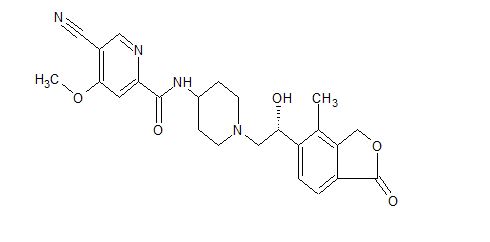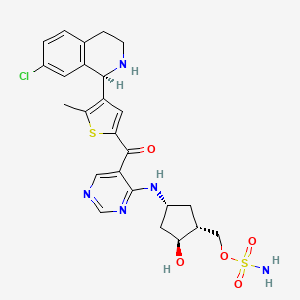
TAK-981
C25 H28 Cl N5 O5 S2, 578.103
[(1R,2S,4R)-4-[(5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl)amino]-2-hydroxycyclopentyl]methyl sulfamate
[(1R,2S,4R)-4-[[5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methyl-thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate
Sulfamic acid, [(1R,2S,4R)-4-[[5-[[4-[(1R)-7-chloro-1,2,3,4-tetrahydro-1-isoquinolinyl]-5-methyl-2-thienyl]carbonyl]-4-pyrimidinyl]amino]-2-hydroxycyclopentyl]methyl ester
| CAS 1858276-04-6 FREE
CAS 1858279-63-6 HYDRATE |
| MW |
578.103 |
- Originator Takeda Oncology
- Class Antineoplastics
- Mechanism of Action Small ubiquitin-related modifier protein inhibitors
- Phase I Lymphoma; Solid tumours
- 01 Oct 2018 Phase-I clinical trials in Solid tumours (Late-stage disease, Metastatic disease) and and Lymphoma (Refractory metastatic disease, Second-line therapy or greater) in USA (IV) (NCT03648372)
- 03 Sep 2018 Takeda Oncology plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) and Lymphoma (Refractory metastatic disease, Second-line therapy or greater) in September 2018 (IV) (NCT03648372)
- 03 Sep 2018 Preclinical trials in Lymphoma in USA (IV) prior to September 2018 (NCT03648372)
Takeda is evaluating TAK-981, a SUMO-Activating Enzyme (SAE) inhibitor, in early clinical trials for the treatment of adult patients with advanced or metastatic solid tumors or with relapsed or refractory lymphomas.

Small ubiquitin-like modifier (SUMO) is a member of the ubiquitin-like protein (Ubl) family that is covalently conjugated to cellular proteins in a manner similar to Ub-conjugation (Kerscher, O., Felberbaum, R., and Hochstrasser, M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 22: 159-80). Mammalian cells express three major isoforms: SUMO l , SUM02 and SUM03. SUM02 and SUM03 share -95% amino acid sequence homology but have -45% sequence homology with SUMO l (Kamitani, T., Kito, K., Nguyen, H. P., Fukuda-Kamitani, T., and Yeh, E. T. 1998. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J Biol Chem. 273( 18): 1 1349-53). SUMO proteins can be conjugated to a single lysine residue of a protein (monosumoylation) or to a second SUMO protein that is already conjugated to a protein forming a SUMO chain (polysumoylation). Only SUM02/3 can form such chains because they possess internal consensus SUMO modification sites (Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H., Naismith, J. H., Hay, R. T. 2001. Polymeric chains of SUMO-2 and SUM 0-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 276(38):35368-74). An additional isoform, SUM04, is found in kidney, lymph node and spleen cells, but it is not known whether SUM04 can be conjugated to cellular proteins.
[0003] SUMO l , SUM02 and SUM03 are activated in an ATP-dependent manner by the SUMO-activating enzyme (SAE). SAE is a heterodimer that consists of SAE 1 (SUMO-activating enzyme subunit 1) and SAE2 (UBA2). SAE, like other El activating enzymes, uses ATP to adenylate the C-terminal glycine residue of SUMO. In a second step, a thioester intermediate is then formed between the C-terminal glycine of SUMO and a cysteine residue in SAE2. Next, SUMO is transferred from the El to the cysteine residue of the SUMO conjugating enzyme (E2), UBC9. Unlike the Ub pathway that contains many E2 enzymes, Ubc9 is currently the only known conjugating enzyme for SUMO and functions with SUMOl , SUM02 and SUM03 proteins. SUMO proteins are then conjugated to the target protein, either directly or in conjunction with an E3 ligase, through isopeptide bond formation with the epsilon amino group of a lysine side chain on a target protein. Several SUMO E3 ligases, including PIAS (protein inhibitor of activated signal transducer and activator of transcription protein) proteins and Ran-binding protein 2 (RanBP2), and polycomb 2 (Pc2), have been identified (Johnson, E. S., and Gupta, A. A. 2001. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 106(6):735-44; Pichler, A., Gast, A., Seeler, J. S., Dejean, A.; Melchior, F. 2002. The nucleoporin RanBP2 has SUMOl E3 ligase activity. Cell. 108(1): 109-20; Kagey, M. H., Melhuish, T. A., and Wotton, D. 2003. The polycomb protein Pc2 is a SUMO E3. Cell. 1 13(1): 127- 37). Once attached to cellular targets, SUMO modulates the function, subcellular localization, complex formation and/or stability of substrate proteins (Miiller, S., Hoege, C, Pyrowolakis, G., and Jentsch, S. 2001. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2(3):202-10). SUMO- conjugation is reversible through the action of de-sumoylating enzymes called SENPs (Hay, R. T. 2007. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 17(8):370-6) and the SUMO proteins can then participate in additional conjugation cycles.
[0004] SAE-initiated SUMO-conjugation plays a major role in regulating diverse cellular processes, including cell cycle regulation, transcriptional regulation, cellular protein targeting, maintenance of genome integrity, chromosome segregation, and protein stability (Hay, R. T. 2005. SUMO: a history of modification. Mol Cell. 18( 1): 1 -12; Gill, G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18(17):2046-59). For example, SUMO- conjugation causes changes in the subcellular localization of RanGAPl by targeting it to the nuclear pore complex (Mahajan, R., Delphin, C., Guan, T., Gerace, L., and Melchior, F. 1997. A small ubiquitin-related polypeptide involved in targeting RanGAPl to nuclear pore complex protein RanBP2. Cell. 88(1):97- 1070). Sumoylation counteracts ubiquitination and subsequently blocks the degradation of Ι Β, thereby negatively regulating NF-κΒ activation (Desterro, J. M., Rodriguez, M. S., Hay, R. T. 1998. SUMO- 1 modification of IkappaB alpha inhibits NF-kappaB activation. Mol Cell. 2(2):233-9). Sumoylation has been reported to play an important role in transcription exhibiting both repressive and stimulatory effects. Many of the transcriptional nodes that are modulated play important roles in cancer. For example, sumoylation stimulates the transcriptional activities of transcription factors such as p53 and HSF2 (Rodriguez, M. S., Desterro, J. M., Lain, S., Midgley, C. A., Lane, D. P., and Hay, R. T. 1999. SUMO- 1 modification activates the transcriptional response of p53. EMBO J. 18(22):6455-61 ; Goodson, M. L., Hong, Y., Rogers, R., Matunis, M. J., Park-Sarge, O. K., Sarge, K. D. 2001. Sumo- 1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J Biol Chem. 276(21 ): 18513-8). In contrast, SUMO-conjugation represses the transcriptional activities of transcription factors such as LEF (Sachdev, S., Bruhn, L., Sieber, H., Pichler, A., Melchior, F., Grosschedl, R. 2001. PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15(23):3088- 103) and c-Myb (Bies, J., Markus, J., and Wolff, L. 2002. Covalent attachment of the SUMO- 1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. / Biol Chem. 277( 1 1):8999-9009). Thus, SUMO-conjugation controls gene expression and growth control pathways that are important for cancer cell survival.
[0005] Altered expression of SAE pathway components have been noted in a variety of cancer types: (Moschos, S. J., Jukic, D. M., Athanassiou, C., Bhargava, R., Dacic, S., Wang, X., Kuan, S. F., Fayewicz, S. L., Galambos, C., Acquafondata, M., Dhir, R., and Becker, D. 2010. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum Pathol. 41(9): 1286-980); including multiple myeloma (Driscoll, J. J., Pelluru, D., Lefkimmiatis, K., Fulciniti, M., Prabhala, R. H., Greipp, P. R., Barlogie, B., Tai, Y. T., Anderson, K. C, Shaughnessy, J. D. Jr., Annunziata, C. M., and Munshi, N. C. 2010. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 1 15(14):2827-34); and breast cancer (Chen, S. F., Gong, C, Luo, M., Yao, H. R., Zeng, Y. J., and Su, F. X. 201 1. Ubc9 expression predicts chemoresistance in breast cancer. Chin J Cancer. 30(9):638-44), In addition, preclinical studies indicate that Myc-driven cancers may be especially sensitive to SAE inhibition (Kessler, J. D., Kahle, K. T., Sun, T., Meerbrey, K. L., Schlabach, M. R., Schmitt, E. M., Skinner, S. O., Xu, Q., Li, M. Z., Hartman, Z. C, Rao, M., Yu, P., Dominguez-Vidana, R., Liang, A. C, Solimini, N. L., Bernardi, R. J., Yu, B., Hsu, T., Golding, I., Luo, J., Osborne, C. K., Creighton, C. J., Hilsenbeck, S. G., Schiff, R., Shaw, C. A., Elledge, S. J., and Westbrook, T. F. 2012. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 335(6066):348-53; Hoellein, A., Fallahi, M., Schoeffmann, S., Steidle, S., Schaub, F. X., Rudelius, M., Laitinen, I., Nilsson, L., Goga, A., Peschel, C, Nilsson, J. A., Cleveland, J. L., and Keller, U. 2014. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood. 124( 13):2081 -90). Since SUMO-conjugation regulates essential cellular functions that contribute to the growth and survival of tumor cells, targeting SAE could represent an approach to treat proliferative disorders such as cancer.
[0006] SAE inhibitors may also be applicable for the treatment of other diseases and conditions outside of oncology. For example, SUMO modifies proteins that play important roles in neurodegenerative diseases (Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C, Slepko, N., Hies, K., Lukacsovich, T., Zhu, Y. Z., Cattaneo, E., Pandolfi, P. P., Thompson, L. M., Marsh, J. L. 2004. SUMO modification of Huntington and Huntington’s disease pathology. Science. 304(5667): 100-4); Dorval, V., and Fraser, P. E. 2006. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 281 ( 15):9919-24; Ballatore, C, Lee, V. M., and Trojanowski, J. Q. 2007. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 8(9):663-72). Sumoylation also has been reported to play important role in pathogenic viral infection, inflammation and cardiac function (Lee, H. R., Kim, D. J., Lee, J. M., Choi, C. Y., Ahn, B. Y., Hayward, G. S., and Ahn, J. H. 2004. Ability of the human cytomegalovirus ΓΕ1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. / Virol. 78(12):6527-42; Liu, B., and Shuai, K. 2009. Summon SUMO to wrestle with inflammation. Mol Cell. 35(6):731-2; Wang, J., and Schwartz, R. J. 2010. Sumoylation and regulation of cardiac gene expression. Circ Rei. l07( l): 19-29). [0007] It would be beneficial therefore to provide new SAE inhibitors that possess good therapeutic properties, especially for the treatment of proliferative, inflammatory, cardiovascular and neurodegenerative disorders.
PATENT
WO 2016004136

https://patents.google.com/patent/WO2016004136A1/en
Example 133: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate I-263a
Step 1: 7-Chloro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-l,2,3,4-tetrahydroisoquinoline
[00714] An oven-dried 2-neck 250 mL round bottom flask under nitrogen was charged with THF (40 mL) and cooled to -74 °C . Added 2.50 M ra-BuLi in hexane (6.92 mL, 17.3 mmol). Added a solution of Int-1 (4.00 g, 16.0 mmol) in THF (60 mL) slowly keeping the internal temperature less than -70 °C . Stirred with cooling 5 min. A second oven-dried 250 mL round bottom flask under nitrogen was charged with THF (60 mL) and Int-50 (2.04 g, 12.4 mmol) and the resulting solution was cooled to 0 °C . Added boron trifluoride diethyl ether complex ( 1.71 mL, 13.6 mmol) slowly and cooled to -30 °C . The contents of the first flask were transferred via cannula to the second flask. Reaction was quenched with saturated aqueous NaHC03 and warmed to rt. Water was added, and the mixture was extracted three times with EtOAc. Combined organic portions were washed with brine, dried over anhydrous Na2S04, filtered, and concentrated in vacuo. Residue was purified via flash column chromatography eluting with a hexane / EtOAc gradient (0 to 100% EtOAc) to afford the title compound as a white solid ( 1.88g, 45%). Ή NMR (400 MHz, Chloroform-d) δ 7.17 – 7.01 (m, 2H), 6.83 – 6.61 (m, 2H), 5.92 (s, 1H), 5.09 (s, 1H), 4.17 – 4.04 (m, 2H), 4.03 – 3.92 (m, 2H), 3.37 – 3.25 (m, 1H), 3.13 – 2.91 (m, 2H), 2.82 – 2.69 (m, 1H), 2.46 (s, 3H). LCMS: (AA) M+l 336.1
Step 2: ieri-Butyl 7-chIoro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3,4-dihydroisoquinoIine -2(lH)-carboxyIate [00715] A 50 mL round bottom flask under nitrogen was charged with 7-chloro-l -[5-(l ,3-dioxolan-2- yl)-2-methyl-3-thienyl]- l ,2,3,4-tetrahydroisoquinoline (5.67 g, 16.9 mmol) and DCM ( 100 mL), to which was added triethylamine (4.71 mL, 33.8 mmol), di-ieri-butyldicarbonate (4.61 g, 21.1 mmol), and N,N-dimethylaminopyridine (23 mg, 0.18 mmol). Reaction was stirred for 1 h at rt and then poured into saturated NaHC03 solution. Mixture was extracted three times with DCM, and the combined organic portions were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford 6.96g (95%) of the title compound. LCMS: (AA) M+ l 436.1
Step 3: tert-Butyl 7-chloro-l-(5-formyl-2-methyl-3-thienyl)-3,4-dihydroisoquinoline -2(1H)- carboxylate
[00716] A 1 L round bottom flask was charged with ferf-butyl 7-chloro-
1 -[5-( 1 ,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3 ,4-dihydroisoquinoline-2( 1 H)-carboxylate (7.30 g, 16.7 mmol), methanol (200 mL), and water (20 mL), to which was added a solution of 12M HC1 (4.00 mL, 130 mmol) in methanol (200 mL), and the reaction was stirred at rt for 1 h. Reaction was quenched via addition of 50mL of saturated NaHC03 and stirred for 5 min. Methanol was removed in vacuo, and the resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.55g, 70%). Ή NMR (400 MHz, Chloroform-d) δ 9.67 (s, 1 H), 7.27 – 7.15 (m, 2H), 7.12 (s, 1 H), 6.98 – 6.94 (m, 1 H), 6.34 (m, l H), 4.15 (s, 1 H), 3.18 – 3.06 (m, 1 H), 3.05 – 2.93 (m, 1H), 2.82 – 2.73 (m, 1 H), 2.69 (s, 3H), 1.50 (s, 9H). LCMS: (AA) M+Na 414.2
Step 4: tert-Butyl 7-chIoro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyI]-2-methyl-3-thienyl}- 3,4-dihydroisoquinoline-2(lH)-carboxylate
[00717] An oven-dried 500 mL 3-neck round bottom flask under nitrogen was charged with 4-chloro- 5-iodopyrimidine (4.08 g, 17.0 mmol) and 2-methyltetrahydrofuran ( 150 mL). An addition funnel containing a solution of rert-butyl 7-chloro- l -(5-formyl-2-methyl-3-thienyl)-3,4- dihydroisoquinoline-2(l H)-carboxylate (4.75 g, 12.1 mmol) in 2-methyltetrahydrofuran (50 mL) was attached, and the contents of the reaction flask were cooled to -75 °C . 2.50 M n-BuLi in hexane ( 14.1 mL, 35.2 mmol) was added in small portions keeping the internal temperature less than -70 °C , at which point the contents of addtion funnel were added in a single portion. Upon completion of addition, the reaction was quenched by adding 20 mL of saturated NaHC03 in small portions and warmed to rt. The aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.85g, 79%). LCMS: (AA) M+Na 528.1
Step 5: tert-Butyl 7-chloro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyl]-2-methyl-3-thienyl}- 3,4- dihydroisoquinoline-2(lH)-carboxylate
[00718] A 1 L round bottom flask was charged with fe/Y-butyl 7-chloro- l – { 5-[(4-chloropyrimidin-5- yl)(hydroxy)methyl]-2-methyl-3-thienyl}-3,4-dihydroisoquinoline-2(l H)-carboxylate (4.85 g, 9.58 mmol) and DCM (300 mL). Manganese (IV) oxide (14.2 g, 163 mmol) was added and the reaction was stirred at rt for 18 h. Mixture was filtered through Celite, and the filter cake was rinsed with hot EtOAc. Filtrate was concentrated in vacuo to afford the title compound (4.47g , 93%). Ή NMR (400 MHz, Chloroform-d) δ 9.09 (s, 1 H), 8.70 (s, 1 H), 7.24 – 7.16 (m, 1 H), 7.16
– 7.07 (m, 1 H), 7.00 – 6.90 (m, 2H), 6.32 (s, 1 H), 4.28 – 3.97 (m, 1H), 3.14 – 2.89 (m, 2H), 2.78
– 2.65 (m, 4H), 1 .53 – 1.43 (m, 9H).
Step 6: tert-Butyl (lR)-7-chloro-l-[5-[4-[[(lR,3R,4S)-3-(hydroxymethyl)-4-triisopropylsiIyloxy- cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dihydro-lH-isoquinoline-2- carboxylate
[00719] A 1 L round bottom flask under nitrogen was charged with iert-butyl 7-chloro- l – { 5-[(4- chloropyrimidin-5-yl)carbonyI]-2-methyl-3-thienyl }-3,4-dihydroisoquinoline-2( l H)-carboxylate (4.47 g, 8.86 mmol), DMF (20.0 mL, 258 mmol), Int-259 (3.06 g, 10.6 mmol), and triethylamine (3.09 mL, 22.2 mmol) and the mixture was stirred at rt for 18 h. Reaction mixture was poured into water and saturated NaHC03, and then extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a 70/30 to 60/40 hexane/EtOAc gradient to afford 0.56g of first-eluting diastereomer 1 (not pictured), 4.3 l g of a mixture of diastereomers, and 1.1 lg ( 17%) of second-eluting diastereomer 2 (the title compound). The mixture of diastereomers thus obtained was resubjected to the described chromatography conditions two additional times to afford a total of 2.62 g of the desired diastereomer. Ή NMR (400 MHz, Methanol-d4) δ 8.54 – 8.46 (m, 2H), 7.27 – 7.19 (m, 2H), 7.09 – 6.99 (m, 2H), 6.37 (s, 1H), 4.87 – 4.75 (m, 1H), 4.38 – 4.29 (m, 1H), 4.20 – 4.09 (m, 1H), 3.66 – 3.52 (m, 2H), 3.28- 3.14 (m, 2H), 3.02 – 2.89 (m, 1 H), 2.89 – 2.78 (m, 1 H), 2.68 (s, 3H), 2.54 – 2.41 (m, 1 H), 2.22 – 2.09 (m, 2H), 1.86 – 1.73 (m, 1H), 1.50 (s, 8H), 1.39 – 1.23 (m, 2H), 1.15 – 1.04 (m, 20H).
LCMS: (AA) M+ 1 755.3
Step 7: tert-Butyl (lR)-7-chloro-l-[2-methyl-5-[4-[[(lR,3R,4S)-3-(sulfamoyloxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3-thienyl]-3,4-dihydro-lH- isoquinoline-2-carboxylate [00720] A solution of ie/t-butyl (lR)-7-chloro-l-[5-[4-[[( lR,3R,4S)-3-(hydroxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dih lH-isoquinoline-2-carboxylate (2.46 g, 3.26 mmol) in 2-methyltetrahydrofuran (25 mL), and DMF (25 mL) was cooled to 0 °C. Triethylamine ( 1.82 mL, 13.0 mmol) and chlorosulfonamide (1.50 g, 13.0 mmol) were added and the reaction was stirred for 10 min. Added methanol (0.53 mL, 13.0 mmol) and stirred for 15 min. Reaction mixture was poured into saturated NaHC03, extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (2.41g, 89%). Ή NMR (400 MHz, Methanol-d4) δ 8.58 – 8.45 (m, 2H), 7.29 – 7.17 (m, 2H), 7.1 1 – 6.98 (m, 2H), 6.36 (s, 1 H), 4.84 – 4.73 (m, 1H), 4.44 – 4.33 (m, 1H), 4.21 – 4.08 (m, 4H), 3.27- 3.17 (m, 1 H),3.02 – 2.89 (m, 1 H), 2.88 – 2.78 (m, 1 H), 2.67 (s, 3H), 2.57 – 2.47 (m, 1 H), 2.41 – 2.30 (m, 1 H), 2.23 – 2.13 (m, 1 H), 1.87- 1.78 (m, 1 H), 1.50 (s, 9H), 1.43 – 1 .33 (m, 1 H), 1 .17 – 1.04 (m, 20H). LCMS: (AA) M+l 834.3
Step 8: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yI]aniino]-2-hydroxy-cyclopentyl]methyl sulfamate
[00721] A solution of f«?r/-butyl ( l R)-7-chloro- l -[2-methyl-5-[4-[[( l R,3R,4S)-3-
(sulfamoyloxymethyl)-4-triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3- thienyl]-3,4-dihydro- l H-isoquinoline-2-carboxylate (2.41 g, 2.89 mmol) in CH3CN ( 10 mL) was cooled in an ice bath to + 1 °C . Phosphoric acid ( 10 mL, 200 mmol) was added dropwise and the reaction was stirred with ice bath cooling for 60 min. The mixture was warmed to rt and stirred for an additional 3 h. Reaction was poured into a stirring mixture of 50 mL water and 50 mL EtOAc, and the the pH was adjusted to ~9 by slowly adding 200 mL of saturated NaHC03 with stirring. Resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a gradient that began with 100% DCM and increased in polarity to 80% DCM / 20% methanol / 2% ammonium hydroxide gradient to afford the title compound (1.50 g, 90%). Ή NMR (400 MHz, Methanol-d4) δ 8.61 (s, 1H), 8.52 (s, 1 H), 7.27 (s, 1 H), 7.18 – 7.13 (m, 2H), 6.73 – 6.68 (m, 1 H), 5.23 (s, 1H), 4.81 – 4.70 (m, 1 H), 4.26 – 4.10 (m, 3H), 3.29 – 3.23 (m, 2H), 3.1 1 – 2.96 (m, 2H), 2.87 – 2.76 (m, 1H), 2.60 (s, 3H), 2.55 – 2.42 (m, 1 H), 2.33 – 2.19 (m, 1H), 2.18 – 2.07 (m, 1H), 1.95 – 1.81 (m, 1H), 1.47 – 1.35 (m, 1 H). LCMS: (AA) M+l 580.0
CLIP

Credit: Tien Nguyen/C&EN
Presenter: Steven Paul Langston, associate director at Takeda Pharmaceuticals International
Target: Sumo activating enzyme
Reporter’s notes: Langston gave the last talk of the morning session, placing him in the “precarious position of being between you and lunch,” he said. Takeda acquired this drug development program, falling under the umbrella of immuno-oncology, along with Millenium Pharmaceuticals in 2008. The team targeted a pathway known as SUMOylation, a protein post translation modification that is implicated in a number of cellular processes including immune response. In SUMOylation, enzymes attach a small protein to another protein. They found that inhibiting this pathway activates a type I interferon response in immune cells. How the molecule, TAK-981, inhibits this pathway is quite complicated, Langston said. TAK-981 forms an adduct with a small ubiquitin like modifier (SUMO) to inhibit a SUMO activating enzyme that catalyzes SUMOylation. While the synthesis of TAK-981 is fairly short, it requires a nonideal chiral chromatography separation after the first step. TAK-981 is in Phase I clinical trials as an intravenous infusion for patients with metastatic solid tumors or lymphomas.
| Patent ID |
Title |
Submitted Date |
Granted Date |
| US2018311239 |
HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME |
2018-03-16 |
|
| US9962386 |
HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME |
2017-04-17 |
|
| US9683003 |
HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME |
2015-06-30 |
2016-01-14 |
//////////TAK-981, TAK 981, Phase I, Lymphoma, Solid tumours, TAKEDA,
Cc3sc(cc3[C@@H]1NCCc2ccc(Cl)cc12)C(=O)c5cncnc5N[C@@H]4C[C@H](COS(N)(=O)=O)[C@@H](O)C4
https://cen.acs.org/pharmaceuticals/drug-discovery/Drug-structures-displayed-first-time-in-Orlando/97/web/2019/04?utm_source=Facebook&utm_medium=Social&utm_campaign=CEN

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


















![2-(4-Chlorophenyl)-N-[[2-(2,6-dioxopiperidin-3-yl)-1-oxo-3H-isoindol-5-yl]methyl]-2,2-difluoroacetamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118647211&t=l)