
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
Quizartinib dihydrochloride
キザルチニブ塩酸塩
| Formula |
C29H32N6O4S. 2HCl
|
|---|---|
| CAS |
1132827-21-4
|
| Mol weight |
633.5891
|
JAPAN, PMDA APPROVED,. 2019/6/18, Vanflyta, To use as part of a treatment regimen for newly diagnosed acute myeloid leukemia that meets certain criteria
Drug Trials Snapshot
fda 2023, 7/20/2023
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor, originated Ambit Biosciences, and acquired by Daiichi Sankyo, that is currently under development for the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.
Clinical trials
It reported good results in 2012 from a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
As of 2017 it has completed 5 clinical trials and another 7 are active.[3]
SYN

References
- ^ Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry. 52 (23): 7808–7816. doi:10.1021/jm9007533.
- ^ Drug Tames Refractory AML. ASH Dec 2012
- ^ Quizartinib studies
 |
|
| Names | |
|---|---|
| IUPAC name
1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea
|
|
| Other names
AC220
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| KEGG | |
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C29H32N6O4S | |
| Molar mass | 560.67 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////Quizartinib dihydrochloride, キザルチニブ塩酸塩, JAPAN 2019
OLD POST

N-(5-tert-butyl-isoxazol-3-yl)-N’-{ 4- [7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea
| CAS | 950769-58-1 (free base) 1132827-21-4 (2HCl) |
| Formula | C29H32N6O4S |
| MW | 560.7 |
| Synonim | AC220, AC-010220 ASP-2689 |


Quizartinib
Ambit Biosciences
Ambit Biosciences (NASDAQ:AMBI) is a biotech company that focuses on treatments that inhibit kinases, which are drivers for diseases such as cancer. Three drugs are in development, with the lead one being quizartinib — a Phase 2B trial treatment for acute myeloid leukemia. However, AMBI’s collaboration agreement with Astellas Pharma is set to expire in September, and if it is not replaced, it could mean a delay in Phase 3 trials for quizartinib. Keep in mind that AMBI generated $23.8 million in collaboration revenues last year.
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor that is currently under development by Ambit Biosciencesfor the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]

AC-220 is an angiogenesis inhibitor that antagonizes several proteins involved in vascularization. It was engineered by Ambit Biosciences using KinomeScan technology to potently target FLT3, KIT, CSF1R/FMS, RET and PDGFR kinases. Ambit is developing oral AC-220 in phase III clinical studies for the treatment of relapsed/refractory acute myeloid leukemia (AML) patients with the FMS-like tyrosine kinase-3 (FLT3)-ITD mutation. Early clinical trials are also ongoing for the treatment of advanced solid tumors, for the treatment of refractory or relapsed myelodysplasia, in combination with induction and consolidation chemotherapy for previously-untreated de novo acute myeloid leukemia, and as a maintenance therapy of AML following hematopoietic stem cell transplantation (HSCT). In 2009, orphan drug designation was received both in the U.S. and in the EU for the treatment of AML. In 2009, Ambit Biosciences and Astellas Pharma have entered into a worldwide agreement to jointly develop and commercialize the drug candidate for the treatment of cancer and non-oncology indications. This agreement was terminated in 2013.
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
| Quizartinib is a small molecule with potential anticancer activity. Quizartinib is a selective inhibitor of class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), stem cell factor receptor (SCFR / KIT), colony-stimulating factor 1 receptor (CSF1R/FMS) and platelet-derived growth factor receptors (PDGFRs .) Able to inhibition of ligand-independent cell proliferation and apoptosis. Mutations in FLT3 are the most frequent genetic alterations in acute myeloid leukemia (AML) and occur in approximately 30% of cases of AML. | |
| Quizartinib представляет собой малую молекулу с потенциальной противораковой активностью. Quizartinib является селективным ингибитором класса III рецепторов тирозин киназ, в том числе FMS-связанных тирозинкиназы 3 (FLT3/STK1), фактор стволовых клеток рецепторов (SCFR / KIT), колониестимулирующий фактор 1 рецепторов (CSF1R/FMS) и тромбоцитарный рецепторов фактора роста (PDGFRs). Способен к торможению лиганд-независимой клеточной пролиферации и апоптоза. Мутации в FLT3 являются наиболее частыми генетическими изменениями в остром миелобластном лейкозе (ОМЛ) и встречаются примерно в 30% случаев ОМЛ. |
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 leading to resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.

Clinical trials
It had good results in a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
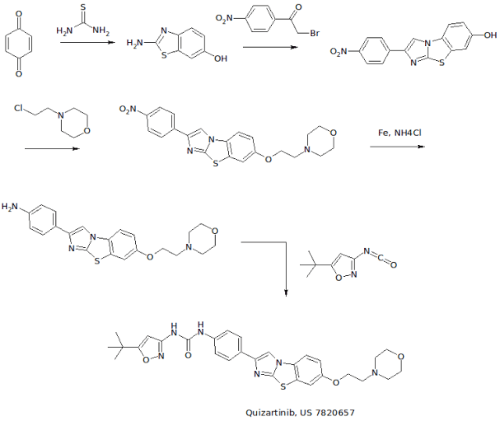

………………………..
WO 2007109120 COMPD B1
EXAMPLE 3: PREPARATION OF N-(5-TERT-BUTYL-ISOXAZOL-3-YL)-N’-{4-[7-(2- MORPHOLIN-4-YL-ETHOXY)IMIDAZO[2,1 -B3[1 ,3]BENZOTHIAZOL-2-YL]PHENYL}UREA [Compound B1]
[00426] A. The intermediate 2-amino-1,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf. J. Org. Chem. 1970, 35, 4103-4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 ml_ ethanol and 9 ml_ concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00427] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2-amino-1 ,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSOd6) £7.6 (m, 2H ), 6.6 (d, 1H).
[00428] B. To prepare the intermediate 2-(4-nitrophenyl)imidazo[2,1- b][1 ,3]benzothiazoI-7-ol, 2-amino-1 ,3-benzothiazol-6-ol, (20.0 g, 0.12 mol) and 2-bromo-4′-nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to 00C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2-(4- nitrophenyl)imidazo[2,1-_D][1,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-CT6) δ 10 (s, 1 H), 8.9 (s, 1H), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, 1 H), 7.4 (s, 1 H), 6.9 (d, 1 H). [00429] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nttro- phenyl)imidazo[2,1-£>][1 ,3]benzothiazo!e intermediate: 2-(4- nitrophenyl)imidazo[2,1-jb][1 ,3]benzothiazol-7-ol, (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-jb][1 ,3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-Cf6) δ 8.97 (s, 1 H), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, 1 H), 7.7 (s, 1 H), 7.2 (d, 1 H), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00430] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4- amino-phenyl)!midazo[2, 1 -b][1 ,3]benzothiazole: To a suspension of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-ib][1 ,3]benzothiazole (3.87g, 9.1 mmol) in 100 ml_ isopropyl alcohol/water (3:1 ) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 ml_). The filtrate was concentrated to approximately 1/3 of the original , volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water — in which case the reaction is virtually complete after 3.5 hours and oniy traces of starting material are observed in the product obtained. NMR (DMSO-d6) δ 8.4 (s, 1 H), 7.8 (d, 1 H), 7.65 (d, 1 H), 7.5 (d, 2H), 7.1 (d, 1 H), 6.6 (d, 2H), 4.1 (t, 2H)1.3.6 (m, 4H), 2.7 (t, 2H).
[00431] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,1-b][1 ,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert- butylisoxazole-3-isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base. Yield: 2.342 g (41 %) NMR (DMSO-Cf6) £9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, 1 H), 7.76 (d, 2H), 7.69 (d, 1 H), 7.51 (d, 2H), 7.18 (dd, 1H), 6.52 (s, 1H), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCI3) £9.3 (br, 1H), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, 1 H), 7.22 (d, 1 H), 7.03 (dd, 1 H)1 5.88 (s, 1 H), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
[00432] F. For the preparation of the hydrochloride salt, N-(5-tert-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2, 1 – b][1 ,3]benzothiazol-2-yl]phenyI}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCI in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrifugation and washed with ethyl ether to give the hydrochloride salt. Yield: 2.44 g (98 %) NMR (DMSO-d6) £11-0 (br, 1 H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1 H), 7.93 (d, 1H), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, 1H), 6.53 (S, 1 H), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H)13.23 (m, 2H)1 1.30 (s, 9H).
[00433] G. Alternatively, Compound B1 may be made by taking the intermediate from Example 4B and reacting it with chloroethyl morpholine hydrochloride under conditions described in Step C. [00434] H . Λ/-(5-tert-butyl-isoxazol-3-yl)-Λ/’-{4-[5-(2-morpholin-4-yl- ethoxy)imidazo[2,1-6][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride, a compound having the general formula (I) where R1 is substituted on the 5 position of the tricyclic ring, was prepared in the manner described in Steps A- F but using the cyciization product 2-amino-benzothiazol-4-ol with 2-bromo-4′- nitroacetophenone in Step A. 1H NMR (DMSO-d6) δ 11.6 (br, 1 H)1 9.78 (br, 1H), 9.56 (br, 1 H), 8.64 (s, 1H)1 7.94 (d, 2H), 7.70 (s, 1H)1 7.56 (d, 2H), 7.45 (t, 1 H), 7.33 (d, 1H), 6.54 (s, 1 H), 4.79 (t, 2H), 3.87 (m, 6H), 3.60 (m, 2H), 3.34 (m, 2H)1 1.30 (s, 9H); LC-MS: ESI 561 (M+H)+. [Compound B11] [00435] I. N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[6-(2-morpholin-4-yl- ethoxy)imidazo[2,1-b][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride [Compound B12] was also prepared by first preparing the benzothiazole starting material, 5 methoxy-benzothiazol-2yl~amine: [00436] To prepare the 5-methoxy-benzothiazol-2-ylamine starting material: To a suspension of (3-methoxy-phenyl)-thiourea (1.822g, 10 mmol) in CH2CI2 (20 ml_) at 0 0C was added dropwise a solution of bromine (1.76 g, 11 mmol) in 10 ml of trichloromethane over a period of thirty minutes. The reaction was stirred for 3 hours at room temperature then heated to 3 hours to reflux for one hour. The precipitate was filtered and washed with dichloromethane. The solid was suspended in saturated NaHCOsand extracted with CH2CI2. The extract was dried over MgSO4 and concentrated to give a white solid (1.716 g, 95%).
………………….
WO 2011056939
N-(5-ieri-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof. N-(5-ieri-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- / ][!, 3]benzo

N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven of the steps of:
(A) converting 2-amino-6-alkoxybenzothiazole (II), wherein R1 is a suitable phenolic hydroxyl protecting ;

(II) (III)
(B) reacting 2-amino-6-hydroxybenzothiazole (III) with compound (IV), wherein X is a leaving group, to yield 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V);

(C) reacting 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V) with compound (VI), wherein X2 is a leaving group, to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[ -b]benzothiazole (VII);


(D) reducing 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2, 1- bjbenzothiazole (VII) to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- am

(E) converting 3-amino-5-£er£-butyl isoxazole (IX) to a 5-?er?-butylisoxazol-3- ylcarbamate derivative (X), wherein R2 is optionally substituted aryl, heteroaryl, alkyl, or cycloalkyl;

(IX) (X)
(F) reacting 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- bjbenzothiazole (VIII) with a 5-£er£-butylisoxazol-3-ylcarbamate derivative (X) to yield N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-

(G) converting N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea to an acid addition salt of N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b] [ 1 ,3]benzo-thiazol-2-yl]phenyl } urea.
[00128] In certain embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea, or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, as depicted in Scheme 1, wherein R1, R2, X1, and X2 are defined herein elsewhere. In specific embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven, of the Steps A, B, C, D, E, F, and G, as depicted in Scheme 1.
Scheme 1 :

A. Preparation of 2-amino-6-hydroxybenzothiazole
1. Example A-l[00252] To a 1-L 3-necked round bottom flask fitted with a condenser, heating mantle, and mechanical stirrer was charged aqueous hydrobromic acid (48%, 632 mL, 5.6 mol, 10 equiv). 2-Amino-6-methoxybenzothiazole (100 g, 0.55 mol, 1 equiv) was added to the above flask over 15 minutes. The reaction temperature was raised slowly to reflux (105-110 °C). A clear dark brown colored solution was observed at about 80 °C. The reflux was continued at 105-110 °C for about 4 hr. The progress of the reaction was monitored by HPLC. When 2-amino-6-methoxybenzothiazole was less than 2%, the reaction was substantially complete.
[00253] The reaction mass was cooled to 0-5 °C and at this point precipitation of a solid was observed. The mixture was maintained at 0-5 °C for 0.5 hr and filtered, and the cake was pressed to remove HBr. The wet cake was transferred to a 2-L round bottom flask fitted with a mechanical stirrer. Saturated aqueous sodium bicarbonate solution (-1500 mL) was added slowly at ambient temperature, whereupon considerable frothing was observed. The pH of the solution was found to be about 6.5 to 7. The mixture was stirred for 0.5 hr at ambient temperature and filtered. The filter cake was washed with water (500 mL), dried on the filter and then under vacuum at 30-35 °C for 10-12 hr to give the product 2-amino-6-hydroxybenzothiazole (82 g, 89% yield, HPLC purity = 99%). JH NMR (DMSO-if6, 500 MHz): δ 7.12 (d, 1H), 7.06 (S, 2H, NH2), 7.01 (d, 1H), 6.64 (dd, 1H); MS (m/z) = 167.1 [M+ + 1].
[00254] Table: Summary of Plant Batches

[00255] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the pH to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 270 nm. The injection volume was 20 μΐ^, and the sample was diluted with a diluent (Mobile Phase A : Mobile Phase B = 70:30). Test solution was prepared by weighing accurately about 25 mg of sample and transferring it into a 100 mL volumetric flask, dissolving with 20-30 mL of diluent, making up the volume to the mark with diluent, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00256] Table: HPLC Method

2. Example A-2
[00257] 2-Amino-6-methoxybenzothiazole was reacted with hot aqueous HBr at a temperature of >70 °C for about 3 hours and then the clear solution was cooled to ambient temperature overnight. The precipitated solids were collected, dissolved in hot water and the pH was adjusted to between 4.5-5.5. The resultant solids were collected, dried and re-crystallized from isopropanol. Second crop material was collected. The solids were vacuum dried to give 2-amino-6-hydroxybenzothiazole.
[00258] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a white solid, with 99.4% purity (HPLC area %). JH NMR (300 MHz, DMSO-if6) was collected, which conformed to structure.
3. Example A-3
[00259] A 22-L 3-neck round bottom flask was equipped with a mechanical agitator, thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with hydrobromic acid (14 L, 123.16 mol, 13.10 equiv). Heating was initiated and 2- amino-6-methoxybenzothiazole was added (1.7 kg, 9.4 mol, 1.00 equiv) over 10 minutes with stirring. The heating of the reaction mixture was continued to reflux, and maintained (>107 °C) for approximately 5 hours. The reaction mixture turned into a clear solution between 75 °C and 85 °C. The reaction progress was monitored by TLC until no starting material was observed (A -0.5 mL reaction mixture aliquot was diluted with -0.5 mL water as a clear solution, neutralized with sodium acetate to pH -5 and extracted with 1 mL dichloromethane. The organic layer was spotted: 5%
methanol/dichloromethane; Rf (product) = 0.35; Rf (starting material) = 0.40).
[00260] The reaction mixture was cooled to – 20 °C (overnight). White solids precipitated. The solids were filtered on a polypropylene filter and pressed to remove as much hydrobromic acid from the solids as possible to facilitate the subsequent pH adjustment step. The slightly wet crude product was dissolved in hot (50 °C) water (5 L). The clear solution was filtered to remove any insoluble material present, and the solids were washed with 50 °C water. The filtrate was cooled to 10 °C. The cooled filtrate was neutralized with sodium acetate (- 1.0 kg) to pH 4.5 to 5.5 with vigorous stirring. A thick white solid precipitated. The solids were collected by filtration, and washed with cool (-10 °C) water (2 x 1.0 L) and air dried.
[00261] The wet crude product was slurried in hot (50 °C) isopropanol (3 L) briefly and allowed to stand in a cool room (-5 °C) overnight. The solids were collected by filtration and washed with methyl ferf-butylether (2 x 500 mL). The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (first crop). Yield: 1068 g (68%), white solid. HPLC: 99.4% (area). JH NMR (300 MHz, DMSO- ) conformed to structure.
[00262] The organic filtrate was collected in a total volume of 1.0 L, cooled to 10 °C. The off-white solids were precipitated and collected by filtration. The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (second crop). Yield: 497 g (32%), off-white solid. HPLC: 99.8% (area).
[00263] The overall yield combining the first crop and the second crop was 1565 g, (99%).
B. Preparation of 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol

1. Example B-l[00264] A 3-L 3-neck round bottom flask fitted with a condenser, a heating mantle, and a mechanical stirrer was charged with H-butanol (1.5 L), followed by 2-amino-6- hydroxybenzothiazole (75 g, 0.45 mol, 1.0 equiv), 2-bromo-4′-nitroacetophenone (121 g, 0.50 mol, 1.1 equiv), and sodium bicarbonate (41.6 g, 0.50 mol, 1.0 equiv). The reaction temperature was gradually raised to reflux and maintained at reflux (110-115 °C) for 2-3 hr. During the temperature increase, the reaction mass turned into a clear solution and then immediately changed into an orange colored suspension at 65-75 °C. The progress of the reaction was monitored by HPLC analysis every 1 hr (reaction mass sample was submitted to QC). When the level of 2-amino-6-hydroxybenzothiazole was less than 2%, the reaction was substantially complete.
[00265] The reaction mass was slowly cooled to 50-60 °C and then further cooled to 0-5 °C and stirred for 15 min. The precipitated solids were collected by filtration, and dried on the filter. The wet cake was transferred in to a 1-L round bottom flask, and water (600 mL) was added. The suspension was stirred for 0.5 hr and filtered, and the solid was dried on the filter. The wet cake was again taken in to a 1-L round bottom flask and stirred with acetone (200 mL). The slurry was filtered and washed with acetone (2 X 100 mL), and the solid was dried on the filter, unloaded and further dried in a vacuum oven at ambient temperature to give the product 2-(4-nitrophenyl)imidazo[2,l- b]benzothiazol-7-ol (V) (120 g, 85.7% yield, HPLC purity = 98.7%). JH NMR (DMSO- d6, 500 MHz): δ 9.96 (s, 1H, OH), 8.93 (s, 1H), 8.27 (d, 2H), 8.06 (d, 2H), 7.78 (d, 1H), 7.38 (d, 1H), 6.97 (dd, 1H); MS (m/z) = 312 [M+ + 1].
[00266] Table: Summary of Plant Batches

* Input of 2-amino-6-hydroxybenzothiazole (III)
[00267] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the H to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 235 nm. The injection volume was 10 μΐ^. The blank was prepared by transferring 200 μΐ. of DMSO and 200 μΐ. of 2M NaOH into a 10 mL volumetric flask, making up the volume to the mark with methanol, and mixing. The test solution was prepared by weighing accurately about 10 mg of sample and transferring it into a 50 mL volumetric flask, dissolving with 1 mL of DMSO and 1 mL of 2M NaOH, sonicating to dissolve, making up the volume to the mark with methanol, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00268] Table: HPLC Method

2. Example B-2
[00269] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with 2-amino-6-hydroxybenzothiazole (1068 g, 6.43 mol, 1.0 equiv) and ethanol (200 proof, 32.0 L), and the suspension was stirred for 10 minutes. 2-Bromo-4- nitroacetophenone (1667 g, 6.49 mol, 1.01 equiv) was added in one portion. The reaction mixture was heated to reflux (78 °C). The reflux was maintained for approximately 25 hours, resulting in a yellow suspension. The reaction progress was monitored by TLC (20% methanol/ethyl acetate; Rf (product) = 0.85; Rf (starting material) = 0.30). TLC indicated -50% 2-amino-6-hydroxybenzothiazole after 24 hours of reflux. Tetrabutylammonium iodide (10 g) was added and reflux was maintained for an additional 12 hours. TLC indicated -50% starting material still present. Coupling was seen to occur at both the thiazole and the amine.
[00270] The reaction mixture was cooled to 0-5 °C. The solids were collected by filtration, and the yellow solid was washed with ethanol (200 proof, 2 X 1.0 L) and diethyl ether (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 930 g (46%), yellow solid. HPLC: 99.5% (area). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
3. Example B-3
[00271] The reaction of Step B was carried out on multiple runs, varying solvents, temperature, and base. The results were summarized in the table below. The product (V) was isolated as yellow or green solids, with 1H NMR consistent with the structure and a purity of greater than about 98% by HPLC analysis.
[00272] Table: Reaction Condition Screening

TBAI = Tetrabutylammonium Iodide
C. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[2, 1 -bjbenzothiazole

1. Example C-l
[00273] To a 2000-L glass-lined (GL) reactor was charged DMF (298 kg), and the agitator was started. Under a nitrogen blanket, the reactor was charged with 2-(4- nitrophenyl)imidazo[2,l-&]benzothiazol-7-ol (36.8 kg, 118.2 mol, 1.0 equiv), 4-(2- chloroethyl)morpholine hydrochloride (57.2-66.0 kg, 307.3-354.6 mol, 2.6-3.0 equiv), tetrabutylammonium iodide (8.7 kg, 23.6 mol, 0.2 equiv) and potassium carbonate (49.0 kg, 354.6 mol, 3.0 equiv). The resulting yellow suspension was heated and stirred at 90 + 5 °C for at least 15 minutes, then the temperature was increased to 110 + 5 °C. The mixture was stirred for at least 1 hour and then sampled. The reaction was deemed complete if 2-(4-nitrophenyl) imidazo[2,l-&]benzothiazol-7-ol was <1% by HPLC. If the reaction was not complete, the heating was continued and the reaction sampled. If the reaction was incomplete after 6 hours, additional 4-(2-chloroethyl)morpholine hydrochloride may be charged. In general, additional charges of 4-(2- chloroethyl)morpholine hydrochloride had not been necessary at the given scale.
[00274] The reactor was cooled to 20 + 5 °C and charged with water (247 kg) and acetone (492 kg). The mixture was agitated at 20 + 5 °C for at least 1 hour. The product (VII) was isolated by filtration or centrifuge, and washed with water and acetone, and then dried in a vacuum oven at 45 °C to constant weight to give a yellow solid (46.2 kg, 92% yield, HPLC purity = 97.4% by area). JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example C-2
[00275] 2-(4-Nitrophenyl)imidazo[2, l-b]benzothiazol-7-ol, 4-(2-chloroethyl)- morpholine hydrochloride, potassium carbonate, and tetrabutylammonium iodide were added to N,N-dimethylformamide forming a yellow suspension that was heated at a temperature of >50 °C for over 3 hours. The reaction was cooled and the solids were collected, slurried into water, filtered, slurried into acetone, filtered and washed with acetone to give yellow solids that were dried under vacuum to give 7-(2-morpholin-4-yl- ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole.
[00276] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %), and a water content of 0.20%. Infrared (IR) spectrum was collected, which conformed to structure.
3. Example C-3
[00277] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a drying tube, a reflux condenser, and a heating mantle. The flask was charged with 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.770 kg, 5.69 mol, 1.0 equiv), N,N-dimethylformamide (18.0 L), 4-(2-chloroethyl)morpholine hydrochloride (2.751 kg, 14.78 mol, 2.6 equiv), potassium carbonate (2.360 kg, 17.10 mol, 3.0 equiv), and tetrabutylammonium iodide (0.130 kg, 0.36 mol, 0.06 equiv) with stirring. The resulting yellow suspension was heated to about 90 °C to 95 °C, maintaining the temperature for approximately 5 hours. The reaction was monitored by TLC until no starting material was observed (20% methanol / ethyl acetate; Rf (product) = 0.15; Rf (starting material) = 0.85).
[00278] The reaction mixture was cooled to -10 °C, and the yellow solids were collected by filtration on a polypropylene filter pad. The solids were slurried in water (2 X 5 L) and filtered. The crude wet product was slurried in acetone (5 L), filtered, and the solids were rinsed with acetone (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 45 °C. Yield: 2.300 kg (95%), yellow solid. TLC: R/ = 0.15 (20% methanol / EtOAc). HPLC: 95.7% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00279] Table: Yields from multiple batch runs

4. Example C-4
[00280] To a reactor were added 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), tetrabutylammonium iodide (0.24 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 85 °C and 95 °C and stirred at between 85 °C and 95 °C for 15 to 30 minutes. The mixture was then heated to between 105 °C and 120 °C and stirred at between 105 °C and 120 °C (e.g. , 115 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2- chloroethyl)morpholine hydrochloride (0.03 kg) may be added and the reaction mixture stirred at between 105 °C and 120 °C (e.g. , 115 °C) until reaction completion.
[00281] The reaction mixture was cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor was added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone was then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture was then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with deionized water (e.g. , about 45x deionized water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). The desired product was obtained in about 89% yield having about 99% purity by HPLC.
5. Example C-5
[00282] To a reactor were added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00283] The reaction mixture was cooled to between 60 °C and 70 °C over a period of at least 60 minutes. Industrial water (6 L) was added to the reactor. The reaction mixture was cooled to between 20 °C and 30 °C. Acetone (6 L) was added to the reactor. The mixture was stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with industrial water (e.g. , about 45 x industrial water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C, until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours).
6. Example C-6
[00284] To a reactor is added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) is added into the reactor. The DMF has a water content of no more than 0.05% w/w. The mixture is stirred for between 15 and 30 minutes to render a homogeneous suspension, which is heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction is complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction is not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00285] The reaction mixture is cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor is added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone is then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture is then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture is filtered, and the solid is washed with deionized water (e.g. , about 45x deionized water) until pH of washes is below 8. The solid is then washed with acetone (9.7 L). The solid is dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer is less than 0.30% w/w and TGA curve shows a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). D. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- aminophenyl)imidazo [2, 1 -bjbenzothiazole

[00286] To a 200-L high pressure (HP) reactor was charged a slurry of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,l-&]benzothiazole (VII) (7.50 kg, 17.7 mol, 1.0 equiv) in methanol (30 kg). The container was rinsed with additional methanol (10 kg) and the rinse was charged to the reactor. The reactor was then charged with THF (67 kg) and methanol (19 kg). The contents were agitated and the reactor was flushed with nitrogen by alternating nitrogen and vacuum. Vacuum was applied to the reactor and Raney Ni catalyst (1.65 kg, 0.18 wt. equiv) was charged through a sample line. Water (1 kg) was charged through the sample line to rinse the line. The reactor was flushed with nitrogen by alternating nitrogen and vacuum. The reactor was then vented and heated to 50 °C. The reactor was closed and pressurized with hydrogen gas to 15 psi keeping the internal temperature below 55 °C. The reactor was vented and re- pressurized a total of 5 times, then pressurized to 150 psi with hydrogen gas. The contents were agitated at 50 °C for at least 4 hours. At this point a hydrogen uptake test was applied: The reactor was re-pressurized to 150 psi and checked after 1 hour. If a pressure drop of more than 5 psi was observed, the process was repeated. Once the pressure drop remained < 5 psi, the reactor was vented and sampled. The reaction was deemed complete when 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,1- 6]benzothiazole (VII) was < 0.5% by HPLC.
[00287] The reactor was flushed with nitrogen as shown above. The 200-L HP reactor was connected to the 2000-L GL reactor passing through a bag filter and polish filter. The bag filter and polish filter were heated with steam. The 200-L HP reactor was pressurized (3 psi nitrogen) and its contents were filtered into the 2000-L reactor. The filtrates were hot. The 200-L reactor was vented and charged with THF (67 kg) and methanol (59 kg), the reactor agitated, and filtered into the 2000-L GL reactor.
[00288] A total of 6 reductions (46.2 kg processed) were carried out and the combined batches were concentrated by vacuum distillation (without exceeding an internal temperature of 40 °C) to a volume of -180 L. The reactor was cooled to 20 °C and charged with heptane (250 kg) and again vacuum distilled to a volume of -180 L. The reactor was charged with heptane (314 kg) and agitated at 20 °C for at least 1 hour, and then the product was isolated by centrifugation or collection on a Nutsche filter, washing with heptanes (2-5 kg per portion for centrifugation, followed by a 10-20 kg heptanes rinse of the reactor; or 94 kg for Nutsche filtration, rinsing the reactor first). The cake was blown dry, transferred to a vacuum oven and dried to constant weight maintaining a temperature < 50 °C to give the desired product (VIII) (34.45 kg, 80% yield, HPLC purity = 97.9%).
2. Example D-2
[00289] 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole was dissolved into methanol and THF and placed in a hydrogenator. Raney nickel was added and the vessel was pressurized with hydrogen and stirred for >24 hours. The reaction mixture was concentrated to a thick paste and diluted with methyl ferf-butyl ether. The resulting solids were filtered and washed with methyl ferf-butyl ether and dried under vacuum to give 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2, 1 -bjbenzothiazole.
[00290] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %). IR was collected, which conformed to structure.
3. Example D-3
[00291] Into a 5-gallon autoclave, 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo[2,l-&]benzothiazole (580 g, 1.37 mol, 1.0 equiv), THF (7.5 L), methanol (7.5 L, AR) and -55 g of decanted Raney nickel catalyst were added. The reaction vessel was purged with nitrogen (3 X 50 psi) and hydrogen (2 X 50 psi), with stirring briefly under pressure and then venting. The autoclave was pressurized with hydrogen (150 psi). The mixture was stirred and the hydrogen pressure was maintained at 150 psi for over 24 hours with repressurization as necessary. The reaction progress was monitored by TLC (10% methanol / chloroform with 1 drop ammonium hydroxide; Rf (product) 0.20; Rf (SM) 0.80). The reaction was substantially complete when the TLC indicated no starting material present, typically after 24 hours of stirring at 150 psi. The hydrogenation was continued at 150 psi for a minimum of 4 hours or until completion if starting material was still present after the initial 4 hours.
[00292] The reaction mixture was filtered through a Buchner funnel equipped with a glass fiber filter topped with a paper filter. Unreacted starting material was removed together with the catalyst. The filtrate was concentrated to a total volume of 0.5 L, and the residue was triturated with methyl ferf-butyl ether (0.5 L). The resultant solids were collected by filtration, and washed with methyl ferf-butyl ether (0.3 L) (first crop).
[00293] The filtrate was concentrated to dryness and the residue was diluted with methyl ferf-butyl ether (2 L). The resultant solids were collected by filtration, washing with methyl ferf-butyl ether (0.5 L) (second crop).
[00294] The solids were dried in a vacuum oven (<10 mm Hg) at 25 °C. Yield: 510 g (95%), beige solid. TLC: R/ 0.2 (10% methanol / chloroform with one drop of ammonium hydroxide). HPLC: 99.0% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00295] Table: Yields from multiple batch runs

4. Example D-4
[00296] The reaction of Step D was carried out in multiple runs under various conditions, such as, e.g. , varying catalyst loading, concentration of reactant, reaction temperature, and/or workup procedures. The results are summarized in the table below.

Description Run # l Run # 2 Run # 3 Run # 4 Run # 5Rxn Temp (°C) RT RT RT RT RT
Rxn Time (Hr) 24 hr 24 hr 24 hr 24 hr 24 hr
Filtered the Filtered the solution
Filtered the Filtered the Filtered the
solution through through celite. The solution through solution through solution through
celite, washed celite filter cake celite, celite, celite,
with THF, refluxed in THF concentrated, concentrated, concentrated,
concentrated, washed with hot solvent exchanged solvent exchanged solvent exchanged
Work Up solvent exchanged THF, concentrated, with heptane, with heptane, with heptane,
with heptane, solvent exchanged stirred the solids stirred the solids stirred the solids
stirred the solids with heptane, stirred and filtered and filtered and filtered
and filtered the solids and washed with washed with washed with
washed with filtered washed with heptane heptane heptane
heptane heptane
Produce (VIII) 1.9 g 3.88 g 1.11 g 2.6 g 4.4 g
Yield 88% 83.4% 56 94.6%
HPLC purity 95.6% 77.5% 91% 93.8%

5. Example D-5
[00297] To a pressure reactor under nitrogen atmosphere was added a slurry of Raney Nickel in water (0.22 kg) (e.g. about 0.14 kg dry catalyst in water) and the charging line was rinsed with deionized water (0.13 L). To another reactor (Reactor B) were added methanol (5.05 L) and 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2, 1- &]benzothiazole (1.0 kg), and the mixture was stirred for between 15 and 30 minutes to render a homogenous suspension. The suspension was transferred to the pressure reactor. Reactor B was washed with methanol (4.88 L) and the wash was transferred to the pressure reactor. Tetrahydrofuran (10.1 L) was added to the pressure reactor.
Hydrogen was charged to the pressure reactor to a pressure of between 2.0 bar and 3.0 bar. The reactor was heated to a temperature of between 45 °C and 55 °C. Hydrogen was then charged to the pressure reactor to a pressure of between 6.0 bar and 7.0 bar. The mixture was stirred at a temperature of between 45 °C and 55 °C (e.g. , 50 °C), while maintaining the hydrogen pressure between 6.0 bar and 7.0 bar until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00298] The mixture was filtered while maintaining the temperature at between 35 °C and 50 °C. The pressure reactor and the filter were washed with a mixture of THF (10.1 L) and methanol (9.93 L) preheated to a temperature of between 45 °C and 55 °C (e.g. , 50 °C). The combined filtrate was concentrated to a volume of between 2.4 L and 2.8 L under vacuum at a temperature of no more than 40 °C, and a precipitate was formed. Methanol (7.5 L) was added. The slurry was cooled to a temperature of between 5 °C and -5 °C (e.g. , over 3 hours) and stirred for at least 1 hour (e.g. , for 3 hours) while maintaining the temperature at between 5 °C and -5 °C. The suspension was filtered. The solid was washed with methanol (2 X 1.2 L). The solid was then dried under vacuum at a temperature of less than 50 °C until the methanol and THF contents were each less than 3000 ppm as analyzed by GC (e.g. , less than 1500 ppm). The desired product was obtained in about 88.5% yield having about 99% purity by HPLC.
E. Preparation of phenyl 5-£er£-butylisoxazol-3-ylcarbamate

[00299] The jacket temperature of a 200-L glass-lined (GL) reactor was set to 20 °C. To the reactor was charged 5-ieri-butylisoxazole-3-amine (15.0 kg, 107.0 mol, 1.0 equiv), then K2C03 (19.5 kg, 141.2 mol, 1.3 equiv) and anhydrous THF (62 kg).
Agitation was started and then phenyl chloroformate (17.6 kg, 112.4 mol, 1.05 equiv) was charged. The charging line was rinsed with additional anhydrous THF (5 kg). The reaction was agitated at 20 + 5 °C for at least 3 hours then sampled. The reaction was deemed complete if 5-£er£-butylisoxazole-3-amine was < 2% by HPLC. If the reaction was not complete after 6 hours, additional K2CO3 and phenyl chloroformate may be added to drive the reaction to completion.
[00300] Once complete, the reaction was filtered (Nutsche). The filter was rinsed with THF (80 kg). The filtrate was vacuum distilled without exceeding an internal temperature of 40 °C until -50 L remained. Water (188 kg) and ethanol (45 L) were charged, and the mixture was agitated for at least 3 hours with a jacket temperature of 20 °C. The resulting solid was isolated by centrifugation or collection on a Nutsche filter, rinsed with water (2-5 kg for each centrifugation portion; 30 kg for Nutsche filtration) and blow-dried. The solid was then dried to constant weight in a vacuum oven (45 °C) to give the desired product (19.4 kg, 92% yield, HPLC purity = 97.4%). On an 800 g scale, 1559 g of the desired product (98% yield) was obtained with a 99.9% HPLC purity. JH NMR (DMSO-i¾) δ 11.17 (s, 1H); 7.4 (m, 2H); 7.2 (m, 3H); 1.2 (s, 9H). LCMS (M+H)+ 261.
F. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea

1. Example F-l
[00301] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- &]benzothiazole (26.7 kg, 67.8 mol, 1.0 equiv), 3-amino-5-?-butylisoxazole phenyl carbamate (19.4 kg, 74.5 mol, 1.1 equiv), DMAP (0.5 kg, 4.4 mol, 0.06 equiv), and DCM (anhydrous, 260 kg). Agitation was started, triethylamine (1.0 kg, 10.2 mol, 0.15 equiv) was charged followed by additional DCM (5 kg) through the charging line. The reaction was heated to reflux (-40 °C) and agitated for at least 20 hours with complete dissolution observed followed by product crystallizing from solution after -30 minutes. The reaction was sampled and deemed complete when HPLC analysis showed a ratio of compound (VIII) to compound (I) < 1%.
[00302] The reactor was cooled to 0 °C and stirred for at least 2 hours. The content of the reactor were isolated by centrifuge. Each portion was rinsed with 2-3 kg of cold (0 °C) DCM and spun dry for at least 5 minutes with a 10 psi nitrogen purge. For the final portion, the reactor was rinsed with 10 kg of cold (0 °C) DCM and transferred to the centrifuge where it was spun dry for at least 5 minutes with a 10 psi nitrogen purge. The combined filter cakes were transferred to a vacuum tray dryer and dried to constant weight at 50 °C and at least >20 inches of Hg to give the desired product (I) (35.05 kg, 92% yield, HPLC purity = 98.8%). Phenol was the major impurity detected (0.99%); and three other impurities (<0.10%) were detected. JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example F-2
[00303] A variety of solvents were used in the reaction of Step F to optimize for better yields and purity profiles. The contents of the symmetrical urea impurity (XI) were compared and summarized in the table below:

http://www.google.com/patents/WO2011056939A1?cl=en SE THIS FOR DELETED CLIPS


4. Example F-4
[00305] To Reactor A under a nitrogen atmosphere was added 7-(2-morpholin-4-yl- ethoxy)-2-(4-aminophenyl)imidazo[2,l-&]benzothiazole (1 kg) and DMAP (0.02 kg). To Reactor B under a nitrogen atmosphere was added 3-amino-5-?-butylisoxazole phenyl carbamate (0.73 kg) and DCM (5.6 L). The DCM used had a water content of less than 0.05 % w/w. The mixture in Reactor B was stirred until dissolution. The solution was transferred into Reactor A (the solution can be filtered into Reactor A to remove any insoluble impurities in the carbamate starting material), and the mixture was stirred in Reactor A. Reactor B was washed with DCM (0.8 L) and the wash was transferred into Reactor A. Reactor A was washed with DCM (0.9 L). To Reactor A was added triethylamine (0.1 L) and the charging vessel and lines were rinsed with DCM (0.1 L) into Reactor A. The mixture was then heated to reflux and stirred at reflux until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00306] The reaction mixture was cooled (e.g. , over 2 to 3 hours) to a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The mixture was then stirred for 2 to 3 hours at a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The suspension was filtered. The solid was washed with cool DCM (2 X 1.5 L) (pre-cooled to a temperature of between -5 °C and 5 °C). The solid was dried under vacuum at a temperature of less than 45 °C until the DCM content was less than 1000 ppm (e.g., below 600 ppm) as analyzed by GC. The desired product was obtained having about 99% purity by HPLC.
G. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-b] [1 ,3]benzothiazol-2-yl]phenyl }urea dihydrochloride

1. Example G-l
[00307] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1-&][1, 3]benzothiazol-2-yl]phenyl}urea (35.0 kg, 62.4 mol, 1.0 equiv) followed by methanol (553 kg). Agitation was started and the reaction mixture was heated to reflux (-65 °C). Concentrated aqueous HC1 (15.4 kg, 156.0 mol, 2.5 equiv) was charged rapidly (<5 minutes) and the charge line was rinsed into the reactor with methanol (12 kg). Addition of less than 2.0 equivalents of HC1 normally resulted in the formation of an insoluble solid. The reaction mixture was heated at reflux for at least 1 hour. Upon HC1 addition, the slurry dissolved and almost immediately the salt started to crystallize, leaving insufficient time for a polish filtration.
[00308] The reactor was cooled to 20 °C and the product was isolated by filtration (Nutsche) rinsing the reactor and then the cake with methanol (58 kg). The solid was then dried in a vacuum oven (50 °C) to constant weight to give the desired
dihydrochloride salt (35 kg, 89% yield, HPLC purity = 99.94%). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
2. Example G-2
[00309] Concentrated HC1 was added to a suspension of N-(5-ieri-butyl-isoxazol-3- yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea in warm methanol forming a solution that slowly began to precipitate. The reaction mixture was refluxed for over 2 hours and then stirred overnight at ambient temperature. The dihydrochloride salt was collected and dried under vacuum.
3. Example G-3
[00310] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a nitrogen inlet, a drying tube, a reflux condenser, an addition funnel, and a heating mantle. The flask was charged with N-(5-ieri-butyl-isoxazol-3-yl)- N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2-yl]phenyl}urea (775 g, 1.38 mol, 1.0 equiv) and MeOH (40 L, AR). The resulting off-white suspension was heated to reflux (68 °C). A clear solution did not form. HC1 (37% aqueous) (228 mL, 3.46 mol, 2.5 equiv) was added over 5 minutes at 68 °C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Heating was continued at reflux for approximately 5 hours. The reaction mixture was allowed to cool to ambient temperature overnight. The off-white solids were collected by filtration on a polypropylene filter, washing with MeOH (2 X 1 L, AR). [00311] Two lots of material prepared in this manner were combined (740 g and 820 g). The combined solids were slurried in methanol (30 L) over 30 minutes at reflux and allowed to cool to the room temperature. The solids were collected by filtration on a polypropylene filter, rinsing with methanol (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 1598 g (84%), off-white solid. HPLC: 98.2% (area). MS: 561.2 (M+l)+. JH NMR (300 MHz, DMSO-i¾) conformed to the structure. Elemental Analysis (EA): Theory, 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual, 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
4. Example G-4
[00312] Into a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, a heating mantle, a condenser and a nitrogen inlet, were charged N-(5-ieri-butyl-isoxazol- 3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (1052.4 g, 1.877 mol, 1.0 equiv) and methanol (21 L). The reactor was heated and stirred. At an internal temperature of > 50 °C, cone. HC1 (398.63 mL, 4.693 mol, 2.5 equiv) was charged over 5 minutes through an addition funnel. During the addition, the mixture changed from a pale yellow suspension to a white suspension. The internal temperature was 55 °C at the conclusion of the addition. The mixture was heated to reflux for 1 hour, then heating was discontinued and the mixture was allowed to cool to room temperature. The mixture was filtered in two portions, and each filter cake was washed with methanol (2 X 1 L), transferred to trays and dried in a vacuum oven (45 °C) to constant weight. The dried trays were combined to produce 1141.9 g of the salt (96% yield, 99.1 % HPLC purity, 10.9% chloride by titration).
H. Analytical Data
1. N-(5-ieri-butyl-isoxazol-3-yl)-N’-{ 4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride
[00314] A batch of about 30 grams of N-(5-ieri-butyl-isoxazol-3-yl)-N’- {4-[7-(2- morpholin-4-yl-ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride was prepared using the methods described herein. This lot was
prepared in accordance with the requirements for production of clinical Active
Pharmaceutical Ingredients (APIs) under GMP conditions. The analytical data for this batch was obtained, and representative data were provided herein. [00315] Summary of analytical data for the dihydrochloride salt.


………………………
EXAMPLE 1. SYNTHESIS OF N-(5-TERT-BUTYL-ISOXAZOL-3-YU- N>-{4-f7-(2-MORPHOLIN-4- YL-ETHOXY)IMID AZO[2,1- BlH,31BENZOTHIAZOL-2-YL|PHENYLiUREA (“COMPOUND Bl”)
[00357] A. The intermediate 2-amino-l,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf: J. Org. Chem. 1970, 35, 4103- 4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 mL ethanol and 9 mL concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00358] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2- amino-l,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSO-^) Sl.6 (m, 2H), 6.6 (d, IH). [00359] B. To prepare the 2-(4-nitrophenyl)imidazo[2,l-b][l,3]benzothiazol-7-ol intermediate, 2-amino-l,3-benzothiazol-6-ol (20.0 g, 0.12 mol) and 2-bromo-4′- nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to O0C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2- (4-nitrophenyl)imidazo[2,l-£][l,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-(I6) δ 10 (s, IH), 8.9 (s, IH), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, IH), 7.4 (s, IH), 6.9 (d, IH).
[00360] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,l-
6][l,3]benzothiazole intermediate: 2-(4-nitrophenyl)imidazo[2,l-6][l,3]benzothiazol-7-ol,
NYI-4144519vl 84 (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2, 1 – b][\, 3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-d6) δ 8.97 (s, IH), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, IH), 7.7 (s, IH), 7.2 (d, IH), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00361] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole: To a suspension of 7-(2-morpholin-4-yl-ethoxy)- 2-(4-nitro-phenyl)imidazo[2,l -b][\ , 3]benzothiazole (3.87g, 9.1 mmol) in 100 mL isopropyl alcohol/water (3:1) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 mL). The filtrate was concentrated to approximately 1/3 of the original volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water – in which case the reaction is virtually complete after 3.5 hours and only traces of starting material are observed in the product obtained. NMR (DMSO-Λfc) δ 8.4 (s, IH), 7.8 (d, IH), 7.65 (d, IH), 7.5 (d, 2H), 7.1 (d, IH), 6.6 (d, 2H), 4.1 (t, 2H), 3.6 (m, 4H), 2.7 (t, 2H).
[00362] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert-butylisoxazole-3- isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over
NYI-4144519vl 85 MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of Compound B 1. Yield: 2.342 g (41 %) NMR (DMSO-J6) £9.6 (br, IH), 8.9 (br, IH), 8.61 (s, IH), 7.86 (d, IH), 7.76 (d, 2H), 7.69 (d, IH), 7.51 (d, 2H), 7.18 (dd, IH), 6.52 (s, IH), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCl3) £9.3 (br, IH), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, IH), 7.22 (d, IH), 7.03 (dd, IH), 5.88 (s, IH), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
6.2 EXAMPLE 2. ALTERNATIVE SYNTHESIS QF N-(5-TERT-BUTYL- ISOXAZQL-3- YL)-N -{4-[7-q-MORPHOLIN-4- YL- ETHOXYUMID AZOf2,l-BUl,31BENZOTHIAZOL-2- YLIPHENYLIUREA (“COMPOUND Bl”)
[00363] A. To a suspension of the intermediate 2-(4-Nitrophenyl)imidazo[2,l- b][l,3]benzothiazol-7-ol from Example IB (2.24 g, 7.2 mmol) in ethanol (40 mL) was added SnCl2 1H2O (7.9Og, 35 mmol) and heated to reflux. Concentrated HCl was added to the reaction mixture and the precipitate formed gradually. The reaction mixture was heated to reflux for 20 hours and then allowed to cool to room temperature. The solution was poured into ice and neutralized with 10% NaOH and adjusted to approximately pH 6. The organic phase was extracted three times with ethyl acetate (80 mL x 3). Extracts were dried over MgSθ4 and concentrated to give a yellow solid. (1.621 g, 80%). The solid was recrystallized from methanol to give a pure product (1.355 g, 67%).
[00364] B. To a suspension of the intermediate from Step 2A (0.563 g, 2 mmol) in toluene (30 mL) was added 5-tert-butylisoxazole-3-isocyanate (0.332g, 2 mmol) and heated to reflux overnight. LC-MS analysis showed presence of the intermediate but no trace of 5- tert-butylisoxazole-3-isocyanate and an additional 0.166 g of the isocyanate was added. The reaction was again heated to reflux overnight. Completion of reaction was verified by LC- MS. The solvent was removed and the resulting mixture was dissolved in methanol which was removed to give the second intermediate as a solid.
[00365] The mixture was dissolved in CH2Cl2 (150 mL) and washed with saturated
NaHCO3. The organic layer was dried over MgSO4, concentrated, and purified by silica gel chromatography three times, first using a methanol/CH2Cl2 gradient, the second time using a
NYI-4144519vl 86 hexane/ethyl acetate gradient followed by a methanol/ethyl acetate gradient, and a third time using a methanol/CH2Cl2 gradient.
[00366] C. To a suspension of the intermediate from Step 2B (0.1 10 g, 0.25 mmol) in
THF (5mL) was added Ph3P (0.079g, 0.3 mmol), diisopropylazodicarboxylate (0.06 Ig, 0.3 mmol) and 4-morpholinoethanol (0.039 g, 0.3 mmol). The reaction mixture was stirred at room temperature overnight. Completion of the reaction was verified by LC-MS. The solvent was removed and the final product was purified using silica gel chromatography, with methanol in CH2Cl2 (0.030g, 21%).
6.3 EXAMPLE 3. BULK SYNTHESIS OF N-(5-TERT-BUTYL- ISOXAZOL-3-YL)-N’-f4-[7-(2-MORPHOLIN-4-YL- ETHOXY^IMID AZO[2α-BUlJlBENZOTHIAZOL-2- YLlPHENYLiUREA (“COMPOUND Bl”)
[00367] A multi-step reaction scheme that was used to prepare bulk quantities of
Compound Bl is depicted in FIG. 66a and FIG. 66b, and is described further below. [00368] Step 1 : Preparation of 2- Amino-6-hydroxybenzothiazole (Intermediate 1). 2-
Amino-6-methoxybenzothiazole is reacted with hot aqueous HBr for about 3 hrs and then the clear solution is cooled to ambient temperature overnight. The precipitated solids are collected, dissolved in hot water and the pH is adjusted to between 4.5-5.5. The resultant solids are collected, dried and recrystallized from Isopropanol. Second crop material is collected. The solids are vacuum dried to give Intermediate 1.
[00369] Step 2: Preparation of 2-(4-Nitrophenyl) imidazo [2J-b]benzothiazol-7-ol
(Intermediate 2). 2-Amino-6-hydroxybenzothiazole, 2-Bromo-4-nitroacetophenone and absolute Ethanol are added together and heated to reflux for approximately 24 hours. Tetrabutylammonium iodide is added and the reaction is refluxed an additional 12 hours. The resulting yellow suspension is cooled and the solids collected and washed with Ethanol and Diethyl ether. The solids are dried under vacuum to give Intermediate 2. [00370] Step 3: Preparation of 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo
[2,1-b] benzothiazole (Intermediate 3). Intermediate 2, 4-(2-Chloroethyl)morpholine hydrochloride, Potassium carbonate and Tetrabutylammonium iodide are added to N,N- Dimethylformamide forming a yellow suspension that is heated for over 3 hours. The reaction is cooled and the solids are collected, slurried into water, filtered, slurried into
NYl-4 l4451′)v l 87 acetone, filtered and washed with Acetone to give yellow solids that are dried under vacuum to give Intermediate 3.
[O0371] Step 4: Preparation of 7-(2-Moφholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2,1 -b] benzothiazole (Intermediate 4). Intermediate 3 is dissolved into Methanol and THF and placed in a Hydrogenator. Raney Nickel is added and the vessel is pressurized with Hydrogen and stirred for >24 hrs. The reaction mixture is concentrated to a thick paste and diluted with Methyl tert-butyl ether. The resulting solids are filtered and washed with Methyl tert-butyl ether and dried under vacuum to give Intermediate 4. [O0372] Step 5: Preparation of {[5-(tert-Butyl) isoxazol-3-vnatnino}-N-{4-r7-(2- morpholin-4-yl-ethoxy)(4-hvdroimidazolo[2J-blbenzothiazol-2-yl)]phenyl|carboxamide (Compound Bl). 3 -Amino- 5 -tert-butyl isoxazole in Methylene chloride is added to a vessel containing toluene which is cooled to approx 0 0C. Triphosgene is then added and the reaction mixture is cooled to below -15 0C. Triethylamine is added, followed by Intermediate 4. The mixture is heated to distill off the Methylene chloride and then heated to over 60 0C for over 12 hours and cooled to 50-60 °C. The resulting solids are filtered, washed with Heptane, slurried with 4% sodium hydroxide solution, and filtered. The solids are then washed with Methyl tert-butyl ether followed by Acetone and dried under vacuum to give Compound Bl.
6.4 EXAMPLE 4. EXAMPLES OF PREPARATION OF COMPOUND Bl HCL SALT
[00373] Example A: For the preparation of a hydrochloride salt of Compound Bl5 N-
(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b][l,3]benzothiazol-2-yl]phenyl}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCl in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrirugation and washed with ethyl ether to give a hydrochloride salt of Compound Bl. Yield: 2.44 g (98 %) NMR (DMSO-^) S X 1.0 (br, IH), 9.68 (s, IH), 9.26 (s, IH), 8.66 (s, IH), 7.93 (d, IH), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, IH), 6.53 (s, IH), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). [00374] Example B: Concentrated HCl is added to a suspension of Compound Bl in warm methanol forming a solution that slowly begins to precipitate. The reaction mixture is
NYI-4144519vl 88 refluxed for over 2 hrs and then stirred overnight at ambient temperature. The HCl salt is collected and dried under vacuum.
[00375] Example C: Materials: {[5-(tert-Butyl) isoxazol-3-yl]amino}-N-{4-[7-(2- morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l-6]benzothiazol-2-yl)] phenyl }carboxamide (775 g, 1.38 mol, 1.0 eq); HCl 37% aqueous (288 mL, 3.46 mol, 2.5 eq); Methanol (MeOH, AR) (40L). Procedure: (Step 1) Equipped a 5OL 3-neck round bottom flask with a mechanical agitator, thermocouple probe, Nitrogen inlet, drying tube, reflux condenser, addition funnel and in a heating mantle. (Step 2) Charged the flask with {[5-(tert-Butyl) isoxazol-3-yl] amino}-N-{4-[7-(2-morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l- b]benzothiazol-2-yl)] phenyl jcarboxamide (775g) and MeOH, AR (40L). Heat the resulting off-white suspension to reflux (680C). A clear solution did not form. (Step 3) Added HCl (37% aqueous) (228 mL) over 5 minutes at 68°C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Continued heating at reflux for approximately 5 hours. Allowed the reaction mixture to cool to ambient temperature overnight. (Step 4) Collected the off-white solids by filtration onto a polypropylene filter, washing the solids with MeOH, AR (2 x 1 L). (Step 5) Combined two lots of material prepared in this manner (74Og and 82Og). Slurried the combined solids in Methanol (30 L) over 30 minutes at reflux and cool to the room temperature. (Step 6) Collected the solids by filtration onto a polypropylene filter, rinsing with Methanol (2 x 1.5L). (Step 7) Dried the solids in a vacuum oven (<10mniHg) at 400C. Yield: 1598 g (84%), off-white solid; HPLC: 98.2% (area); MS: 561.2 (M+l); IH NMR: conforms (300 MHz, DMSO-d6); Elemental Analysis (EA): Theory = 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual = 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
NYl-4I44519v! 89 [00376] Examples of Compound Bl HCl salt synthesis

[00377] Example D: In a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, heating mantle, condenser and nitrogen inlet was charged Compound Bl (1052.4 g, 1.877 mol, 1.00 equiv.) and methanol (21 L). The reactor was heated and stirred. At an internal temperature > 50 0C, cone. HCl (398.63 mL, 4.693 mol, 2.5 equiv.) was charged over 5 minutes through an addition funnel. With the addition, the reaction changed from a pale yellow suspension to a white suspension. The internal temperature was 55 0C at the conclusion of the addition. The reaction was heated to reflux for 1 hour, then heating discontinued and the reaction allowed to cool to room temperature. The reaction was filtered in two portions, each filter cake washed with methanol (2 x 1 L), transferred to trays and dried in a vacuum oven (45 0C) to constant weight. The dried trays were combined to produce 1141.9 g, 96% yield, 99.1 % HPLC purity, 10.9% chloride by titration.
Solid Forms Comprising the HCl Salt of Compound Bl 6.6.2.1 Preparation of Solid Forms

6.6.2.2 Cold Precipitation Experiments

NYl-4144519vl 102 6.6.2.3 Slurry Experiments
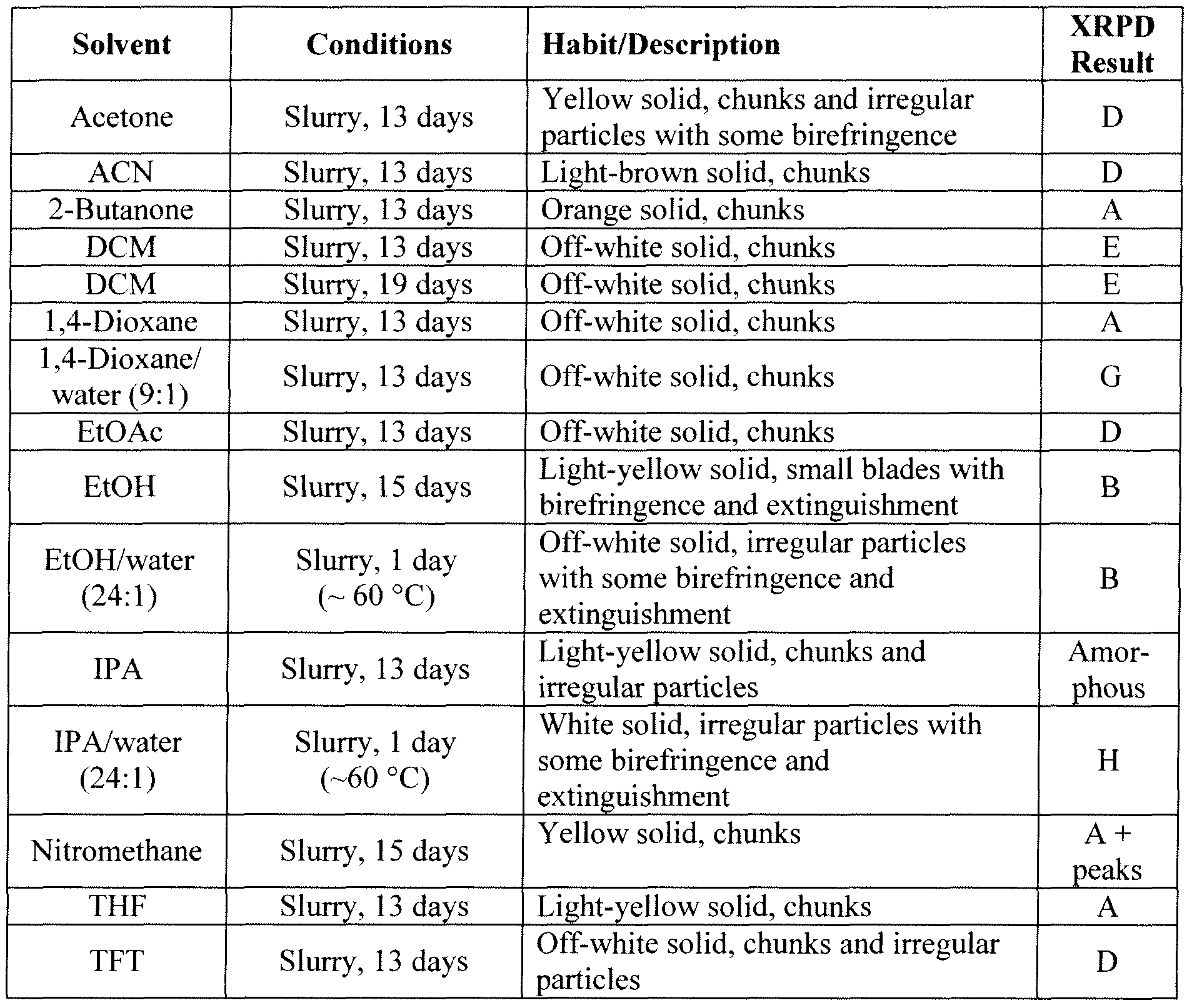
NYI-41445 l9vl 103 6.6.2.4 Additional Preparation of Solid Forms Comprising the HCI Salt of Compound Bl

NYl-4144519v l 104

NYM 144519vl 105

N Y l -4 1 4 4 5 1 9 v l 1 0 6

NYI-4I44519vi 107

N V I 4 1 4 4 5 1 9 1 0 8

“Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, VD = vapor diffusion, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
6.6.2.5 Scale-up Experiments of Involving Crystal Forms Comprising the HCl Salt of Compound Bl

NYI-4144519v l 109

Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
……………………
Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor
J Med Chem 2009, 52(23): 7808
http://pubs.acs.org/doi/full/10.1021/jm9007533

General Procedure E for Preparation of Hydrochloride Salt
References
- Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry 52 (23): 7808–7816.
- Drug Tames Refractory AML. ASH Dec 2012
- NMR……….http://file.selleckchem.com/downloads/nmr/S152601-AC-220-HNMR-Selleck.pdf
- HPLC………http://file.selleckchem.com/downloads/hplc/S152601-AC-220-HPLC-Selleck.pdf



