was spin-dried, petroleum ether (10 ml) was slurried, and 0.2 g of the product was obtained by suction filtration with a yield of 77%.
[0216]
Preparation of intermediate C (1-aminoprop-2-yl-1,1,3,3,3-d 5) carbamate.
[0218]
Use the following route for synthesis:
[0217]
WO2020011141 / pic / bZLBsYoBZtulvxpYYI8e5PX_miQYNGkgLgTUstJSMH5SqupQ2PJkQONEOn2GgxHGWmCDZMa-2G5AAvETeF0Qc5Isx_T67ZCJL4_fm2
[0218]
Use the following route for synthesis:
[0219]
WO2020011141 / pic / NoYKNLy2Fhdd3EaVaPfdnESILNKxV3p8R23Zhj7ewo2iRP1aX1fafA7EijayZQiw1sBGSuhkSMC5kcA3OJoo4VaSIpow2Qpww2wwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwwzw
[0220]
Step 1 Synthesis of compound C-3
[0221]
A mixture of compound C-2 (4.6 g, 61.8 mmol), compound C-1 (11.5 g, 67.6 mmol) and sodium hydroxide (7.16 g, 67.7 mmol) in water (60 ml) was stirred and reacted at 0 ° C for 3 h. After the reaction was completed, water (60 ml) was added, and the mixture was extracted with ethyl acetate (60 ml x 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 10: 1), 11.2 g of an oily substance was obtained, and the yield was 88%.
[0222]
Step 2 Synthesis of Compound C-4
[0223]
Under a nitrogen atmosphere, DMSO (4.8 g, 61.5 mmol) was slowly added to a solution of oxalyl chloride (6.0 g, 47.2 mmol) in DCM (60 ml) at -78 ° C, and the mixture was stirred at -78 ° C for half an hour. Then, a solution of compound C-3 (8.0 g, 38.2 mmol) in DCM (20 ml) was added to the mixture, and the mixture was further stirred at -78 ° C for 1 h, and then triethylamine (16 ml) was added to the mixture, and the mixture was raised to At room temperature, it was washed with 1N hydrochloric acid (50ml) and sodium bicarbonate aqueous solution (50ml) successively. The organic phase was dried over anhydrous sodium sulfate, heat-shrinked, and then slurried with a volume ratio of PE: EA = 8: 1 to obtain 5.3g of a white solid product. The rate is 87%.
[0224]
Step 3 Synthesis of Compound C-5
[0225]
1,5,7-Triazabicyclo [4.4.0] dec-5-ene (0.27 g, 1.9 mmol) was added to a solution of compound C-4 (4.0 g, 19.3 mmol) in deuterated chloroform (30 ml) After the reaction solution was stirred at room temperature for 30 hours, water (10 ml) was added to quench the reaction, and the organic phase was separated and washed with saturated sodium chloride. The organic phase was dried and spin-dried to obtain 3.9 g of an oil with a yield of 98%.
[0226]
Step 4 Synthesis of compound C-6
[0227]
Under a nitrogen atmosphere, compound C-5 (4.0 g, 18.9 mmol) and tert-butylsulfinamide (2.7 g, 22.6 mmol) were added to the THF (60 ml) solution, and tetraisopropyl titanate was added at room temperature. Ester (11.8 g, 41.5 mmol), and then heated to 60 ° C for 3 h. The reaction solution was cooled to room temperature, quenched by adding water, and extracted with ethyl acetate (60 ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate ( v / v) = 4: 1), 3.5 g of product was obtained with a yield of 58%. LC-MS (APCI): m / z = 315.80 (M + 1) + .
[0228]
Step 5 Synthesis of compound C-7
[0229]
At -50 ° C, NaBH 4 (0.73 g, 19.1 mmol) was added to a solution of compound C-6 (2.0 g, 6.3 mmol) in methanol (20 ml), and then the reaction was continued at low temperature for 1 h. 1M hydrochloric acid was added to quench the reaction, and the mixture was extracted with dichloromethane (30 ml × 2). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and spin-dried to obtain 2.1 g of an oily product.
[0230]
Step 6 Synthesis of compound C-8
[0231]
A solution of 4M hydrochloric acid in dioxane (10 ml) was slowly added to a solution of compound C-7 (2.0 g, 6.3 mmol) in dichloromethane (20 ml) at 0 ° C, and the reaction was continued at 0 ° C for 6 h. After the reaction is completed, the solvent is spin-dried and directly used in the next step without further processing.
[0232]
Step 7 Synthesis of compound C-9
[0233]
Triethylamine (1.43 g, 14.1 mmol) was added to a solution of compound C-8 (1.5 g, 7.0 mmol) in dichloromethane (20 ml) at 0 ° C, and methyl chloroformate was added dropwise to the mixture. (0.8g, 8.5mmol), and react at room temperature for 5 hours after the addition. After the reaction is complete, water (10ml) is added to quench the reaction. The reaction solution is extracted with dichloromethane (20ml × 2). Sodium was dried, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 4: 1) to obtain 1.1 g of a white solid product with a yield of 58%.
[0234]
Step 8 Synthesis of intermediate compound C
[0235]
Under a hydrogen atmosphere, Pd-C (0.2g, 10%) was added to the compound C-9 (1.0g, 3.7mmol) in ethanol (5ml) and a 1N hydrochloric acid solution (5ml), and the reaction was stirred for 5h. After the reaction was completed, It was filtered and the filtrate was directly concentrated to obtain 0.4 g of the product.
[0236]
Example 1 (S) -methyl- (1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1- (propan-2-yl -d 7) Preparation of 1H-pyrazol-4-yl) pyrimidin-2-yl) amino) propan-2-yl) carbamate (compound L-1).
[0238]
Use the following route for synthesis:
[0237]
WO2020011141 / pic / 3xtiuTx657XV12_fky8oaKP_xXwX4wCXzmrOFYj-6WrLGfn7RokqPCy3lz6vK0t_oUjqoYktURzPEI8R4Z4fga0Yw0QXQQWYQZYUZTYWYQT
[0238]
Use the following route for synthesis:
[0239]
WO2020011141 / pic / kZCwkP7P-x1L3nCmUBMv9tcq80zMDMHYE9GLLB13iwjtMkE58H7GHYCHtBFrk_OoAPcX1xuC9dLyLTpjsyBA2GaUqv2D2XU2C2R2C2R2C2R2C2B2C2D2C2C2B2
[0240]
Step 1 Synthesis of Compound 2
[0241]
Under nitrogen protection, intermediate compound A (1.0 g, 2.8 mmol), compound 1 (0.52 g, 3.1 mmol), and sodium carbonate (1.2 g, 11.2 mmol) were sequentially added to the DMSO (20 ml) solution, and the temperature was raised to 90 ° C. The reaction was stirred at this temperature for 16h. After the reaction was completed, DCM (30ml × 3) was extracted, the organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was separated by column (eluent: petroleum ether / ethyl acetate (v / v) = 1: 2), 0.8 g of product was obtained with a yield of 63%. LC-MS (APCI): m / z = 452.33 (M + 1) + .
[0242]
Step 2 Synthesis of Compound 4
[0243]
Under nitrogen protection, compound 2 (0.5 g, 1.11 mmol), compound 3 (0.5 g, 1.33 mmol), sodium carbonate (0.47 g, 4.43 mmol), and Pd (dppf) Cl2 (0.09 g, 0.11 mmol) were added in this order. Into a mixed solution of toluene (20 ml) and water (4 ml), heated to 80 ° C. for 2 h. The reaction solution was cooled to room temperature, extracted with ethyl acetate (30 ml × 3), the organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 1: 1) 0.2 g of product was obtained with a yield of 31%. LC-MS (APCI): m / z = 569.09 (M + 1) + .
[0244]
Step 3 Synthesis of Compound 5
[0245]
At 0 ° C, a solution of 4M hydrochloric acid in dioxane (4ml) was slowly added to a solution of compound 4 (0.2g, 0.35mmol) in DCM (10ml), and the reaction mixture was warmed to room temperature for 6h. After the reaction is complete, the solution is spin-dried and directly sent to the next step without further processing. LC-MS (APCI): m / z = 469.27 (M + 1) + .
[0246]
Step 4 Synthesis of Compound L-1
[0247]
Compound 5 (0.15 g, 0.32 mmol) and triethylamine (0.16 g, 1.6 mmol) were sequentially added to the DCM (10 ml) solution. After the temperature was lowered to 0 ° C, MsCl (0.11 g, 1.0 mmol) was slowly added dropwise. After the addition was completed, the reaction temperature was raised to room temperature for 5 hours. After the reaction was completed, the reaction solution was spin-dried to obtain a residue. Toluene (9 ml), methanol (1 ml), water (10 ml), and sodium carbonate (2 g) were sequentially added to the residue. The reaction temperature was raised to 85 ° C for 10 hours, cooled to room temperature, and extracted with ethyl acetate (20ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: dichloromethane / methanol (v / v) = 20: 1), 50 mg of product was obtained with a yield of 35%. LC-MS (APCI): m / z = 547.31 (M + 1) + . 1 H NMR (400MHz, CDCl 3 ) δ 8.08 (d, J = 11.4 Hz, 2H), 7.61 (d, J = 6.3 Hz, 1H), 7.42 (d, J = 5.6 Hz, 1H), 6.48 (d , J = 5.1 Hz, 1H), 5.32 (d, J = 18.8 Hz, 1H), 5.17 (s, 1H), 4.59 (d, J = 13.2 Hz, 1H), 3.79 (s, 1H), 3.61 (s , 3H), 3.24 (s, 1H), 2.98 (d, J = 16.6Hz, 3H), 2.01 (s, 1H), 1.31 (s, 3H).
[0248]
Example 2 (S)-(methyl-d 3)-(1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1-iso Preparation of propyl-1H-pyrazol-4-yl) pyrimidin-2-yl) amino) propan-2-yl) carbamate (compound L-2).
[0249]
WO2020011141 / pic / tVDfDEoOqWI5X7v8Kaju3q5h9JqkTve6llLuavobFC_1bh4Bp_PcG7AbdlZy5eFwRexqa8OY2mQ_WQBTMQu5Ce-x7qWisFmuvIijUJGQ7JhMqHf6vDSCLDW8ySQjx0v3LUA6YMGFZwOYZJznC59drnUBFfVdu6tdIqqvonWRiGg “>
[0250]
Use the following route for synthesis:
[0251]
WO2020011141 / pic / m9mXD-mrSGFj20R47ROzFF6keVQ70kCzBace3esKjuDXwTUrjQQweunbgPzPIPpGrRj1It6FgZXqv5ywjyC2eHI6VD0F0D0f0FJ1DKfY1D1KVFY1D1F1D1F2D2F2D2F2D2D2D2F2D2D2D2D2D2D2D2D2D2D2D2D2D2D2D2D2D2D2D2d2d2d2d2d2ddffd1d2d2dffd2d2dffd2ddfffd1d2d2dffd1ddffj1nKixYeQ2ohmGYVDVF7F7R2
[0252]
Step 1 Synthesis of compound 7
[0253]
Under nitrogen protection, compound 6 (0.5 g, 1.5 mmol), intermediate compound B (0.2 g, 1.5 mmol), and sodium carbonate (0.63 g, 6.0 mmol) were sequentially added to the DMSO (20 ml) solution, and the temperature was raised to 90 ° C. The reaction was stirred at this temperature for 16h. After the reaction was completed, DCM (30ml × 3) was extracted, the organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was separated by column (eluent: petroleum ether / ethyl acetate (v / v) = 1: 2), 0.42 g of product was obtained in a yield of 65%. LC-MS (APCI): m / z = 447.80 (M + 1) + .
[0254]
Step 2 Synthesis of Compound 8
[0255]
Under nitrogen protection, compound 7 (0.4 g, 0.90 mmol), compound 3 (0.4 g, 1.07 mmol), sodium carbonate (0.38 g, 3.6 mmol), and Pd (dppf) Cl2 (0.07 g, 0.09 mmol) were added in this order. Into a mixed solution of toluene (20 ml) and water (4 ml), the mixture was heated to 80 ° C. and reacted for 2 h. The reaction solution was cooled to room temperature, extracted with ethyl acetate (30 ml × 3), the organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 1: 1) 0.2 g of product was obtained with a yield of 40%. LC-MS (APCI): m / z = 565.03 (M + 1) + .
[0256]
Step 3 Synthesis of Compound 9
[0257]
At 0 ° C, a solution of 4M hydrochloric acid in dioxane (4 ml) was slowly added to a solution of compound 8 (0.2 g, 0.35 mmol) in DCM (10 ml), and the reaction mixture was warmed to room temperature and continued to react for 6 h. After the reaction is complete, the solution is spin-dried and directly sent to the next step without further processing. LC-MS (APCI): m / z = 465.27 (M + 1) + .
[0258]
Step 4 Synthesis of Compound L-2
[0259]
Compound 9 (0.2 g, 0.43 mmol) and triethylamine (0.22 g, 2.1 mmol) were sequentially added to the DCM (10 ml) solution. After lowering to 0 ° C, MsCl (0.15 g, 1.3 mmol) was slowly added dropwise. After the addition was completed, the reaction temperature was raised to room temperature for 5 hours. After the reaction was completed, the reaction solution was spin-dried to obtain a residue. Toluene (9 ml), methanol (1 ml), water (10 ml), and sodium carbonate (2 g) were sequentially added to the residue. The reaction temperature was raised to 85 ° C for 10 hours, cooled to room temperature, and extracted with ethyl acetate (20ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: dichloromethane / methanol (v / v) = 20: 1), 70 mg of product was obtained with a yield of 30%. LC-MS (APCI): m / z = 543.21 (M + 1) + . 1 H NMR (400MHz, CDCl 3 ) δ 8.01 (d, J = 11.4 Hz, 2H), 7.63 (d, J = 6.3 Hz, 1H), 7.40 (d, J = 5.8 Hz, 1H), 6.58 (d , J = 6.1 Hz, 1H), 5.47 (d, J = 18.8 Hz, 1H), 5.17 (s, 1H), 4.59 (d, J = 12.2, Hz, 1H), 3.80 (s, 1H), 3.61 ( s, 1H) 3.24 (s, 1H), 3.10 (d, J = 16.6 Hz, 3H), 2.21 (s, 1H), 1.35 (s, 3H), 1.27 (d, 6H).
[0260]
Example 3 (S)-(methyl-d 3)-(1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1- ( Preparation of prop-2-yl-d 7) -1H-pyrazol-4 -yl) pyrimidin-2-yl) amino) prop-2-yl) carbamate (compound L-3).
[0261]
WO2020011141 / pic / iqj6pvdjjM4HOwS5mON3pOQ9HR7saOazmNYNpzaiXojjcGBiI6WDlFm3cKezb4yS-LlWgLP3UOsiRLU-U82AHxNXxfErtH82vSuy7aRZyypOhFxSIKcmsU1IrgUTfZfHvHyV7GUrqgilmX3Uhs5HqB4J8lAtCQzt3Usg8oMeezs “>
[0262]
Take the following synthetic route:
[0263]
WO2020011141 / pic / YwVS_N4uouPkEHjeYuqZOHrNDrfCXIg0xzYvgPjs2CnKzWkQFiTy2WMm9EsgMfhElppKsKCS5sgXcDsnhYWWYWWYWWYVWYWYWW
[0264]
Step 1 Synthesis of compound 10
[0265]
Under nitrogen protection, intermediate compound A (0.6 g, 1.7 mmol), intermediate compound B (0.23 g, 1.7 mmol), and sodium carbonate (0.71 g, 6.8 mmol) were added to the DMSO (20 ml) solution in this order, and the temperature was raised to The reaction was stirred at 90 ° C for 16h at this temperature. After the reaction was completed, DCM (30ml × 3) was extracted, the organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate ( v / v) = 1: 2), 0.65 g of product was obtained with a yield of 84%. LC-MS (APCI): m / z = 454.92 (M + 1) + .
[0266]
Step 2 Synthesis of Compound 11
[0267]
Under nitrogen protection, compound 10 (0.6 g, 1.3 mmol), compound 3 (0.59 g, 1.6 mmol), sodium carbonate (0.56 g, 5.3 mmol), and Pd (dppf) Cl2 (0.10 g, 0.13 mmol) were added in this order. Into a mixed solution of toluene (20 ml) and water (4 ml), heated to 80 ° C. for 2 h. The reaction solution was cooled to room temperature, extracted with ethyl acetate (30 ml × 3), the organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 1: 1) 0.32 g of the product was obtained with a yield of 43%. LC-MS (APCI): m / z = 572.10 (M + 1) + .
[0268]
Step 3 Synthesis of Compound 12
[0269]
At 0 ° C, a solution of 4M hydrochloric acid in dioxane (4 ml) was slowly added to a solution of compound 11 (0.3 g, 0.52 mmol) in DCM (10 ml), and the reaction mixture was warmed to room temperature and continued to react for 6 h. After the reaction is complete, the solution is spin-dried and directly sent to the next step without further processing. LC-MS (APCI): m / z = 472.09 (M + 1) + .
[0270]
Step 4 Synthesis of Compound L-3
[0271]
Compound 12 (0.25 g, 0.53 mmol) and triethylamine (0.27 g, 2.6 mmol) were sequentially added to the DCM (10 ml) solution. After dropping to 0 ° C, MsCl (0.18 g, 1.6 mmol) was slowly added dropwise. After the addition was completed, the reaction temperature was raised to room temperature for 5 hours. After the reaction was completed, the reaction solution was spin-dried to obtain a residue. Toluene (9 ml), methanol (1 ml), water (10 ml), and sodium carbonate (2 g) were sequentially added to the residue. The reaction temperature was raised to 85 ° C for 10 hours, cooled to room temperature, and extracted with ethyl acetate (20ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: dichloromethane / methanol (v / v) = 20: 1), 75 mg of product was obtained with a yield of 26%. LC-MS (APCI): m / z = 550.29 (M + 1) + . 1 H NMR (400 MHz, CDCl 3 ) δ 8.13 (d, J = 11.4 Hz, 2 H), 7.63 (d, J = 6.3 Hz, 1 H), 7.40 (d, J = 5.8 Hz, 1 H), 6.65 (d , J = 6.1 Hz, 1H), 5.47 (d, J = 18.8 Hz, 1H), 5.17 (s, 1H), 4.63 (d, J = 12.2, Hz, 1H), 3.70 (s, 1H), 3.54 ( s, 1H), 3.16 (d, J = 16.6 Hz, 3H), 2.11 (s, 1H), 1.38 (s, 3H).
[0272]
Example 4 (1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1- (prop-2-yl) -1H-pyrazole- 4- yl) pyrimidin-2-yl) amino ) propan-2-yl -1,1,3,3,3-d 5) carbamate (compound L-4),
[0273]
(S)-(1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1- (prop-2-yl) -1H-pyrazole 4-yl) pyrimidin-2-yl) amino) propan-2- yl-1,1,3,3,3-d 5) methyl carbamate (compound L-4-S) and
[0274]
(R)-(1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1- (prop-2-yl) -1H-pyrazole 4-yl) pyrimidin-2-yl) amino) propan-2- yl-1,1,3,3,3-d 5) Preparation of methyl carbamate (compound L-4-R).
[0275]
WO2020011141 / pic / m0IN31dnhItfm5H-dGFizFalHv9quUKvHfmY4zFpAaHFgTp-0iUzxdHuZwlvRxqTStKdio_PlNaIPfHi8pthED3hbMalT8GyFmZ1tCDOIKmutZCiuLJ4FJW4WY
[0276]
Take the following synthetic route:
[0277]
WO2020011141 / pic / fjV2PIKmugqfUgshQfiVwrkjSTGfhIl9ZWz96JIiDMEhwjAlTOxFStuhxFFooUqAr0FVv7GXsyKUDxeLYZl-uQQWMt1C9_9Zi9U9U9Zi9U9U
[0278]
Step 1 Synthesis of compound 13
[0279]
Under nitrogen protection, compound 6 (0.4 g, 1.1 mmol), intermediate compound C (0.16 g, 1.1 mmol), and sodium carbonate (0.50 g, 4.6 mmol) were sequentially added to the DMSO (15 ml) solution, and the solution was raised to The reaction was stirred at 90 ° C at this temperature for 16 hours. After the reaction was completed, the reaction solution was extracted with dichloromethane (30 ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether). / Ethyl acetate (v / v) = 1: 2) to obtain 0.40 g of the product in a yield of 75%. LC-MS (APCI): m / z = 449.53 (M + 1) + .
[0280]
Step 2 Synthesis of Compound 14
[0281]
Under a nitrogen atmosphere, compound 13 (0.4 g, 0.9 mmol), compound 3 (0.5 g, 1.4 mmol), sodium carbonate (0.40 g, 3.56 mmol), and Pd (dppf) Cl 2 (0.08 g, 0.1 mmol) were added in this order. Into a mixed solution of toluene (20 ml) and water (4 ml), heated to 80 ° C. for 2 h. The reaction solution was cooled to room temperature, and then extracted with ethyl acetate (30 ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 1: 1), 0.28 g of product was obtained with a yield of 55%. LC-MS (APCI): m / z = 567.12 (M + 1) + .
[0282]
Step 3 Synthesis of Compound 15
[0283]
At 0 ° C, a solution of 4M hydrochloric acid in dioxane (2ml) was slowly added to a solution of compound 14 (0.28g, 0.50mmol) in dichloromethane (10ml), and the reaction was continued at room temperature for 6h. After the reaction is completed, the solution is directly spin-dried and directly sent to the next step without further processing. LC-MS (APCI): m / z = 467.29 (M + 1) + .
[0284]
Step 4 Synthesis of compound L-4
[0285]
Triethylamine (0.13 g, 1.28 mmol) was added to a solution of compound 15 (0.2 g, 0.43 mmol) in dichloromethane (10 ml). After the solution was lowered to 0 ° C, methanesulfonyl chloride (0.15 g, 1.3 mmol) was slowly added dropwise to the upper solution. The reaction solution was reacted at room temperature for 5 hours. After the reaction was completed, the reaction solution was spin-dried. To the residue were added toluene (9 ml), methanol (1 ml), and water (10 ml). Sodium carbonate (2g), the solution was reacted at 85 ° C for 10h, the reaction solution was cooled to room temperature, and then extracted with ethyl acetate (20ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation ( Eluent: dichloromethane / methanol (v / v) = 20: 1) to obtain 65 mg of the product with a yield of 27%. LC-MS (APCI): m / z = 545.08 (M + 1) + . 1 H NMR (400MHz, CDCl 3 ) δ8.05 (d, 2H), 7.61 (d, 1H), 7.45 (d, 1H), 6.40 (d, 1H), 5.29 (d, 1H), 5.18 (s, 1H), 4.62 (d, 1H), 3.89 (d, 1H), 3.58 (s, 3H), 3.10 (d, 3H), 2.05 (s, 1H), 1.29 (d, 6H).
[0286]
Step 5 Synthesis of compounds L-4-S and L-4-R
[0287]
The racemic compound L-4 was separated using a chiral preparative column to obtain compounds L-4-S and L-4-R.
[0288]
Example 5 (1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1- (prop-2-yl-d 7))-1H -Pyrazol-4-yl) pyrimidin-2-yl) amino) propan-2-yl-1,1,3,3,3-d 5) methyl carbamate (compound L-5),
[0289]
(S)-(1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1- (prop-2-yl-d 7))- 1H-pyrazol-4-yl) pyrimidin-2-yl) amino) propan-2-yl-1,1,3,3,3-d 5) methyl carbamate (compound L-5-S) and
[0290]
(R)-(1-((4- (3- (5-chloro-2-fluoro-3- (methylsulfonylamino) phenyl) -1- (prop-2-yl-d 7))- 1H-pyrazol-4-yl) pyrimidin-2-yl) amino) propan-2-yl-1,1,3,3,3-d 5) Preparation of methyl carbamate (compound L-5-R) .
[0291]
WO2020011141 / pic / 4br07jLUTScNPUcnWdxxTyAAMGS9P15P0yXUsyhcCny-ABv5BZExa5YOY-Hj3wTZWdByUUB-EQbGG-h4QuoddgCTRMClBcl1WY1TjnTsnDDYTZxC6-taMQZYW1Z1WY
[0292]
WO2020011141 / pic / By6lfXwpBcoklf-47-VujG_XNVWV7ZjYOo73wMiKwo9v4cKff0K2As3lqLKG1kFOYG87EWp6SIobdq2gtEFMnxfVVVJVYVZGYZFYZVYG-ZVY-ZFY-ZF
[0293]
Take the following synthetic route:
[0294]
WO2020011141 / pic / dMfm7g9kIiR87Eo-VsdQ2-2wcdHuYsfKuUWOyKuR4SUJ3Kmoy907w2C1tLHvEDhc4vBBT2l48TSysgdivcFJmRqGQNZWYQZNYWQD
[0295]
Step 1 Synthesis of compound 16
[0296]
Under nitrogen protection, intermediate compound A (0.5 g, 1.5 mmol), intermediate compound C (0.2 g, 1.5 mmol), and sodium carbonate (0.63 g, 6.0 mmol) were added to the DMSO (20 ml) solution in this order. The temperature was raised to 90 ° C, and the reaction was stirred at this temperature for 16 hours. After the reaction was completed, the reaction solution was extracted with dichloromethane (30ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: Petroleum ether / ethyl acetate (v / v) = 1: 2) to obtain 0.45 g of the product with a yield of 68%. LC-MS (APCI): m / z = 456.68 (M + 1) + .
[0297]
Step 2 Synthesis of Compound 17
[0298]
Under a nitrogen atmosphere, compound 16 (0.45 g, 0.98 mmol), compound 3 (0.55 g, 1.54 mmol), sodium carbonate (0.42 g, 3.95 mmol), and Pd (dppf) Cl 2 (0.08 g, 0.1 mmol) were sequentially added Into a mixed solution of toluene (20 ml) and water (4 ml), heated to 80 ° C. for 2 h. The reaction solution was cooled to room temperature, and then extracted with ethyl acetate (30 ml × 3). The organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation (eluent: petroleum ether / ethyl acetate (v / v) = 1: 1), 0.40 g of the product was obtained in a yield of 70%. LC-MS (APCI): m / z = 574.16 (M + 1) + .
[0299]
Step 3 Synthesis of Compound 18
[0300]
A solution of 4M hydrochloric acid in dioxane (2 ml) was slowly added to a solution of compound 17 (0.40 g, 0.70 mmol) in dichloromethane (10 ml) at 0 ° C, and the reaction was allowed to proceed to room temperature for 6 h. After the reaction is completed, the solution is directly spin-dried and directly sent to the next step without further processing. LC-MS (APCI): m / z = 474.21 (M + 1) + .
[0301]
Step 4 Synthesis of Compound L-5
[0302]
Triethylamine (0.23 g, 2.21 mmol) was added to a solution of compound 18 (0.35 g, 0.74 mmol) in dichloromethane (10 ml). After the solution was lowered to 0 ° C, methanesulfonyl chloride (0.25 g, 2.2 mmol) was slowly added dropwise to the upper solution. The reaction solution was reacted at room temperature for 5 hours. After the reaction was completed, the reaction solution was spin-dried. To the residue were added toluene (9 ml), methanol (1 ml), and water (10 ml). Sodium carbonate (2g), the solution was reacted at 85 ° C for 10h, the reaction solution was cooled to room temperature, and then extracted with ethyl acetate (20ml x 3), the organic phases were combined, dried over anhydrous sodium sulfate, and the concentrated solution was subjected to column separation Eluent: dichloromethane / methanol (v / v) = 20: 1) to obtain 120 mg of the product in a yield of 30%. LC-MS (APCI): m / z = 552.33 (M + 1) + . 1 H NMR (400MHz, CDCl 3 ) δ 8.02 (d, 2H), 7.61 (d, 1H), 7.45 (d, 1H), 6.40 (d, 1H), 5.22 (d, 1H), 5.18 (s, 1H), 4.59 (d, 1H), 3.58 (s, 3H), 2.98 (d, 3H), 2.05 (s, 1H).
[0303]
Step 5 Synthesis of compounds L-5-S and L-5-R
[0304]
The racemic compound L-4 was separated using a chiral preparative column to obtain compounds L-5-S and L-5-R.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


