
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
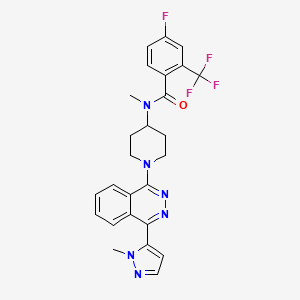
Taladegib
LY2940680; 1258861-20-9; Taladegib; LY-2940680; UNII-QY8BWX1LJ5; QY8BWX1LJ5
4-fluoro-N-methyl-N-[1-[4-(2-methylpyrazol-3-yl)phthalazin-1-yl]piperidin-4-yl]-2-(trifluoromethyl)benzamide
| Molecular Formula: | C26H24F4N6O |
|---|---|
| Molecular Weight: | 512.513 g/mol |
Taladegib is an orally bioavailable small molecule antagonist of the Hedgehog (Hh)-ligand cell surface receptor smoothened (Smo) with potential antineoplastic activity. Taladegib inhibits signaling that is mediated by the Hh pathway protein Smo, which may result in a suppression of the Hh signaling pathway and may lead to the inhibition of the proliferation of tumor cells in which this pathway is abnormally activated. The Hh signaling pathway plays an important role in cellular growth, differentiation and repair; constitutive activation of this pathway is associated with uncontrolled cellular proliferation and has been observed in a variety of cancers.
Taladegib has been used in trials studying the treatment of Solid Tumor, COLON CANCER, BREAST CANCER, Advanced Cancer, and Rhabdomyosarcoma, among others.
- Originator Eli Lilly
- Developer Eli Lilly; Ignyta
- Class Antineoplastics; Benzamides; Fluorobenzenes; Phthalazines; Piperidines; Pyrazoles; Small molecules
- Mechanism of Action Hedgehog cell-signalling pathway inhibitors; SMO protein inhibitors
Highest Development Phases
- Phase I/II Oesophageal cancer; Small cell lung cancer
- Phase I Ovarian cancer; Solid tumours
- Preclinical Basal cell cancer
- No development reported Cancer
Most Recent Events
- 04 Nov 2017 No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Second-line therapy or greater) in Japan (PO, Tablet)
- 02 Jun 2017 Adverse events data from a phase I/II trial in Ovarian cancer (Solid tumours) presented at the 53rd Annual Meeting of the American Society of Clinical Oncology (ASCO-2017)
- 23 Mar 2017 Ignyta amends its license, development and commercialisation agreement with Eli Lilly for taladegib
SYN






PATENT


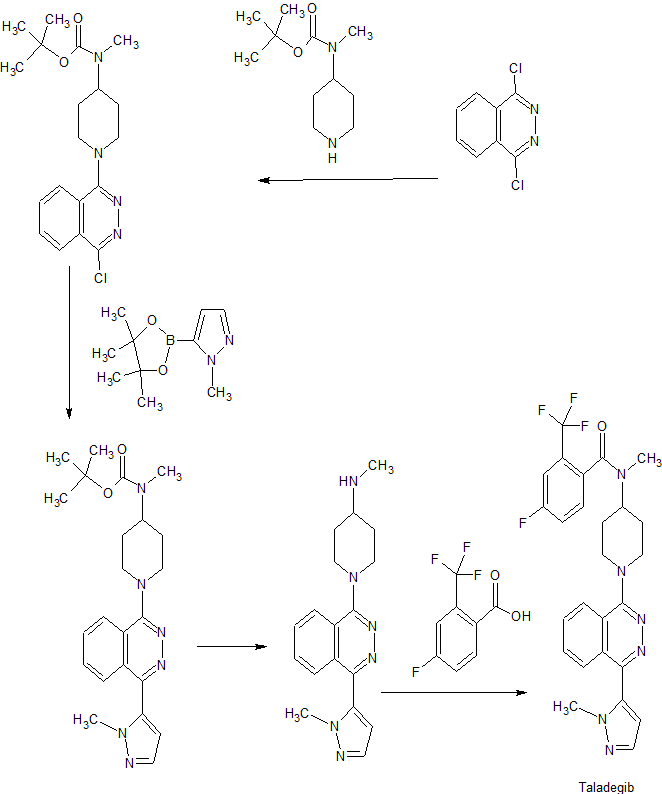
Preparation 1 ter?-Butyl 1 -(4-chlorophthalazin- 1 -yl)piperidin-4-yl(methyl)carbamate

Heat a mixture of potassium carbonate (21.23 g, 153.6 mmol), 1,4-dichlorophthalazine (26 g, 128 mmol) and methyl-piperidin-4-yl carbamic acid ter?-butyl ester (30.01 g, 134.4 mmol) in N-methylpyrrolidine (200 mL) at 80 0C overnight. Pour the reaction mixture into water, extract with dichloromethane, dry over Na2SC”4, and concentrate under reduced pressure. Add diethylether and filter off the resulting solid (4-chlorophethalazin-1-ol from starting material impurity). Concentrate the filtrate. Purify the resulting residue by flash silica gel chromatography (hexane : ethyl acetate = 2 : 1) to X-18698
-9- provide the title compound as a white solid (17.66 g, 37%). ES/MS m/z (37Cl) 377.0 (M+ 1).
Preparation 2 fer?-Butyl 1 -(4-chlorophthalazin- 1 -yl)piperidin-4-ylcarbamate

Prepare the title compound by essentially following the procedure described in Preparation 1 , using piperidin-4-yl-carbamic acid tert-butyl ester. Cool the reaction mixture and pour into water (500 mL). Extract with ethyl acetate, wash with water, dry over Na2SC”4, and remove the solvents under reduced pressure to provide the title compound as a yellow solid (36 g, 97%). ES/MS m/z 363.0 (M+l).
Preparation 3 ter?-Butyl methyl( 1 -(4-( 1 -methyl- lH-pyrazol-5 -yl)phthalazin- 1 -yl)piperidin-4- yl)carbamate

Place sodium carbonate (3.82 g, 36.09 mmol), tert-butyl 1 -(4-chlorophthalazin- 1-yl) piperidin-4-yl(methyl)carbamate (6.8 g, 18.04 mmol) and 1 -methyl- lH-pyrazole-5-boronic acid pinacol ester (5.63 g, 27.1 mmol) in a flask with a mixture of toluene (50 mL), ethanol (17 mL), and water (17 mL). Degas the mixture for 10 min with nitrogen gas. Add tetrakis(triphenylphosphine)palladium (0.4 g, 0.35 mmol) and heat the mixture at 74 0C overnight. Cool the mixture to ambient temperature and dilute with dichloromethane. Wash the organic portion with brine, dry over Na2SC”4, and concentrate under reduced pressure. Purify the resulting residue by flash silica gel chromatography X-18698
-10-
(hexane : ethyl acetate : 2 M NH3 in MeOH = 20 : 5 : 1) to provide the title compound as a yellow foam (5.33 g, 70%). ES/MS m/z 423.2 (M+ 1).
Alternate procedure to prepare tert-butyl methyl(l-(4-(l-methyl-lH-pyrazol-5-yl)phthalazin-l-yl)piperidin-4-yl)carbamate: Preparations 4 – 6
Preparation 4
1 ,4-Dibromophthalazine

Charge a pressure tube with phosphorus pentabromide (24.5 g, 54.1 mmol) and
2,3-dihydro-phthalazine-l,4-dione (5.00 g, 30.8 mmol). Seal the tube and heat at 140 0C for 6-7 h. Allow to cool overnight. Carefully open the tube due to pressure. Chisel out the solid and pour into ice water. Allow to stir in ice water and collect the resulting solid by vacuum filtration. Dry in a vacuum oven to obtain the final product (8.31 g, 93%). ES/MS (79Br, 81Br) m/z 288.8 (M+). Ref: Can. J. Chem. 1965, 43, 2708.
Preparation 5 ter?-Butyl 1 -(4-bromophthalazin- 1 -yl)piperidin-4-yl(methyl)carbamate

Combine 1 ,4-dibromophthalazine (0.70 g, 2.38 mmol), N-methylpyrrolidone (7.0 mL), potassium carbonate (395 mg, 2.86 mmol), and methyl-piperidin-4-yl-carbamic acid ter?-butyl ester (532 mg, 2.38 mmol). Heat at 80 0C overnight. Cool and pour into water. Collect the solid and dry in a vacuum oven at ambient temperature overnight to obtain the final product (0.96 g, 95%). ES/MS m/z (81Br) 421.0 (M+ 1).
X-18698
-11-
Preparation 6 fer?-Butyl methyl (l-(4-(l -methyl- lH-pyrazol-5-yl)phthalazin-l-yl)piperidin-4- yl)carbamate

Charge a reaction tube with fer?-butyl l-(4-bromophthalazin-l-yl)piperidin-4-yl(methyl)carbamate (500 mg, 1.2 mmol), 1 -methyl- lH-pyrazole-5-boronic acid pinacol ester (370 mg, 1.8 mmol), sodium carbonate (252 mg, 2.4 mmol), toluene (3.75 mL), ethanol (1.25 mL), and water (1.25 mL). Degas the reaction mixture with nitrogen for 10 min. Add tetrakis (triphenylphosphine) palladium (137.1 mg, 118.7 μmol). Bubble nitrogen through the reaction mixture for another 10 min. Cap the reaction vial and heat at 90 0C overnight. Cool the reaction and filter through a silica gel pad eluting with 5% MeOH : CΗ2CI2. Concentrate the fractions under reduced pressure. Purify the resulting residue using silica gel chromatography (2% 2 N NH3 in MeOHiCH2Cl2) to obtain the final product (345.6 mg, 69%). ES/MS m/z 423.2 (M+ 1).
Preparation 7 ter?-Butyl 1 -(4-( 1 H-pyrazol-5 -yl)phthalazin- 1 -yl)piperidin-4-yl(methyl)carbamate

Prepare the title compound by essentially following the procedure described in Preparation 3, using tert-buty\ l-(4-chlorophthalazin-l-yl)piperidin-4-yl(methyl)carbamate and lH-pyrazole-3-boronic acid pinacol ester to provide 580 mg,
(67%). ES/MS m/z 409.2 (M+ 1).
Preparation 8 X-18698
-12- tert- Butyl 1 -(4-(I -methyl- lH-pyrazol-5-yl)phthalazin- 1 -yl)piperidin-4-ylcarbamate

Prepare the title compound by essentially following the procedure described in Preparation 3, using tert-bυXy\ 1 -(4-chlorophthalazin- 1 -yl)piperidin-4-ylcarbamate to provide 5.92 g (94%). ES/MS m/z 308.8 (M+).
Preparation 9 iV-methyl- 1 -(4-( 1 -methyl- lH-pyrazol-5-yl)phthalazin- 1 -yl)piperidin-4-amine

Dissolve tert-bvAyl methyl(l-(4-(l-methyl-lH-pyrazol-5-yl)phthalazin-l-yl)piperidin-4-yl)carbamate (7.77 g, 18.39 mmol) in dichloromethane (100 mL). Add an excess of 1 M hydrogen chloride in diethyl ether (20 mL, 80 mmol) to the solution and stir at ambient temperature for 2 h. Concentrate under reduced pressure. Purify the resulting residue by flash silica gel chromatography (dichloromethane : 2 M NΗ3 in MeOH = 10 : 1) to provide the title compound as a yellow foam (5.83 g, 98%). ES/MS m/z 323.2 (M+ 1).
Example 1
4-Fluoro-N-methyl-N-(l-(4-(l-methyl-lH-pyrazol-5-yl)phthalazin-l-yl)piperidin-4-yl)-2- (trifluoromethyl)benzamide

Treat a solution of N-methyl-1 -(4-(I -methyl- lH-pyrazol-5-yl)phthalazin-l-yl)piperidin-4-amine (2.8 g, 8.68 mmol) and triethylamine (3.36 mL, 26.1 mmol) in CH2Cl2(30 mL) with 4-fluoro-2-(trifluoromethyl)benzoyl chloride (2.14 mL, 10.42 mmol). Stir for 3 h at ambient temperature. Concentrate the reaction mixture under reduced pressure. Purify the resulting residue by flash silica gel chromatography (hexane : ethyl acetate : 2 M ΝH3 in MeOH = 20 : 5 : 1) to provide the free base as a yellow foam (3.83 g, 86%). ES/MS m/z 513.0 (M+ 1).
Example Ia
4-Fluoro-N-methyl-N-(l-(4-(l-methyl-lH-pyrazol-5-yl)phthalazin-l-yl)piperidin-4-yl)-2- (trifluoromethyl)benzamide hydrochloride X-18698
-14-
Dissolve 4-fluoro-N-methyl-N-(l -(4-(I -methyl- lH-pyrazol-5-yl)phthalazin-l- yl)piperidin-4-yl)-2-(trifluoromethyl)benzamide (7.13 g, 13.91 mmol) in dichloromethane (100 mL) and add excess 1 N HCl in diethyl ether (30 mL, 30 mmol). Remove the solvents under reduced pressure to provide the title compound (7.05 g, 92%). ES/MS m/z 513.0 (M+ 1). NMR showed a 2:l mixture of amide rotamers. Major rotamer; 1H NMR (400 MHz, DMSOd6): δ 8.34 (m, IH), 8.26 (m, 2H), 7.95 (m, IH), 7.75 (m, IH), 7.64 (m, 2H), 7.55 (m, IH), 6.72 (d, IH, J=2Hz), 5.15 (br, IH), 4.71 (m, IH), 4.22 (m, 2H), 3.84 (s, 3H), 3.48 (m, 2H), 2.65 (s, 3H), 2.19 (m, 2H), 1.89 ( m, 2H). Minor rotamer; 1H NMR (400 MHz, DMSOd6): δ 8.27 (m, IH), 8.24 (m, 2H), 7.94 (m, IH), 7.73 (m, IH), 7.63 (m, 3H), 6.70 (d, IH, J=2Hz), 5.15 (br, IH), 4.71 (m, IH), 4.07 ( m, 2H), 3.81 (s, 3H), 3.16 (m, 2H), 2.92 (s, 3H), 1.90 (m, 2H), 1.62 ( m 2H).
PATENT
Example 5 Preparation of title compound LY-2940680 [0061] Embodiment
[0062] Compound 10 (0.2g, 0.429mmo 1,1 eq.) Was dissolved in a mixed solution of 18mL of toluene, 6 mL of ethanol, 6 mL of water was added to a solution of 0.091g (0.858mmol, 2eq.) Sodium carbonate which ester (CAS No. 847818-74-0) and 0.098g (0.472mmol, 1 · leq.) in 1-methyl -1H- pyrazole-5-boronic acid, degassed with nitrogen for 20min after addition of 60mg of four (triphenylphosphine) palladium, degassed with nitrogen for lOmin, homogeneous reaction was stirred at reflux for 12h at 74 ° C; after completion the reaction was cooled to room temperature, diluted with methylene chloride, the organic phase washed three times with brine, dried no over anhydrous sodium sulfate, and concentrated under reduced pressure to give a crude product, purified by column chromatography (eluent dichloromethane / methanol, a volume ratio of 30: 1) to give the desired product as a pale yellow foam LY-2940680 (0 · 202g, 92% yield).
[0063] The title compound of detection data LY-2940680:
[0064] 1 ^: 951 ^ 4 ^^ (3001 ^, 0) (: 13) 38.09 ((1 (1,1 = 7.7 ^ 11 (17.74 ^, 210,7.85 (111,210, 7.65 (d, J = 1.80 hz, 1H), 7.47-7.28 (m, 3H), 6.59 (d, J = 1.77Hz, 1H), 4.93 (m, lH), 4.21-4.08 (m, 2H), 4.05 (s, 3H), 3.44 -3.35 (m, 2H), 2.76 (s, 3H), 2.35-2.11 (m, 2H), 2.04-1,88 (m, 2H) ppm; 13C NMR (300Mz, CDC13) S168.0,163.8,159.9,147.4 , 138.2,136.7,132.0,131.9, 131.5,129.4,129.0,128.0,126.3,124.6,121.4,119.5,114.5,109.1,56.9,51.4,38.3, 31.8,29.7,28.4ppm; MS (ESI) m / z: [M + H] + = 513.20181.
PATENT
PATENT
CN 106831718
Paper
A novel and efficient route for synthesis of Taladegib
Taladegib (LY-2940680), a small molecule Hedgehog signalling pathway inhibitor, was obtained from N-benzyl-4-piperidone via Borch reductive amination, acylation with 4-fluoro-2-(trifluoromethyl)benzoyl chloride, debenzylation, substitution with 1,4-dichlorophthalazine and Suzuki cross-coupling reaction with 1-methyl-1H-pyrazole-5-boronic acid. The advantages of this synthesis route were the elimination of Boc protection and deprotection and the inexpensive starting materials. Furthermore, the debenzylation reaction was achieved with simplified operational procedure using ammonium formate as hydrogen source that provided high reaction yield. This synthetic procedure was suitable for large-scale production of the compound for biological evaluation and further study.
Synthesis of Taladegib (LY-2940680)
purified by flash silica gel chromatography (dichloromethane/MeOH, 30:1) to provide Taladegib as a yellow foam. Yield 0.20 g, 92%; m.p. 95 °C;
1 H NMR (300 MHz, CDCl3 ) δ 8.09 (dd, J = 7.6, 7.7 Hz, 2H), 7.90–7.80 (m, 2H), 7.65 (d, J = 1.8 Hz, 1H), 7.47–7.28 (m, 3H), 6.59 (d, J = 1.8 Hz, 1H), 4.97–4.89 (m, 1H), 4.21–4.08 (m, 2H), 4.05 (s, 3H), 3.44–3.35 (m, 2H), 2.76 (s, 3H), 2.35–2.11(m, 2H), 2.04–1.88 (m, 2H);
13C NMR (75 MHz, CDCl3 ) δ 168.0, 163.8, 159.9, 147.4, 138.2, 136.7, 132.0, 131.9, 131.5, 129.4, 129.0, 128.0, 126.3, 124.6, 121.4, 119.5, 114.5, 109.1, 56.9, 51.4, 38.3, 31.8, 29.7, 28.4; MS calcd for C26H24F4 N6 O [M + H]+: 513.2026; found: 513.2018.
////////////PHASE 2, Taladegib, LY-2940680,
CN1C(=CC=N1)C2=NN=C(C3=CC=CC=C32)N4CCC(CC4)N(C)C(=O)C5=C(C=C(C=C5)F)C(F)(F)F

