
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Metyrosine, Metirosine
メチロシン метирозин , ميتيروسين , 甲酪氨酸 ,
Metyrosine (USP);
Metirosine (JAN/INN);
Demser (TN)
CAS 672-87-7
- Molecular FormulaC10H13NO3
- Average mass195.215 Da
APPROVED P[MDA JAPAN 2019/1/8
- (-)-alpha-Methyl-L-tyrosine
- (–)-α-methyl-L-tyrosine
- (S)-alpha-Methyltyrosine
- Methyltyrosine
Synthesis ReferenceUS2868818
An inhibitor of the enzyme tyrosine 3-monooxygenase, and consequently of the synthesis of catecholamines. It is used to control the symptoms of excessive sympathetic stimulation in patients with pheochromocytoma. (Martindale, The Extra Pharmacopoeia, 30th ed)
Metirosine (INN and BAN; α-Methyltyrosine, Metyrosine USAN, AMPT) is an antihypertensive drug. It inhibits the enzyme tyrosine hydroxylase and, therefore, catecholamine synthesis, which, as a consequence, depletes the levels of the catecholamines dopamine, adrenaline and noradrenaline in the body.
For use in the treatment of patients with pheochromocytoma, for preoperative preparation of patients for surgery, management of patients when surgery is contraindicated, and chronic treatment of patients with malignant pheochromocytoma.

Pharmacodynamics
In patients with pheochromocytoma, who produce excessive amounts of norepinephrine and epinephrine, administration of one to four grams of metyrosine per day has reduced catecholamine biosynthesis from about 35 to 80 percent as measured by the total excretion of catecholamines and their metabolites (metanephrine and vanillylmandelic acid). The maximum biochemical effect usually occurs within two to three days, and the urinary concentration of catecholamines and their metabolites usually returns to pretreatment levels within three to four days after metyrosine is discontinued. Most patients with pheochromocytoma treated with metyrosine experience decreased frequency and severity of hypertensive attacks with their associated headache, nausea, sweating, and tachycardia. In patients who respond, blood pressure decreases progressively during the first two days of therapy with metyrosine; after withdrawal, blood pressure usually increases gradually to pretreatment values within two to three days.
Mechanism of action
Metyrosine inhibits tyrosine hydroxylase, which catalyzes the first transformation in catecholamine biosynthesis, i.e., the conversion of tyrosine to dihydroxyphenylalanine (DOPA). Because the first step is also the rate-limiting step, blockade of tyrosine hydroxylase activity results in decreased endogenous levels of catecholamines and their synthesis. This consequently, depletes the levels of the catecholamines dopamine, adrenaline and noradrenaline in the body,usually measured as decreased urinary excretion of catecholamines and their metabolites. One main end result of the catecholamine depletion is a decrease in blood presure
Clinical use
Metirosine has been used in the treatment of pheochromocytoma.[1] It is contra-indicated for the treatment of essential hypertension.
However it is now rarely used in medicine, its primary use being in scientific research to investigate the effects of catecholamine depletion on behaviour.[2] Info on how catecholamine depletion from this medicine affects behavior needed in this srticle.
SYN
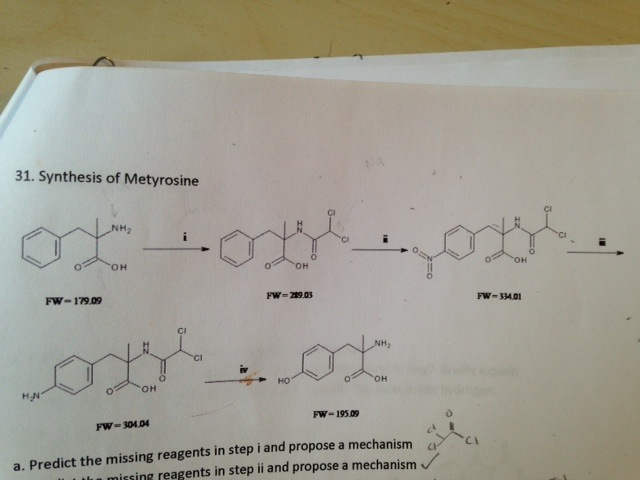

PAPER
Saito, Hiroyuki; Agricultural and Biological Chemistry 1988, 52(9), PG 2349-50
PATENT
https://patents.google.com/patent/WO2011053835A1
Metyrosine, which has the structure of Formula:

is useful in reducing elevated levels of catecholamines associated with pheochromocytoma, and preventing hypertension. Metyrosine, as shown, is a chiral compound. The synthesis of metyrosine in pure or substantially pure enantiomeric form requires a process that involves using substantially diastereomerically and/or enantiomerically pure intermediates. The Applicant has discovered, surprisingly, certain compounds that are substantially
diastereomerically or enantiomerically pure and processes to prepare them, and which compounds may be converted to metyrosine.
Scheme 1


Lower diastereomeric purity Higher diastereomeric purity

Example 1. (R)-Phenylglycinamide«HCl

[0091] To a 500 mL flask were charged (i?)-phenylglycinamide (20.0 g, 133 mmol, 1 eq. Amplachem ref: Aa-33365) and MeOH (160 mL). 4 M HCl/dioxane (50 mL, 200 mmol, 1.5 eq.) was then added dropwise resulting in the formation of a white precipitate. The mixture was stirred for 30 min, was filtered and was washed with MeOH (20 mL) and diethyl ether (20 mL). Drying in vacuo provided (i?)-phenylglycinamide.HCl (21.9 g, 89%) as a white solid. Ή NMR (D20, 400 MHz) 4.97 (s, 1 H); 7.36-7.41 (m, 5 H).
[0092] Example 2. 2-[l-(S)-Cyano-2-(4-methoxyphenyl)-l-methylethylamino]-2-
(R)-phenylacetamide 2

[0093] Method A. To a 500 mL flask were charged (j )-phenylglycinamide’HCl
(15.0 g, 80.6 mmol, 1 eq.), MeOH (104 mL), H20 (17 mL) and -methoxyphenylacetone (12.4 mL, 80.6 mmol, 1 eq, Aldrich, ref: 19917-6). To this mixture was added a solution of NaCN (3.95 g, 80.6 mmol, 1 eq.) in H20 (10 mL). The resulting solution was stirred for 4 days at room temperature while a white precipitate formed. The precipitate was filtered and washed with H20/MeOH (7:3) to provide 2-[l-(5)-cyano-2-(4-methoxyphenyl)-l- methylethylamino]-2-(i?)-phenylacetamide 2 (11 ,0 g) as a white solid. The filtrate was stirred for 3 d more at room temperature and the solid formed was filtered to provide 2 (3.30 g). The filtrate was stirred for 1 d more to provide 2 (1.70 g). The filtered solids were combined and dried in vacuo to provide 2 (16.0 g, 61 %, dr 98/2) as a white solid.
[0094] Method B. In a sealed tube were charged (i?)-phenylglycinamide.HCl (1.2 g,
6.45 mmol, 1 eq.), MeOH (4 mL), H20 (7 mL) and 7-methoxyphenylacetone (991 μL, 6.45 mmol, 1 eq.). A solution of NaCN (316 mg, 6.45 mmol, 1 eq.) in H20 (1 mL) was added. The mixture was stirred for 20 hours at 40°C resulting in the formation of a white precipitate. The precipitate was filtered, was washed with H20/MeOH (7:3 v/v, 2 x 2 mL) and was dried in vacuo to provide 2 (1.59 g, 76%, dr 98/2) as a white solid. Ή NMR (CDC13, 400 MHz) 1.14 (s, 3 H); 2.90 (d, J= 13.6 Hz, 1 H); 2.99 (d, J= 13.6 Hz, 1 H); 3.20 (bs, 1 H); 3.80 (s, 3 H); 4.51 (s, 1 H); 5.45 (bs, 1 H); 5.75 (bs, 1 H); 6.90 (d, J= 8.6 Hz, 2 H); 7.27 (d, J= 8.6 Hz, 2 H); 7.30-7.50 (m, 5 H). dr determination: Ή NMR comparing integration of peaks of 2 at 2.90/2.99 (1.98 H, formally 2 H) with those of its diastereoisomer or diastereomer
(prepared from racemic phenylglycinamide) at 2.82/2.85 (0.04 H, formally 2 H). Ή NMR diastereoisomer of 2 (CDC13, 400 MHz) 1.49 (s, 3 H); 2.82 (d, J = 13.8 Hz, 1 H); 2.85 (d, J = 13.8 Hz, 1 H); 3.78 (s, 3 H); 4.52 (s, 1 H); 5.55 (bs, 1 H); 6.60 (bs, 1 H); 6.84 (d, J = 8.6 Hz, 2 H); 7.17 (d, J = 8.6 Hz, 2 H); 7.30-7.40 (m, 5 H).
[0095] Example 3. 2-[(R)-(Carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(S)-methylpropionamide 3

[0096] To a 500 mL flask were added nitrile 2 (10.0 g, 30.95 mmol) and CH2C12(130 mL). The solution was cooled to -10°C and H2S04 (10 mL) was added dropwise over 15 min. The mixture was stirred for 2 h at 0°C. Ice (200 g) was added and the mixture was stirred for 1 h. The mixture was basified with 32% aq NH3 to pH 8-9, EtOAc (400 mL) was added and the phases were separated. The aqueous phase was extracted with EtOAc (2 x 250 mL). The combined organic phases were dried over MgS04 and were concentrated to provide 3 (1 1.1 g (9.99 g theoretical, the sample contains 10% w/w of EtOAc by NMR) 94%, 92.9% chemical purity, 97% de by HPLC/MS) as a white solid. HPLC/MS tR – 3.04 min; m/z = 342.1 (M+l) de determination: HPLC/MS comparing integration of peaks at tR = 3.04 min (98.5 area%) and tR = 2.92 min (1.5 area% the other diastereoisomer of 3) HPLC/MS diastereoisomer of 3 tR = 2.92 min; m/z = 342.1 (M+l)
[0097] Example 4. Purification of 3 by Crystallization. Amide 3 (8.10 g, 23.7 mmol) and methyl isobutyl ketone (124 mL) were heated to reflux temperature until the solid was dissolved and the solution was cooled to room temperature. The solid formed was filtered, was washed with methyl isobutyl ketone (2 x 20 mL) and was dried in vacuo to provide 3 (5.45 g, 67%, >99.5% purity by HPLC/MS) as a white solid. 1H NMR (DMSO, 200 MHz) 1.00 (3H, s); 2.35 (bs, 1 H); 2.75 (d, J = 13.3 Hz, 1 H) 2.85 (d, J = 13.3 Hz, 1 H); 3.61 (s, 3 H); 4.17 (s, 1 H); 6.60 (d, J= 8.4 Hz, 2 H); 691 (d, J= 8.4 Hz, 2 H); 6.93 (bs, 1 H), 7.01 bs (1 H); 7.20-7.50 (m, 6 H); 7.6 (bs, 1 H). HPLC/MS m z = 342.1 (M+l ) Example 5. 2-(S)-Amino-3-(4-methoxyphenyl)-2-methyIpropionamide 4

[0099] Method A, Hydro genolysis: Amine 3 (6.00 g, 17.6 mmol, 1 eq) was dissolved in MeOH (60 mL) and 10% Pd/C (2.15 g, 56% moisture content, 16 wt%) was added. The mixture was stirred under H2 (3 bar) at 50°C for 16 h. The mixture was filtered through Celite, the filter pad was washed with MeOH (20 mL) and the filtrates were concentrated to provide 5.50 g of a 1 : 1 mixture of 4 (3.37 g, 91%) and phenylacetamide as a white solid.
[0100] Method B, Transfer hydrogenolysis: Amine 3 (500 mg, 1 ,47 mmol, 1 eq) was dissolved in i-PrOH (5 mL) under Argon. 10% Pd/C (200 mg, 56% moisture content, 18 wt%) and ammonium formate (601 mg, 9,56 mmol, 6.5 eq.) were added. The mixture was stirred at reflux temperature for 1 h. The mixture was filtered through Celite, the filter pad was washed with EtOH (10 mL) and the filtrates were concentrated to provide 480 mg of a 1 : 1 mixture of desired compound (293 mg, 96%) and phenylacetamide as a white solid. Ή NMR (DMSO, 400 MHz) 1.14 (s, 3 H); 2.10 (bs, 2 H); 2.50 (d, J= 13.1 Hz, 1 H); 2.95 (d, J = 13.1 Hz, 1 H); 3.69 (s, 3 H); 6.80 (d, J = 8.5 Hz, 2 H); 6.81 (bs, 1 H); 6.90 (bs, 1 H); 7.09 (d, J= 8.5 Hz, 2 H). (Phenylacetamide 3.34 (s, 2 H); 7.15-7.28 (m, 6 H); 7.42 (bs, 1 H).). HPLC/MS 4 tR = 1.96 min. MS (ESI (+)) m/z = 164.2 (M-CONH2). (Phenylacetamide tR = 1.76 min. MS (ESI (+)) m/z = 136.2 (M+l)).
[0101] Example 6. Metyrosine 1

[0102] To a 100 mL flask with a reflux condenser was charged amide 4 (5.50 g of a mixture containing 4 (3.37 g, 16.1 mmol) and phenylacetamide (2.13 g)) and 48% HBr (30 mL). The solution was heated for 5 h at 120 °C and was cooled to room temperature. H20 (60 mL) was added and the solution was washed with EtOAc (3 35 mL). The aqueous phase was concentrated in vacuo to provide a beige paste. The paste was dissolved in H20 (15 mL) and the resulting mixture was heated to 65 °C. Activated carbon (300 mg, Type NORIT SX) was added and the mixture was stirred for 15 min, was filtered and the filter pad was washed with water (2 4 mL). The combined filtrates were heated to 55 °C and the pH was adjusted to 5-6 using 32% aq. NH3. The mixture was cooled to 0 °C, was stirred for 15 min and was filtered. The collected solids were washed with cold water (2 x 5 mL) and were dried in vacuo to provide (-)-Q!-methyl-L-tyrosine (or metyrosine) 1 (2.65 g, 84%) as a white solid. HPLC (Zorbax C18, NaH2P04 10 mM pH = 3 / MeCN (100:0) 10 min, (100:0) to (0: 100) 15 min, 0: 100 5 min) tR = 10.1 min. Chiral HPLC (Nucleosil Chiral-1, CuS04 10 mM / MeCN 10:1) tR = 16.9 min. m.p. – 320-321°C. [a]5 6 = +201° (c – 0.5 Copper complex solution)(lit.2 + 185-190°) Copper complex solution preparation: Solution A
(anhydrous NaOAc dissolved in H20 (150 mL) in 250 mL volumetric flask, glacial acetic acid (50 mL) added and diluted to volume with H20) mixed with Solution B (cupric sulfate (62.5 g) diluted to volume with H20 in a 200 mL volumetric flask) in a 1 L volumetric flask and was diluted to volume with H20. Metyrosine solution (5 mg/mL) was prepared in this solution.
[0103] To obtain an NMR spectrum (taking into account the low solubility of the product), a small sample (10 mg) was transformed into its HC1 salt. The sample was dissolved in 2 M HC1 and the solution was evaporated to dryness. Ή NMR (D20, 400 MHz) 1.49 (s, 3 H); 2.90 (d, J= 14.5 Hz, 1 H); 3.16 (d, J = 14.5 Hz, 1 H); 6.75 (d, J= 8.2 Hz, 2 H), 7.01 ((d, J= 8.2 Hz, 2 H). 13C NMR (D20, 100.6MHz) 21.6; 41.6; 61.0; 116.0, 125.0; 131.7; 155.5; 173.8. [0104] Preparation of Metyrosine using (S)-phenylethylamine
Scheme 4

(S)~Phenylethy la mine. HCl


[0105] Example 7. (S)-Phenylethylamine hydrochloride.

[0106] To a 500 mL flask were added (5)-phenylethylamine (BASF, ref: UN2735,
40.0 g, 333 mmol, 1 eq.) and MeOH (160 mL). The solution was cooled to 0 °C and 37% HCl (40 mL, 480 mmol, 1.44 eq.) were added dropwise. Concentration of the reaction mixture gave a white solid. Diethyl ether (300 mL) was added and the suspension was stirred for 15 min. The solid was filtered and was washed with diethyl ether (2 x 60 mL) to provide (^-phenylethylamine hydrochloride (39.1 g, 75%) as a white solid. Ή NMR (D20, 400 MHz) 1.52 (d, J= 7.2 Hz, 3 H); 4.42 (q, J= 7.2 Hz, 1 H); 7.35-7.40 (m, 5 H).
Example 8. 3-(4-Methoxyphenyl)-2-methyl-2-(l-(S)-phenylethylamino)-

To a 500 mL flask were added (^-phenylethylamine.HCl (25.0 g, 159.2 mmol, 1 eq.), MeOH (125 mL), NaCN (7.80 g, 159.2 mmol, 1 eq.) and 4-methoxyphenylacetone (Aldrich, ref: 19917-6. 24.5 mL, 159.2 mmol, 1 eq.). The mixture was stirred for 14 h at room
temperature. The mixture was filtered, the filter cake was washed with MeOH (30 mL) and the filtrates were concentrated to an oil which was dissolved in CH2C12(370 mL) and washed with water (250 mL). The organic phase was dried (MgS04) and concentrated in vacuo to provide 3-(4-methoxyphenyl)-2-methyl-2-(l-(5 -phenylethylamino)-propionitrile (47.2 g, 100% as a 6/4 mixture of diastereoisomers (S,S)/(R,S)), containing 5% of 4- methoxyphenylacetone) as a yellow oil. Ή NMR (CDC13, 400 MHz) 1 ,05 (s, 0.62 x 3 H); 1.27 (d, J- 6.4 Hz, 0.6 x 3 H); 1.40-1.44 (m, 0.4 x 6 H); 2.47 (d, J= 13.6 Hz, 0.4 x 1 H); 2.74 (d, J= 13.6 Hz, 0.4 x 1 H); 2.84 (d, J= 14 Hz, 0.6 x 1 H) 2.94 (d, J= 14 Hz, 0.6 x 1 H); 3.78 (s, 0.4 x 3 H); 3.82 (s, 0.6 x 3 H); 4.02 (q, J= 6.4 Hz, 0.6 x 1 H); 4.16 (q, J= 6, 4 Hz, 0.4 x 1 H); 6.80-7.40 (m, 9H). [0108] Example 9. 3-(4-Methoxyphenyl)-2-methyl-2-(l-(S)-phenylethylam o)- propionamidc 6.

[0109] Method A. To a 1 L flask with mechanical stirring under argon was added 3-
(4-methoxyphenyl)-2-methyl-2-(l-(5)-phenylethylamino)-propionitrile 5 (40.0 g, 136.1 mmol) dissolved in CH2C12 (400 mL). The solution was cooled to -5 °C (using an ice salt bath) and cone. H2S04 (40 mL) was added dropwise maintaining the temperature between – 5°C and 5°C. The mixture was warmed to RT over 2 h and was stirred for 16 h. Ice (400 g) was added and the mixture was stirred for 40 min. The two phases were separated and the aqueous phase was neutralized to pH 8-9 with 32% aq. NH3. The aqueous phase was extracted with EtOAc (3 x 350 mL). The combined organic layers were dried (MgS04) and were concentrated in vacuo to provide 6 (14.2 g, 34%, 98% chemical purity by HPLC/MS) as a 6/4 mixture of diastereoisomers (S,S)/(R, S)) as a yellow oil.
[01 10] Method B. In a 250 mL flask was dissolved 3-(4-methoxyphenyl)-2-methyl-
2-(l-(«S -phenylethylamino)-propionitrile 5 (5.0 g, 17.0 mmol) in CH2C12 (50 mL). The solution was cooled to 0°C and cone. H2S04 (2.5 mL) was added dropwise. The mixture was stirred at 40 °C for 28 h, was cooled to RT and ice (50 g) was added. The mixture was stirred for 1 h and the phases separated. The aqueous phase was basified to pH 8-9 using 32% aq. NH3 and was extracted with EtOAc (3 x 50 mL). The combined organic layers were dried (MgS04) and concentrated in vacuo to provide 6 (3.32 g, 62%, 97% purity by HPLC/MS) as a 6/4 mixture of diastereoisomers (S,S)/(R,S)) as a yellow oil. Ή NMR (CDC13, 400 MHz) 1.13 (s, 0.4 x 3 H); 1.15 (s, 03 x 3 H) 1.24 (d, J= 6.6 Hz, 0.4 x 3 H); 1.30 (d, J = 6.6 Hz 0.6 x 3 H); 2.75 (d, J= 13.4 Hz, 0.6 x 1 H); 2.78 (d, J= 13.6 Hz, 0.4 x 1 H); 2.84 (d, J= 13.4 Hz, 0.6 x 1 H) 3.32 (d, J = 13.6 Hz, 0.4 x 1 H); 3.78 (s, 0.6 x 3 H); 3.80 (s, 0.4 x 3 H); 3.85 (q, J = 6.6 Hz, 0.6 x 1 H); 4.16 (q, J = 6.6 Hz, 0.4 x 1 H); 6.80-7.40 (m, 9 H). HPLC/MS tR = 4.21 min [(S,S)-6 MS (ESI (+)) m/z 313.2 (M+l )] and 4.34 min [(R,S)-6 MS (ESI (+)) m/z 313.2 (M+l)].
[01 1 1 ] Example 10. 3-(4-Methoxyphenyl)-2-(5)-methyl-2-(l-(S)- phenylethylamino)-propionamide hydrochloride.

[01 12] In a 500 mL flask was dissolved amide 6 (14.2 g, 45.5 mmol, 1 eq.) in z‘-PrOH
(140 mL). Cone. HCl (5.7 mL, 68.3 mmol, 1.5 eq.) was added dropwise and the mixture was stirred for 20 min. The solvent was evaporated in vacuo and methyl isobutyl ketone (200 mL) was added. The mixture was heated to reflux temperature, was cooled to room temperature and was stirred for 72 h. The solids were collected by filtration, washed with methyl isobutyl ketone (20 mL) and dried in vacuo to provide 6«HC1 (13.6 g, 86%, diasteremeric ratio (dr) 63/37( i.e., 63% diastereomeric purity of the S,S diastereomer)) as a white solid.
[01 13] Example 11. Purification (Enhancing Diastereomeric Purity) of 6.HC1 by
Crystallization. In a 250 mL flask were placed amide hydrochloride 6·ΗΟ (13.6 g, dr 37/63) and /-BuOH (136 mL). The mixture was heated to reflux temperature and z‘-BuOH (95 mL) was distilled. The mixture was cooled to room temperature and was stirred overnight. The solids were collected by filtration and were washed with z‘-BuOH to provide 6»HC1 (1 1.6 g, 85%, dr 73/27 as a white solid.
[01 14] This solid was dissolved in /-BuOH (139 mL) and was heated to reflux temperature. z‘-BuOH (70 mL) was distilled and the mixture was cooled to room temperature and was stirred for 3 h. Filtration provided 6»HC1 (7.5 g, 65%, dr 88/12) as a white solid. [01 15] This solid was dissolved in /-BuOH (130 mL) and was heated to reflux temperature. z‘-BuOH (65 mL) was distilled and the mixture was cooled to room temperature and was stirred for 3 h. Filtration provided 6·ΗΟ (6.0 g, 80%, dr 99/1) as a white solid.
[01 16] This solid was dissolved in z‘-BuOH (105 mL) and was heated to reflux temperature. z‘-BuOH (53 mL) was distilled and the mixture was cooled to room temperature and was stirred for 16 h. Filtration provided 6·ΗΟ (5.4 g, 90%, dr >99/l, 100% purity by HPLC/MS, 40% overall yield (67% theoretical yield)) as a white solid. Ή NMR (DMSO, 400 MHz) 1.06 (s, 3 H); 1.57 (d, J= 6.4 Hz, 3 H); 2.84 (d, J= 13.2 Hz, 1 H); 3.27 (d, J = 13.2 Hz, 1 H); 3. 69 (s, 3 H) 4.40 (bs, 1 H); 6.83 (d, J= 8.4 Hz, 2 H); 6.98 (d, J= 8.4 Hz, 2 H); 7.37-45 (m, 2 H); 7.50-7,65 (m, 2 H); 7.80 (bs, 1 H); 9.40 (bs, 2 H). HPLC/MS tR = 4.21 min [(S,S)- 6»HC1. MS (ESI (+)) m z 313.2 (M+l)].
Example 12. 2-(S)-Amino-3-(4-methoxyphenyl)-2-methyl-propionamide

[01 18] Amine 6-HC1 (5.40 g, 15.5 mmol, 1 eq) was dissolved in MeOH (60 mL) and
10%Pd/C (2.0 g, 56% moisture content, 16% w/w) was added. The mixture was stirred under H2 (3 bar) at 50 °C for 80 min. The mixture was filtered through celite and the filter pad was washed with MeOH (20 mL). The filtrates were concentrated in vacuo to provide 2-(S)- amino-3-(4-methoxyphenyl)-2-methyl-propionamide hydrochloride (3.80 g, 100%, 99.5% purity by HPLC/MS) as a yellow solid. Ή NMR (DMSO, 400 MHz) 1.46 (s, 3 H); 3.02 (d, J = 14 Hz, 1 H); 3.10 (d, J- 14 Hz, 1 H), 3.72 (s, 3 H); 6.87 (d, J= 8.8 Hz, 2 H); 7.15 (d, J = 8.8 Hz, 2 H); 7.64 (s, 1 H); 7.94 (s, 1 H); 8.08 (bs, 2 H). HPLC/MS tR = 1.95 min. MS (ESI (+)) m z = 164.2 (M – CONH2). [01 19] Example 13. Metyrosine 1

[0120] To a 100 mL flask with a reflux condenser were added 2-(iS -amino-3-(4- methox phenyl)-2-methyl-propionamide hydrochloride (3.80 g, 15.6 mmol) and 48% HBr (20 mL). The solution was heated for 4 h at 120 °C, was cooled to room temperature and was concentrated in vacuo to give a beige paste. The paste was dissolved in water (15 ml) and the solution was again concentrated under vacuum. The paste was dissolved in H20 (15 mL), the solution was heated to 65 °C and 300 mg of activated carbon were added. The mixture was stirred for 15 min, was filtered and the filter pad was washed with water (2 x 4 mL). The solution was heated to 55 °C and the pH was adjusted to 5-6 using 32% aq. NH3. The mixture was cooled to 0 °C and was stirred for 15 min. Filtration, washing with cold water (2 x 5 mL) and drying in vacuo provided Metyrosine 1 (2.55 g, 83% yield, 99.6% HPLC purity, >99,5% ee) as a white solid. HPLC ((Zorbax C18, NaH2P04 10 mM pH = 3/MeCN (100:0) 10 min, (100:0) to (0: 100) 15 min, 0: 100 5 min), tR = 10.1 min. Chiral HPLC (Nucleosil Chiral-1, CuS04 10 mM/MeCN 10: 1), tR = 16.9 min. m.p. = 321-322 °C. [α]546 = +187° (c = 0.5, Copper complex solution) Copper complex solution preparation: Solution A (anhydrous NaOAc dissolved in H20 (150 mL) in 250 mL volumetric flask, glacial acetic acid (50 mL) added and diluted to volume with H20) mixed with Solution B (cupric sulfate (62.5 g) diluted to volume with H20 in a 200 mL volumetric flask) in a 1 L volumetric flask and diluted to volume with H20. Sample prepared 5 mg/mL in this solution.
[0121] In order to obtain an NMR spectrum and taking into account the low solubility of the product, a small sample (10 mg) was transformed into its HC1 salt. The sample was dissolved in 2 M HC1 and the solution was evaporated to dryness. Ή NMR (D20, 400 MHz) 1.49 (s, 3 H); 2.90 (d, J= 14.5 Hz, 1 H); 3.16 (d, J= 14.5 Hz, 1 H); 6.75 (d, J- 8.2 Hz, 2 H), 7.01 (d, J= 8.2 Hz, 2 H). [0122] Preparation of Metyrosine using L-alanine tert-butyl ester:
Scheme 5

[0123] Example 14. Synthesis of Aldimine. 4-Chlorobenzaldehyde (3.87 g, 27.5 mmol) was dissolved in methanol (50 mL) and treated with triethylamine (3.87 g, 38.3 mmol, 1.39 equiv). The mixture was stirred for 7 min at ambient temperature followed by addition of L-alanine tert-butyl ester hydrochloride (5.00 g, 27.5 mmol). Magnesium sulfate (6.63 g, 55.1 mmol, 2 equiv) was added to this solution and the slurry was stirred for 17 h at ambient temperature. The solid was filtered and washed with methanol (6 mL). The filtrate was evaporated to dryness to result in an oily solid. This solid was dissolved in a biphasic MTBE/water (70 mL/20 mL) mixture. The organic phase was separated and washed with water (20 mL). The organic phase was dried over MgS04, the solid was filtered, and the filtrate was evaporated to dryness to afford aldimine 7 [7.09 g; 96.2%] as a clear oil, which became a solid when stored in a refrigerator. 1H NMR (500 MHz, CDC13): δ 8.25 (br. s, 1H, ArCH), 7.71 (d, J= 8.5 Hz, 2H, Ar), 7.38 (d, J= 8.5 Hz, 2H, Ar), 4.04 (dq, J, = 0.6 Hz, J2 = 6.8 Hz, 1H, CH), 1.48 (d, J= 6.8 Hz, 3H, CH3), 1.47 (s, 9H, 3 CH3).
[0124] Example 15. Synthesis of ferf-Butyl 2-Amino-3-(4-methoxyphenyl)-2- methylpropanoate. Aldimine (2.00 g, 7.47 mmol) and O-allyl-N-benzylcinchonidinium bromide (0.38 g, 0.75 mmol, 0.10 equiv) were mixed with toluene (20 mL) at ambient temperature. The mixture was stirred for 30 min and then was cooled to 0°C. Powdered KOH (2.10 g, 37.35 mmol, 5 equiv) was added at once to convert the thin slurry into a yellow solution. The mixture was stirred for 5 min and 4-methoxybenzyl bromide (7.51 g, 37.35 mmol, 5 equiv) was added at 0 to 1°C. The solution was allowed to warm and was stirred at ambient temperature for 16 h. The reaction mixture was sequentially washed with water (20 mL) and brine (20 mL), separated, and treated with a 5-6 N HC1 solution in IPA (7 mL) for 1 h at ambient temperature. The reaction mixture was washed with water (20 mL). The aqueous phase was separated and treated with toluene (20 mL). The aqueous phase was separated, treated with a 2 N NaOH solution until basic, and the product was extracted with toluene (20 mL). The toluene phase was washed with brine (20 mL), separated, and dried over Na2S04. The solid was filtered and the solvent was stripped to dryness to afford tert- butyl 2-amino-3-(4-methoxyphenyl)-2-methylpropanoate; 1.70 g; 85.8% as a clear oil. Ή NMR (500 MHz, CDC13): δ 7.13 (d, J= 8.7 Hz, 2H, Ar), 6.81 (d, J= 8.7 Hz, 2H, Ar), 3.78 (s, 3H, CH3), 3.05 (d, J= 13.3 Hz, 1H, CH2), 2.71 (d, J- 13.3 Hz, 1H, CH2), 1.62 (br. s, 2H, NH2), 1.45 (s, 9H, 3xCH3), 1.32 (s, 3H, CH3). Ή NMR analysis, carried out in the presence of 1.2 equiv of BINOL, resulted in 47.6% ee. Optical rotation (Q?5D, chloroform, c = 1.38): – 9.06°.
[0125] Example 16. Synthesis of 2-Amino-3-(4-methoxyphenyl)-2- methylpropanoic Acid Hydrochloride. Intermediate
2-amino-3-(4- methoxyphenyl)-2-methylpropanoate (0.60 g, 2.26 mmol) was mixed with toluene (6 mL) and a 5-6 N HC1 solution in IPA (2 mL). A clear yellow solution was heated to reflux and kept at that temperature for 7 h. The resulting slurry was cooled to ambient temperature and filtered. The solid was washed with toluene (3 mL) on a filter and air-dried to afford 2- amino-3-(4-methoxyphenyl)-2-methylpropanoic acid hydrochloride [0.37 g; 67%] as a white solid [HPLC 71.8% (AUC; ¾= 3.71 ]. Ή NMR (500 MHz, DMSO-i¾): <5 13.96 (br. s, 1H, COOH), 8.44 (br. s, 3H, NH3), 7.16 (d, J= 8.7 Hz, 2H, Ar), 6.90 (d, J= 8.7 Hz, 2H, Ar), 3.74 (s, 3H, CH3), 3.08 (s, 2H, CH2), 1.48 (s, 3H, CH3). Optical rotation (Λ, DMSO, c = 1.10) +7.27°. [0126] Example 17. Synthesis of Metyrosine 1. tert-Butyl 2-amino-3-(4- methoxyphenyl)-2-methylpropanoate (0.30 g, 1.13 mmol) was dissolved in CH2C12(3 mL) and BBr3 (0.85 g, 3.39 mmol, 3 equiv) was added at room temperature. The reaction mixture was stirred for 1.5 h and treated with a NaHC03 solution to a basic pH. The aqueous phase was isolated. Solid started to precipitate in the aqueous phase in 30 min. Solid was filtered in 16 h and was washed on a filter with CH2C12(2 mL) and water (2 mL). The solid was air- dried to afford metyrosine [0.10 g; 45.4%] as a white solid [HPLC 80.7% (Metyrosine; AUC; fR= 2.32 & 2.57]. Ή NMR (500 MHz, TFA-d): δ 8.60 (d, J= 8.7 Hz, 2H, Ar), 8.39 (d, J = 8.7 Hz, 2H, Ar), 4.91 (d, J= 15.0 Hz, 1H, CH2), 4.67 (d, J= 15.0 Hz, 1H, CH2), 3.30 (s, 3H, CH3). Optical rotation (ο?0ο, c = 1.080, 1 = 10 mm, NaOAc/CuS04/H20/AcOH) +148.1.
[0127] Larger scale (e.g. >100 g) Synthesis Metyrosine using (R)- phenylglycinamide (Examples 18-24). Scheme 6 provides the general synthetic outline.
Scheme 6

l) NaCN, MeOH/H20, 24 h, 44 °C; 2) H2S04 cone, DCM, 15 °C to RT, 1.25 h: 3) a. 3 atm H2 6 wt% 10% Pd/C, MeOH, 55 °C, 16 h, b. HBr aq; 4) HBr 48%, 105 °C, 17 h; 5) NaOH aq; 6) 5 wt% 10% Pd/C, HC02H/H20 MeOH, 55 °C, 6 h
[0128] Example 18. 2-[l-(S)-Cyano-2-(4-methoxyphenyl)-l-methylethylamino]-
2-(R)-phenylacetamide 2. In a 5 L reactor equipped with anchor stirrer were charged (i?)-phenylglycinamide»HCl (330 g, 1.77 mol, 1 eq.), MeOH (1.1 L), H20 (1.9 L) and
7-methoxyphenylacetone (290 g, 1.77 mol, 1 eq.). A solution of NaCN (86.7 g, 1.77 mol, 1 eq.) in H20 (300 mL) was added over 15 min at room temperature. The mixture was stirred for 24 hours at 44 °C resulting in the formation of a yellow precipitate. The mixture was cooled to room temperature. The precipitate was filtered, was washed with H20/MeOH (7:3 v/v, 2 x 750 mL) and -PrOH (2 x 500 mL). The solid was dried in vacuo (3 days) at 35°C to provide 2-[l-(5)-Cyano-2-(4-methoxyphenyl)-l-methylethylamino]-2-( ?)-phenylacetamide 2 (460 g, 80%, dr 97/3) as a yellow solid. Ή NMR (400 MHz, CDC13) 1.14 (s, 3 H), 2.90 (d, J = 13.6 Hz, 1 H), 2.99 (d, J = 13.6 Hz, 1 H), 3.20 (bs, 1 H), 3.80 (s, 3 H), 4.51 (s, 1 H), 5.45 (bs, 1 H), 5.75 (bs, 1 H), 6.90 (d, J= 8.6 Hz, 2 H), 7.27 (d, J= 8.6 Hz, 2 H), 7.30-7.50 (m, 5 H). Ή NMR (R,R and S^-diastereoisomers of 2 (400 MHz, CDC13) 1.49 (s, 3 H), 2.82 (d, J = 13.8 Hz, 1 H), 2.85 (d, J= 13.8 Hz, 1 H), 3.78 (s, 3 H), 4.52 (s, 1 H), 5.55 (bs, 1 H), 6.60 (bs, 1 H), 6.84 (d, J = 8.6 Hz, 2 H), 7.17 (d, J= 8.6 Hz, 2 H), 7.30-7.40 (m, 5 H). dr determination: Ή NMR comparing integration of peaks of 2 at 2.90/2.99 (1.00 H, formally 2 H) with those of its (^.ii/S’^-diastereoisomeric pair (prepared from nearly rac- phenylglycinamide) at 2.82/2.85 (0.03 H, formally 2 H).
[0129] Example 19. 2-[(R)-(Carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(S)-methylpropionamide 3. Into a 10 L reactor equipped with anchor stirrer was charged CH2C12 (1.64 L). The solvent was cooled to 15°C, then 95% H2S04 (492 mL) and 2-[l-(5)-cyano-2-(4-methoxyphenyl)-l-methylethylamino]-2-( ?)-phenylacetamide 2 (410 g, 1.27 mol) were added alternately in 9 portions over approximately 45 min
(specifically: 164 mL of H2S04 then 82 g of 2; subsequently, at approximately 5 min intervals, 8 x [41 mL of H2S04 then immediately 41 g of 2]). On addition of each portion, the suspension of 2 in the dense oily phase slowly dissolved (1-2 min) to provide a biphasic mixture. The resulting biphasic mixture (a red-brown dense oil with a pale yellow
supernatant CH2C12 layer) was stirred for 0.5 h at 25°C. Ice-cold water (4.1 L) was added over 30 min, very slowly initially (200 mL dropwise over 15 min) due to a violent exotherm, and the biphasic mixture was stirred for 0.5 h. The phases were separated and the organic phase discarded. The combined aqueous phases were washed with CH2C12 (450 mL), and residual CH2C12 was stripped from the aqueous phase by distillation under vacuum at 55°C (20-30 mBar). The aqueous solution was then cooled to 20°C and was basified with 32% aq NH3 (1 150 mL) to pH 8-9 at such a rate that the temperature was kept below 28°C
(approximately 120 min). The suspension was stirred for 30 min to ascertain a stable pH. The white solid which formed was separated by filtration, washed with H20 (2 x 2050 mL), and was thoroughly drained of water (but was not dried) to provide 2-[(R)- (carbamoylphenylmethyl)-amino]-3-(4-methoxyphenyl)-2-(5)-methylpropionarnide 3 (1087 g (391 g theoretical, the sample contains 64% w/w of H20), yield 90%, 97% HPLC purity, 96% de) as a wet white solid. HPLC (Luna C18, H20 / MeCN 95:5 to 0: 100 30 min, 254 nm, sample 2 mg/mL in MeOH). tR (3) = 15.3 min, 96% de. (2 degrades under these conditions: 3 peaks are detected at 17.7, 18.9 and 19.9 min). HPLC (#R/S,S)-diastereoisomer of 3, tR = 1 5.0 min. de determination: HPLC comparing integration of peaks at tR = 15.3 min (97.3 area% 3) and tR = 15.0 min (1 .6 area% ( ?, ?)-diastereoisomer of 3 (reference (R,R/S,S) prepared from nearly rac-phenylglycinamide).
[0130] Example 20. Purification of 2-[(R)-(carbamoylphenylmethyl)-amino]-3-
(4-methoxyphenyl)-2-(S)-methylpropionamide 3. 2-[( ?)-(Carbamoylphenylmethyl)- amino]-3-(4-methoxyphenyl)-2-(5)-methylpropionamide 3 (321 g, 941 mmol) and methyl isobutyl ketone (4173 mL) were heated to 72°C until the solid was dissolved and the biphasic mixture (the minor lower aqueous layer is only visible on stopping stirring) was allowed to cool to room temperature with constant stirring. Stirring was maintained for 2 h. The solid formed was filtered at room temperature, was washed with methyl isobutyl ketone (2 x 320 mL) and was dried in vacuo to provide 2-[( ?)-(carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(5 methylpropionamide 3 (268 g, 84%, >99.5% purity by HPLC) as a white solid. Ή NMR (400 MHz, DMSO-d6) 1.04 (s, 3H), 2.35 (bs, 1 H), 2.79 (d, J- 12.8 Hz, 1 H), 2.95 (d, J = 12.8 Hz, 1 H), 3.65 (s, 3 H), 4.22 (s, 1 H), 6.65 (d, J – 8.4 Hz, 2 H), 6.96 (d, J = 8.4 Hz, 2 H), 7.02 (bs, 1 H), 7.05 (bs, 1 H), 7.33-7.30 (m, 3 H), 7.48 (d, J= 7.2 Hz, 2 H), 7.52 (bs, 1 H), 7.64 (bs, 1 H). HPLC (Luna CI 8, H20 / MeCN 95:5 to 0: 100 30 min, 254 nm, sample 2 mg/mL in MeOH) tR = 15.3 min, >99.5% purity. Mp: 106-108°C.
[0131 ] Example 21. Hydrogenolysis to provide 2-(S)-Amino-3-(4- methoxyphenyl)-2-methyl-propionamide hydrogen bromide salt 4»HBr. To a 1 L hydrogenation reactor were added 2-[( ?)-(carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(5)-methylpropionarnide 3 (183.0 g, 537 mmol, 1 eq), MeOH (549 mL) and 10% Pd/C (19.4 g, 5 wt%). The mixture was stirred under H2 (3 bar) at 51 °C for 8 h. Further 10% Pd/C (3,88 g, 1 wt%) was added and the mixture was stirred for a further 8 h at 53°C. The mixture was cooled to room temperature, was filtered through Celite and the filter pad was washed with MeOH (2 x 50 mL). The combined filtrates were concentrated at 30°C under reduced pressure (rotary evaporator) to a dense white “stirrable” paste (250 mL) containing 2-(,S)-amino-3-(4-methoxyphenyl)-2-methyl-propionamide 4 and 5.
[0132] H20 (75 mL) and 48% HBr (75 mL, 667 mmol, 1 ,25 eq.) were then added resulting in a white suspension. Residual MeOH was stripped from the mixture (45 mL distilled) by distillation at 100°C (bath temperature) at reduced pressure (20-30 mBar). The resulting aqueous solution was cooled to room temperature and filtered; the solid was washed with H20 (50 mL). The white solid was discarded (containing phenylacetamide 5 and 4% 4-HBr by NMR) and the resulting solution of 4»HBr (approximately 400 mL, containing 23% 5 with respect to 4 by NMR) was used directly. 1H NMR (400 MHz, DMSO-6d) 4 1.14 (s, 3 H), 2.10 (bs, 2 H), 2.50 (d, J= 13.1 Hz, 1 H), 2.95 (d, J= 13.1 Hz, 1 H), 3.69 (s, 3 H), 6.80 (d, J= 8.5 Hz, 2 H), 6.81 (bs, 1 H), 6.90 (bs, 1 H), 7.09 (d, J- 8.5 Hz, 2 H).
Phenylacetamide 5 3.34 (s, 2 H), 7.15-7.28 (m, 6 H), 7.42 (bs, 1 H). HPLC (Kromasil C8, H2O/0.1 % TFA / MeCN/0.07% TFA 95:5 to 0: 100 30 min, 254 nm, 1 mg/mL in MeOH) phenylacetamide 5 tR = 1 1.21 min. 4 tR = 9.10 min. (3 tR = 1 1 ,95 min.).
[0133] Example 22. (-)-a-Methyl-L-tyrosine, Metyrosine 1. To a 2 L flask equipped with an anchor stirrer was charged the 4»HBr solution (519 mmol obtained from hydrogenolysis) and 48% HBr (648 mL) was added. The solution was heated for 17 h at 105°C and was cooled to room temperature. The solution was washed with CH2C12 (8 x 80 mL, to remove traces of phenylacetic acid) and the aqueous phase was stripped of residual CH2C12 by distillation at 65°C at reduced pressure (20-30 mBar). Activated carbon (10.5 g) was added and the mixture was stirred for 30 min at 60°C, was filtered at 60°C and the filter pad was washed with water (2 x 35 mL) at RT. The combined filtrates were cooled to room temperature and were basified with 12.5 M NaOH (430 mL) to pH 6-7 at such a rate as to maintain the temperature below 30°C (over approximately 2 h). The white solid formed was separated by filtration, was washed with H20 (2 x 315 mL) and was dried in vacuo to provide (-)-a-methyl-L-tyrosine, metyrosine 1 (87 g, 86%, >99.9% HPLC purity, no impurities detected, >99.9% ee the other enantiomer is not detected) as a white solid. HPLC (Zorbax CI 8, NaH2P04 10 mM pH = 3 / MeCN (100:0) 10 min, (100:0) to (0:100) 15 min, 0: 100 5 min, , 225 nm, sample 1 mg/mL in 0.1 M HC1) tR (1) = 7.6 min, tR (4) = 13,95 min. Chiral HPLC (Nucleosil Chiral-1 , CuS04 10 mM / MeCN 9: 1 , 254 nm, sample 1 mg/mL in eluant) tR = 14.4 min. tR enantiomer = 8.4 min. Mp: 309-313°C.
[0134] In order to obtain an NMR spectrum (taking into account the low solubility of the product), a small sample (10 mg) was transformed into its HC1 salt. The sample was dissolved in 2 M HC1 and the solution was evaporated to dryness. Ή NMR (400 MHz, D20) 1.49 (s, 3 H), 2.90 (d, J = 14.5 Hz, 1 H), 3.16 (d, J = 14.5 Hz, 1 H), 6.75 (d, J = 8.2 Hz, 2 H), 7.01 (d, J = 8.2 Hz, 2 H).
[0135] Example 23. Transfer hydrogenolysis to provide 2-(S)-Amino-3-(4- methoxyphenyl)-2-methylpropionamide formic acid salt 4-HCOOH. In a 5 L reactor equipped with anchor stirrer and oil bubbler, 2-[(i?)-(carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(5)-methylpropionamide 3 (261.8 g, 766.8 mmol) was dissolved in MeOH (1570 mL). A first batch of 10% Pd/C (22.2 g, 4% w/w) was added and the mixture was heated to 56 °C. HCOOH (217 mL, 5.75 mol) was dissolved in H20 (393 mL) and 480 mL of the resulting solution were added to the mixture dropwise over 4.5 h, and the temperature was maintained between 54 and 60°C. When gas development ceased (as determined from the oil bubbler, approximately 30 min after complete addition of HC02H aq), the mixture was cooled to room temperature and a second batch of 10% Pd/C (5.6 g, 1 % w/w) was added. The mixture was heated again to 55°C and the remaining HCOOH solution (130 mL) was added dropwise over 45 min. Stirring was maintained for a further 30 min. The mixture was cooled to room temperature, was filtered over a pad of Celite and the filter pad was washed with MeOH (2 x 100 mL). The combined filtrates were concentrated under reduced pressure (rotary evaporator) to a white “stirrable” paste (approximate volume 300 mL) containing 4’HCOOH and phenylacetamide 5.
[0136] H20 (100 mL) was added and residual MeOH was stripped by distillation at reduced pressure (20-30 mBar) at 70 °C. The resulting solution (approximately 360 mL) was filtered and was washed with 100 mL of H20. The white solid (5 containing 1 % of
4«HCOOH by NMR) was discarded and the resulting solution of 4»HCOOH (containing 13% of 5 with respect to 4 by NMR) was used directly. Ή NMR (400 MHz, DMSO-d6) 1.43 (s, 3 H), 2.93 (d, J = 13.6 Hz, 1 H), 3.08 (d, J = 13.6 Hz, 1 H), 3.73 (s, 3 H), 6.88 (d, J = 8.8 Hz, 2 H), 7.17 (d, J = 8.8 Hz, 2 H), 7.56 (s, 1 H), 7.83 (s, 1 H). Phenylacetamide 5 (200 MHz, DMSO-d6) 3.36 (s, 2 H), 6.87 (bs, 1 H), 7.20-7.33 (m, 5 H), 7.45 (bs, 1 H). HPLC (Kromasil C8, H2O/0.1% TFA / MeCN/0.07% TFA 95:5 to 0:100 30 min, 254 nm, 1 mg/mL in MeOH) Phenylacetamide 5 tR = 1 1.38 min. 4.HC02H tR = 9.11 min. (3 tR = 11,95 min).
[0137] Example 24. (-)-a-Methyl-L-tyrosine, Metyrosine 1. Into a 2 L reactor equipped with anchor stirrer, a solution of 4»HCOOH (760 mmol from transfer
hydrogenolysis) and H20 (200 mL) was mixed with 48% aqueous HBr (948 mL, 8.43 mol). The resulting solution was heated to 105°C for 17 h. The mixture was cooled to room temperature and was washed with CH2C12 (6 x 125 mL, to remove traces of phenylacetic acid), the aqueous phase was stripped of residual CH2C12 at 60 °C at reduced pressure (20-30 mBar). The solution was mixed with activated carbon (15.6 g, 10% w/w) and was heated to 60°C for 30 min. The mixture was filtered at 60°C and the residue was rinsed with H20 (2 x 60 mL). The combined filtrates were cooled to 15°C and were basified with 12.5 M NaOH (730 mL) to pH 6-7 at such a rate as to maintain the temperature below 30°C (over approximately 100 min). The white solid formed was separated by filtration, was washed with H20 (2 x 460 mL) and IPA (490 mL and 245 mL) and was dried to provide (-)-ot- methyl-L-tyrosine, Metyrosine 1 (121.1 g, 82%, >99.9% HPLC no impurities >0.1% detected; >99.9% ee., the other enantiomer is not detected) as a white solid. HPLC (Zorbax CI 8, NaH2P04 10 mM pH = 3 / MeCN (100:0) 10 min, (100:0) to (0:100) 15 min, 0: 100 5 min, 225 nm, sample 1 mg/mL in 0.1 M HC1) tR (1) = 7.6 min; t (4) = 13,95 min. Chiral HPLC (Nucleosil Chiral-1, CuS04 10 mM / MeCN 9:1, 254 nm, sample 1 mg/mL in eluant) tR – 14.3 min. tR enantiomer = 8.4 min. Mp: 308-313 °C.
Patent
IN 2010/CHE/1204, IN 1204/CHE/2010,








EXAMPLE-1:
Step- (a): Preparation of (S)- [1-(3,5-Dichloro-phenylcarbamoyl)-ethyl]-carbamic acid tert-butyl ester(32).
Dissolved N-BOC-L-AIanine (200 gm, 1.057 mol) in methylene chloride (800 ml) under stirring. Cooled the resulting reaction mixture to -15 to -10°C, added N-methyl morpholine (128.3 gm, 1.268 mol) then further cooled to -35°C, added ethylchloroformate (131.9 gm, 1.215 mol) followed by 3,5-dichloroaniline (171.2 gm, 1.057 mol). Stirred the above reaction mixture at 0 to 5°C for a period of 16-18 hours. Quenched the reaction mixture with water (500 ml), stirred for 10-15 minutes and separated the organic layer. The organic Iayerwas washed with water (2×200 ml), dried with anhydrous sodium sulfate and filtered. The organic layer was distilled off under reduced pressure, added hexane (400 ml) and stirred for 30 minutes at 0-5°C. Filtered the resulting solid and washed with hexane (100ml). Dried until constant weight is reached. Dry weight of obtained (S)- [1-(3,5-Dichloro-phenylcarbamoyl)-ethyl]-carbamic acid tert-butyl ester is 300.0 gm.
Yield: 85.1%;
Melting point of the resulting compounds ranges from 138.1-140.5°C;
IR spectra (cm’1): 3321, 2981, 1671, 1589, 1539,1446, 1317, 1255, 1165, 1117, 1072, 858, 802;
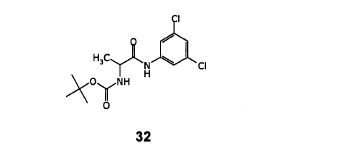
32
1H NMR (400 MHz, CDCI3): 59.27(br.s, 1H), 7.37(s, 2H), 6.96(s, 1H), 5.42 (br.d, J=5.16HZ, 1H), 4.39(br.s, 1H),1.47 (s, 9H), 1.41(d, J=7.00HZ, 3H).
Mass (m/z): 334.2 [M+H]+.
Step- (b): Preparation of (S)- 2-Amino-N-(3,5-dichloro-phenyl)-propionamide (33).
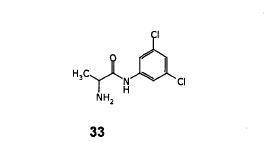
Dissolved (S)- [1-(3,5-Dichloro-phenylcarbamoyl)-ethyl]-carbamic acid tert-butyl ester (200.0 gm, 0.599 mol) obtained from step-(a) in methanol (300 ml). To the resulting mixture Cone, hydrochloric acid (500 ml) and water (320 ml) are added at 20-25°C. The resulting reaction mixture was stirred for 16-18 hours at 20-25°C. Added water (200 ml) and toluene (400 ml), cooled to 5-100C1 then basified with 50% sodium hydroxide solution. Stirred for 15 minutes and separated the layers. Aqueous layer was washed with toluene (3x200ml). Combined organic layers and washed with water, dried with anhydrous sodium sulfate and filtered. The organic layer was distilled off under reduced pressure to get the title compound as brown colored syrup. Weight: 135.0 gm.
Yield: 96.4%;
IR spectra (cm-1): 3281, 2969, 2930, 1681, 1585, 1515, 1445, 1409, 1372, 1258, 1185, 1112, 925, 843, 798, 669;\
1H NMR (400 MHz, CDCI3) : 59.67(br.s, 1H), 7.50(s, 2H), 7.00(s, 1H), 3.51 (q,J=7.03HZ, 1H), 1.65(br.s, 2H),1.34(d, J=7.04HZ, 3H).
Mass (m/z): 234.09 [M+H]+.
Step- (c): Preparation of (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one(34).

Dissolved (S)- 2-Amino-N-(3,5-dichloro-phenyl)-propionamide (135 gm, 0.579 mol) obtained in step-(b) in toluene (925 ml) under stirring. Cooled the resulting reaction mixture to 20°C, added isobutyraldehyde (83.5 gm, 1.157mol) over a period of 50 to 60 minutes. Stirred the above reaction mass at 50 to 55°C for a period of 18-19 hours. The organic layer was distilled off under reduced pressure, added hexane (270 ml) and stirred for 1.45 hours at 0-5°C. Filtered the resulting solid and washed with hexane (50ml). Dried until constant weight is reached. Dry weight of obtained (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one is 138.0 gm.
Yield: 82.98%;
Melting point ranges from 128.9-131.8°C;
IR spectra (cm”1): 3305, 3051, 2962, 2877, 1689, 1587, 1450, 1383, 1278, 1223, 1055, 846, 798, 743;
1H NMR (400 MHz, CDCI3) : 57.40 (s, 2H), 7.15 (s, 1H), 5.00 (s, 1H), 3.68 (q, J=6.83HZ, 1H), 1.98 (br.s, 1H), 1.35 (d, J=6.84HZ, 3H), 0.96(d, J=6.87HZ, 3H), 0.76 (d, J=6.70HZ, 3H).
Mass (m/z): 288.1 [M+H]+.
Step- (d): Preparation of (2R, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1 -(2,2,2-trifluoroacetyl)-imidazolidin-4-one (35).
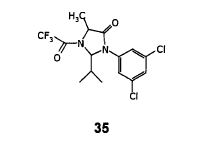
Dissolved (S)-3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one (135 gm, 0.470 mol) obtained in step-(c) in methylene chloride (1080 ml) under stirring. Cooled the resulting reaction mixture to 0 to 5°C, added triethyl amine (66.48 gm, 0.658 mol) followed by trifluoroacetic anhydride (132.3 gm, 0.658 mol) over a period of 30 minutes. Stirred the above reaction mass at 0 to 5°C for a period of 2-3 hours. Quenched the reaction mixture with water (405 ml), stirred for 20-25 minutes and separated the organic layer. The organic layer was washed with water (2×270 ml), dried with anhydrous sodium sulfate and filtered. The organic layer was distilled off under reduced pressure, to get crude product as semi solid. The obtained semi solid mass was recrystalised using isopropyl alcohol to get pure product. Dried until constant weight is reached. Dry weight of obtained (2R, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1-(2,2,2-trifluoroacetyl)-imidazolidin-4-one is 153.0 gm.
Yield: 84.93%;













Step- (c): Preparation of (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one (34).
Dissolved (S)- 2-Amino-N-(3,5-dichloro-phenyl)-propionamide (104 gm, 0.446 mol) obtained in step-(b) in toluene (715 ml) under stirring. Cooled the resulting reaction mixture to 20°C, added isobutyraldehyde (64.34 gm, 0.892mol) over a period of 50 to 60 minutes. Stirred the above reaction mass at 50 to 55°C for a period of 18-19 hours. The organic layer was distilled off under reduced pressure, added hexane (200 ml) and stirred for 3 hours at 0-5°C. Filtered the resulting solid and washed with hexane (25ml). Dried until constant weight is reached. Dry weight of obtained (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one is 101.0 gm. Yield: 78.90%;
Step- (d): Preparation of (2R, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1-(2,2,2-trifluoroacetyl)-imidazolidin-4-one (35).
Dissolved (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one (100 gm, 0.348 mol) obtained in step-(c) in methylene chloride (800 ml) under stirring. Cooled the resulting reaction mixture to 0 to 5°C, added triethyl amine (42.28 gm, 0.417 mol) followed by trifluoroacetic anhydride (87.73 gm, 0.417 mol) over a period of 30 minutes. Stirred the above reaction mass at 0 to 5°C for a period of 2-3 hours. Quenched the reaction mixture with water (300 ml), stirred for 20-25 minutes and separated the organic layer. The organic layer was washed with water (2×200 ml), dried with anhydrous sodium sulfate and filtered. The organic layer was distilled off under reduced pressure, to get crude product as semi solid. The obtained semi solid mass was recrystalised using isopropyl alcohol to get pure product. Dried until constant

reached. Dry weight of obtained (2S, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1-(2,2,2-trifluoroacetyl)-5-(4-methoxy benzyl)-imidazolidin-4-one is 6.6 gm.
Yield: 61.10%;
Melting point ranges from 114.2-116.4°C;
IR spectra (cm’1): 2977, 1731, 1692, 1609, 1585, 1572, 1513, 1426, 1348, 1250, 1197, 1048, 890, 748;
1H NMR (400 MHz, CDCI3) : 57.29 (s,1H), 6.84(d, J=8.5HZ, 2H), 6.80(d, J=1.5HZ, 2H), 6.73(d, J=8.1HZ, 2H), 5.07(s, 1H), 3.99(q, J=6.9Hz, 2H), 3.66(d, J=13.8HZ, 1H), 2.99(d, J=13.8HZ, 1H), 2.05(t, J=6.94HZ, 1H), 1.95(s, 3H), 1.41 (t, J=6.94HZ, 3H), 0.91 (d, J=6.61HZ, 3H), 0.52(d, J=7.30HZ, 3H).
Mass (m/z): 517.3 [M+H]+.
Step- (f): Preparation of (S)-2-amino-N-(3,5-Dichloro-phenyl)-2-methyl-3-(4-ethoxy phenyi)-propionamide (45).
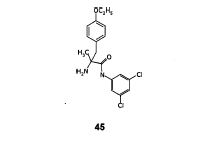
To the suspension of potassium hydroxide (1.12 gm, 0.0169 mol) in isopropyl alcohol (25 ml) , (2S, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1 -(2,2,2-trifluoroacetyl)- 5-(4-ethoxy benzyl)-imidazolidin-4-one (5.0 gm, 0.009 mol) obtained in step-(e) was added at 25 to 30°C under stirring. The resulting reaction mixture was stirred at 40 to Cl 45 45°C for 3-4 hours. Cooled to 10 to 15°C,added 3M sulfuric acid (15 ml) over a period of 30 minutes. The resulting reaction mixture was heated to 70 to 75°C, stirred for 2 to 3 hours at same temperature. Distilled solvent completely under reduced pressure, added water (25 ml) and ethyl acetate (50 ml). Stirred for 15 minutes and basified with 20% sodium hydroxide solution. Stirred for 15 minutes at 25 to 30°C, separated the organic layer. Aqueous layer washed with ethyl acetate (50 ml). Combined the organic layers and washed with saturate sodium chloride solution (25 ml), dried over anhydrous sodium sulfate, filtered. Removed solvent completely under reduced pressure to get the title compound as brown colored syrup. Weight of (S)-2-amino-N-(3,5-Dichloro-phenyl)- 2-methyl-3-(4-ethoxy phenyl)-propionamide is 2.8 gm. Yield: 79.0%; IR spectra (cm”1 ): 2981, 1732, 1689, 1682, 1575, 1513, 1446, 1302, 1244, 1179, 1116, 1048, 843; 1H NMR (400 MHz, CDCI3) : 59.79(br.s, 1H), 7.52(s, 2H), 7.05(d, J=8.68HZ, 3H), 6.79(d, J=8.53HZ, 2H), 3.94(q, J=6.98, 2H), 3.38(d, J=13.25HZ, 1H), 2.57(d, J=13.56HZ, 1H), 2.03(s, 1H), 1.58(br.s, 2H), 1.43(s, 3H),1.39(t, J=5.42HZ, 3H), Mass (m/z): 368.2 [M+H]+ .
Step- (g): Preparation of (2S)-2-amino-3-(4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine). Dissolved the (S)-2-amino-N-(3,5-Dichloro-phenyl)-2-methyl-3-(4-ethoxy phenyl)- propionamide (2.8 gm, 0.007 mol) obtained in step-(f) in aqueous HBr (50 ml) under stirring. The resulting reaction mixture was heated to 120-125°C and stirred for 24 hours. Cooled to 50°C, added water (100 ml) stirred for 15 minutes then further cooled 54 to 10-15°C , pH adjusted to 5-6 with ammonium hydroxide solution. Stirred for 30 minutes at 10-15°C, filtered and cake washed with water (2×5 ml). Dried until constant weight is reached. Dry weight of obtained crude (2S)-2-amino-3- (4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine) of formula-1 is 1.8 gm.
Step- (h): Purification of (2S)-2-amino-3-(4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine). The crude (2S)-2-amino-3-(4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine) obtained in step ( g) 1.8 gm) was dissolved in water (180 ml) by heating the reaction mixture to 90°C. Darco (Charcoal) was added and stirred for 10-15 minutes at same temperature. The resulting reaction mixture was filtered through celite bed. The filtered reaction mass concentrated up to half volume reached under reduced pressure. Cooled to IO0C and stirred for a period of 30 minutes at same temperature. Filtered the resulting solid, washed with water. Dried until constant weight is reached. Dry weight of obtained pure (2S)-2-amino-3- (4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine) of formula-1 was 0.8 gm. The product is matching in all respects with compounds of Metyrosine obtained from EXAMPLE-1 (Step-h). Purity: 99.98%. Chiral purity by HPLC: 100.0%.
IR FROM NET


13 C NMR
References
- ^ Green KN, Larsson SK, Beevers DG, Bevan PG, Hayes B (August 1982). “Alpha-methyltyrosine in the management of phaeochromocytoma”. Thorax. 37 (8): 632–3. doi:10.1136/thx.37.8.632. PMC 459390. PMID 7179194.
- ^ O’Leary OF, Bechtholt AJ, Crowley JJ, Hill TE, Page ME, Lucki I. Depletion of serotonin and catecholamines block the acute behavioral response to different classes of antidepressant drugs in the mouse tail suspension test. Psychopharmacology. 2007 Jun;192(3):357-71. PMID 17318507
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Demser |
| AHFS/Drugs.com | Consumer Drug Information |
| ATC code | |
| Pharmacokinetic data | |
| Elimination half-life | 3.4–3.7 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.010.546 |
| Chemical and physical data | |
| Formula | C10H13NO3 |
| Molar mass | 195.215 g/mol |
| 3D model (JSmol) | |
////////////Metyrosine, Metirosine, Demser, メチロシン , JAPAN 2019, α-Methyl-p-tyrosine, метирозин , ميتيروسين , 甲酪氨酸 ,

