The method has the problems of more reaction steps, small batch size, post-treatment method using thin layer chromatography purification, low yield, etc., wherein the yield of the second step reaction is 22.4%, and the yield of the product prepared in the last step is only 11.3. % is not conducive to industrial expansion of production, it is necessary to improve its preparation method.
Example 1. Preparation of (R)-4-methyl-5-(oxiran-2-yl)isobenzofuran-1(3H)-one
First step, preparation of compound of formula (h)
Sodium borohydride (57.8 g) was dissolved in tetrahydrofuran (2000 mL), argon-protected, cooled to 0 ° C, material i (130.0 g) was added portionwise, and stirred at 5-10 ° C for 1 hour, 5-10 ° C Add boron trifluoride diethyl ether (237 mL) dropwise, stir at room temperature for 4 hours, stop the reaction, add methanol (800 mL) to quench the reaction, stir, add 1N hydrochloric acid (1000 mL) solution, stir at 0-20 ° C for 1 hour, decompress The organic solvent was evaporated, and the residue was evaporated. mjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj Concentration gave the title product (95 g).
The second step, the preparation of the compound of formula (g)
The raw material h (120.0 g) and trifluoroacetic acid (64 mL) were dissolved in acetonitrile (1 L), stirred, and cooled to 0-5 ° C under ice bath, and solid N-bromosuccinimide (147.0 g) was added portionwise. The reaction temperature was controlled at 0-8 ° C. After the reaction was completed, the reaction was quenched by adding 200 mL of potassium carbonate aqueous solution (containing 66.0 g of potassium carbonate) under ice-cooling, and concentrated under reduced pressure, water (200 mL) and ethyl acetate (800 mL) ×1,400 mL×2), and the organic phase was combined with EtOAc EtOAc (EtOAc m. Drying gave 150.0 g of product.
The third step, the preparation of the compound of formula (f)
The cuprous cyanide (123.0 g) was added to N,N-dimethylformamide (500 mL), and the material g (150.0 g) was dissolved in N,N-dimethylformamide (250 mL), and added to the dropping funnel. Under an argon atmosphere, after heating to 140-150 ° C, the N,N-dimethylformamide solution of the raw material g was added dropwise, and the reaction was stirred at 145 ° C for 2 hours. After the reaction was completed, the temperature was lowered to 90-95 ° C, and the mixture was added dropwise. Ionized water (62 mL), reacted for 18 hours, stopped the reaction, and cooled to room temperature. The reaction solution was added to a mixed solvent of isopropyl acetate/methanol (V/V = 4:1, 1500 mL), stirred for 30 minutes, and padded with silica gel and silicon. The mixture was filtered with celite, and the filter cake was washed with isopropyl acetate/methanol (V/V = 4:1, 100 mL×3), and the filtrate was concentrated under reduced pressure. The residue was slowly added to deionized water (3 L) and stirred for 1 hour. Filtration, the filter cake was washed with ethanol (50 mL×3), and the filter cake was dried to give 133.0 g of crude product. The crude product was added to ethyl acetate/methanol (V/V=4:1, 2.0L) and heated to reflux. After filtration, the cake was washed with ethyl acetate /methanol (EtOAc/EtOAc (EtOAc)
The fourth step, the preparation of the compound of formula (e)
The starting material f (26.0 g) was dissolved in dichloromethane (520 mL), triethylamine (33 mL) was added, and the mixture was cooled to -5-0 ° C and added trifluoromethanesulfonic anhydride (29.2 mL), 0-10 After reacting at ° C for 2 hours, the reaction was stopped. Under ice-cooling conditions, water (250 mL) was added dropwise to the reaction mixture to quench the reaction, and the mixture was separated, and the aqueous phase was extracted with dichloromethane (100 mL×2). The sodium solution (300 mL) was washed with EtOAc EtOAc (mjjHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH After dissolving at 70 ° C, the supernatant liquid was separated, and the lower layer of the oil was dissolved in a mixed solution of petroleum ether and ethyl acetate (V/V = 5:1) (300 mL × 2), and the organic phases were combined and concentrated under reduced pressure. (41.0 g), EtOAc (EtOAc m.
The fifth step, the preparation of the compound of formula (d)
The starting material e (50.1 g) was dissolved in isopropanol (500 mL), and ethylene trifluoroborate (29.5 g) and 1,1′-bisdiphenylphosphinoferrocene palladium dichloride (1.25 g) were added. Further, triethylamine (71 mL) was added, and the reaction was refluxed for 1.5 hours under an argon atmosphere. The reaction was stopped, cooled to room temperature, filtered, and the filtrate was washed with ethyl acetate (20 mL×3), and the filtrate was concentrated and concentrated through silica gel column. The title product (29.0 g) was obtained (yield: ethyl acetate: petroleum ether = 1:5-1:3).
The sixth step, the preparation of the compound of formula (c)
Potassium ferricyanide (279.0 g) was added to the reaction flask, followed by potassium carbonate (116.0 g) and hydrogenated quinidine 1,4-(2,3-naphthyridinyl)diether ((DHQD) 2 PHAL , 1.1g) and potassium citrate dihydrate (103mg), add 2L of deionized water, stir for 30 minutes, add tert-butanol (1.5L) under argon atmosphere, stir for 15 minutes, 0-5 ° C raw material d ( 49.0g) was added in portions, stirred at 0-5 ° C for 4 hours, warmed to room temperature and stirred for 18 hours, the reaction was stopped, saturated sodium sulfite solution (800 mL) and ethyl acetate (1000 mL) were added, stirred until fully dissolved, layered, The aqueous layer was extracted with EtOAc (EtOAc (EtOAc) (EtOAc (EtOAc) The mixture was cooled to rt.
The seventh step, the preparation of the compound of formula (a)
The raw material c (54.0 g) was added to dichloromethane (600 mL), and the mixture was white turbid. Under argon atmosphere, b (46.9 g) was added, stirred at room temperature for 10 minutes, cooled to 0 ° C, and trimethylchlorosilane was added dropwise. (54.0g), stirring at 0 ° C for 30 minutes, the solution became clear, warmed to room temperature for 1 hour, then cooled to 0 ° C, added b (23.0g), raised to room temperature for 30 minutes, stop the reaction, the reaction solution Concentration under reduced pressure gave the crude title product which was used in the next step without purification.
The eighth step, the preparation of the compound of formula (VI)
The raw material a (69.6 g) was added to methanol (1000 mL), and potassium carbonate (90.0 g) was added, and the mixture was stirred at room temperature for 2 hours, the reaction was stopped, and the mixture was evaporated under reduced pressure. ethyl acetate (500 mL) and water (200 mL) The aqueous phase was extracted with EtOAc (EtOAc (EtOAc) (EtOAc) The title compound (35.0 g) was obtained in vacuo.
Example 2 Preparation of 5-cyano-4-methoxypyridinecarboxylic acid hydrochloride
First step, preparation of the compound of formula (p)
Raw material n (110.0g), o (150.0g), acetic anhydride (151.5g) was added to the reaction flask and refluxed for 4 hours, the reaction was stopped, concentrated under reduced pressure, and the obtained residue was controlled at a temperature of 0-10 ° C to add ammonia water and Water (V / V = 1:1, 600mL) mixed solution, when a large amount of solids were formed, add ice water (400mL), drip, stir for 30 minutes, adjust to pH 2-3 with concentrated hydrochloric acid, stir 30 After a minute, the mixture was filtered, and the filter cake was dried, and then filtered with anhydrous ethanol (500 mL) for 1 hour, filtered, filtered, washed with cold anhydrous ethanol (100 mL×3), and the filter cake was dried to give the title product (80.0 g) The yield was 59%.
The second step, the preparation of the compound of formula (q)
Sodium hydroxide (43.6 g) was added to water (800 mL) under ice bath, and the starting material p (79.8 g) was added portionwise to the above aqueous sodium hydroxide solution, the ice bath was removed, and the mixture was heated to reflux for 2 hours to terminate the reaction. The reaction solution was cooled to room temperature with ice water, 2M hydrochloric acid solution was added dropwise to adjust the pH to 2-3, stirred for 30 minutes, filtered, and the filter cake was washed with ice water (100 mL) and cold ethanol (100 mL), and the obtained solid was dried. The title product (71.2 g), yield 100%.
The third step, the preparation of the compound of formula (r)
The raw material q (70.3 g) was dissolved in phosphorus oxychloride (210 mL), stirred at 110 ° C for 2 hours under reflux, concentrated under reduced pressure to remove phosphorus oxychloride, and the residue was added to acetonitrile (350 mL). Add diisopropylethylamine (117.0 g), dilute the solution to a black suspension, add the suspension to the ammonia water (350 mL) under ice bath, drop the reaction for 30 minutes, ethyl acetate (500 mL × 3) Extraction, the organic phase was combined, washed with saturated sodium chloride (500 mL), dried over anhydrous sodium sulfate, filtered and evaporated. (44.7 g), yield 51%.
The fourth step, the preparation of the compound of formula (s)
The raw material r (44.3 g) was added to dichloromethane (440 mL) under an argon atmosphere, and the temperature was controlled to 0-5 ° C under ice-cooling, triethylamine (58.6 g) was added dropwise, and the mixture was stirred for 10 minutes. Trifluoroacetic anhydride (58.5g) was added dropwise, the addition was completed, and the reaction was carried out for 1 hour in an ice bath. The reaction was stopped, the pH of the reaction mixture was 7-8, and the reaction was quenched by adding water (400 mL), and the mixture was separated. The organic phase was extracted with EtOAc (EtOAc) (EtOAc) (HHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH g), yield 91%.
The fifth step, the preparation of the compound of formula (t)
The raw material s (25.6 g) and cesium carbonate (49.2 g) were dissolved in N,N-dimethylformamide (260 mL), cooled to 0 ° C in an ice bath, and methanol (9.5 g) was added dropwise in an ice bath, 0 ° C After reacting for 6 hours, the mixture was stirred at 20-25 ° C for 12 hours, and the reaction was stopped. The reaction mixture was quenched with water (650 mL), and extracted with ethyl acetate (200 mL×3). The title compound (16.8 g) was obtained. The title compound (16.8 g) was obtained from EtOAc (EtOAc). The rate is 67%.
The sixth step, the preparation of the compound of formula (II-1)
Add t (22.0g), palladium acetate (1.46g), 1,3-bis(diphenylphosphino)propane (2.68g), triethylamine (36mL) to the mixed solution, pressurize with carbon monoxide to 10bar, heat up The reaction was stopped at 70 ° C for 18 hours, the reaction was stopped, the organic solvent was removed by concentration, the aqueous phase was added with saturated sodium chloride solution and dichloromethane (300 mL×3), and the organic phase was combined, decolorized by activated carbon, filtered, and the organic phase was adjusted to pH with concentrated hydrochloric acid. =1, a solid precipitated, and after adding 50 mL of isopropanol, the dichloromethane was concentrated to remove the product, which was filtered and dried to give a product (23.6 g).
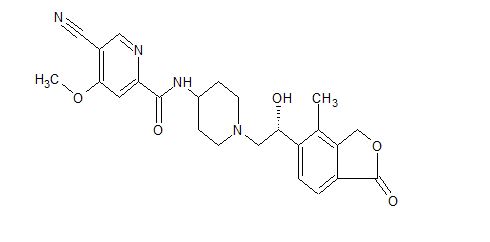
Example 3, (R)-5-cyano-N-(1-(2-hydroxy-2-(4-methyl-1-oxo-1,3-dihydroisobenzofuran-5-yl) Preparation of ethyl)piperidin-4-yl)-4-methoxypyridinecarboxamide (formula (I))
First step, synthesis of intermediate (IV)
Into the reaction flask, 4.0 L of absolute ethanol was added, and (R)-4-methyl-5-(oxiran-2-yl)-benzoisofuran-1(3H)-one (274.8) was added under stirring. g), 4-Boc-aminopiperidine (341.2 g), heated to 65-70 ° C, stirred for 18-20 h, and the heating was stopped. Naturally cooled to 50-55 ° C, 8.0 L of n-hexane was added under stirring, stirred until the temperature naturally dropped to 20-25 ° C, a large amount of solids were precipitated, the temperature of the ice bath was lowered to 0-5 ° C, stirred, suction filtered, filter cake It was washed twice with n-hexane (250 ml × 2) and dried to give a solid (354.3 g).
The second step, the synthesis of intermediate (III-1)
5.2 L of ethyl acetate was placed in a glass bottle, and the temperature was lowered to 0 to 5 ° C under stirring. The stirring was stopped, hydrogen chloride gas (0.48 kg) was introduced, and the temperature of the reaction liquid was controlled to be lower than 5 ° C during the passage of hydrogen chloride. The above product (349.3 g) was added to the reaction mixture with slow stirring. After the addition, the reaction was stirred for 3-4 hours, and the reaction temperature was naturally raised to 20-25 ° C, and the stirring was stopped. After suction filtration, the filter cake was washed three times with ethyl acetate (1.0 L×3), and the filter cake was dried under vacuum at 40-45 ° C for 6-8 h to give a solid (322.8 g) in a yield of 99.3%; The ratio of hydrochloric acid was determined by silver nitrate titration to be 20.5%.
The third step, the synthesis of the compound of formula (I)
Into the reaction flask, 4.0 L of N, dimethylformamide was added, and the product of the above step (317.8 g), 5-cyano-4-methoxypyridinecarboxylate II-1 (205.9 g) was sequentially added with stirring. Triethylamine (528.2 g), 1-hydroxybenzotriazole (152.7 g), N,N-diisopropylcarbodiimide (142.6 g). After the addition, the argon gas was replaced three times, and the mixture was heated to 40-45 ° C to stir the reaction for 16-18 h. The heating was stopped, and the reaction liquid was poured into ice water (30 L), and stirred for 1 hour. After suction filtration, the filter cake was washed three times with purified water, dried, and then pulverized with anhydrous ethanol (3.0 L) at 20-25 ° C for 1 h. Filtering, drying 10-12h to obtain crude (290.4g), yield 73.7%, purity: 97.76%;
N,N-dimethylformamide (2.0 L) was added to the crude product (290.4 g) with stirring. The reaction solution was heated to 70-75 ° C, 20.3 g of activated carbon (7% w/w) was added, and the mixture was stirred for 1 h. Heat filtration, wash the filter residue with hot N,N-dimethylformamide (70-75 ° C, 200 mL), combine the filtrate, heat the filtrate to 70-75 ° C, add hot (65-70 ° C, 5 L) with stirring Anhydrous ethanol to the reaction liquid in the previous step, stirring and crystallization, until the temperature naturally drops to 20-25 ° C, the reaction bottle is transferred to an ice water bath and stirring for 1 h, suction filtration, the filter cake is washed with absolute ethanol, dried to obtain a solid 219.5 g, total yield 55.7%, purity: 99.69%.
1 H-NMR (400 MHz, DMSO-d 6 ) δ 8.88 (s, 1H), 8.75 (d, 1H), 7.77 (s, 1H), 7.71-7.69 (m, 2H), 5.43-5.40 (m, 2H), 5.35 (s, 1H), 5.08 (s, 1H), 4.09 (s, 3H), 3.78 (s, 1H), 2.95 (s, 3H), 2.38 (s, 1H), 2.27 (s, 3H) ), 2.25 (s, 2H), 1.72 (s, 4H).

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....