
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

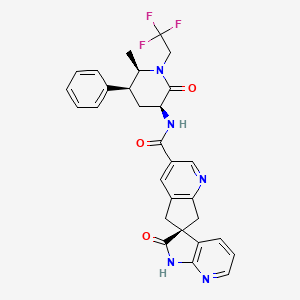
Ubrogepant, MK-1602
(S)-N-((3S,5S,6R)-6-methyl-2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)piperidin-3-yl)-2′-oxo-1′,2′,5,7-tetrahydrospiro[cyclopenta[b]pyridine-6,3′-pyrrolo[2,3-b]pyridine]-3-carboxamide
CAS: 1374248-77-7
Chemical Formula: C29H26F3N5O3
Molecular Weight: 549.5542
UNII-AD0O8X2QJR
CAS TRIHYDRATE 1488325-95-6
CAS MONOHYDRATE 1488327-13-4
- Originator Merck & Co
- Class Amides; Antimigraines; Fluorine compounds; Small molecules; Spiro compounds
- Mechanism of Action Calcitonin gene-related peptide receptor antagonists
- Phase III Migraine, Allergan
Most Recent Events
- 01 Sep 2016 Allergan initiates a phase III extension trial for Migraine in USA (PO, Tablet) (NCT02873221)
- 12 Aug 2016 Allergan plans a phase III trial for Migraine in USA (PO) (NCT02867709)
- 01 Aug 2016 Allergan initiates a phase III trial for Migraine in USA (PO) (NCT02867709)
Process for making piperidinone carboxamide indane and azainane derivatives, which are CGRP receptor antagonists useful for the treatment of migraine. This class of compounds is described in U.S. Patent Application Nos. 13/293,166 filed November 10, 2011 , 13/293, 177 filed November 10, 2011 and 13/293,186 filed November 10, 2011, and PCT International Application Nos. PCT/US11/60081 filed November 10, 2011 and PCT/US 11/60083 filed November 10, 2011.
CGRP (Calcitonin Gene-Related Peptide) is a naturally occurring 37-amino acid peptide that is generated by tissue-specific alternate processing of calcitonin messehger RNA and is widely distributed in the central and peripheral nervous system. CGRP is localized predominantly in sensory afferent and central neurons and mediates several biological actions, including vasodilation. CGRP is expressed in alpha- and beta-forms that vary by one and three amino acids in the rat and human, respectively. CGRP-alpha and CGRP-beta display similar biological properties. When released from the cell, CGRP initiates its biological responses by binding to specific cell surface receptors that are predominantly coupled to the activation of adenylyl cyclase. CGRP receptors have been identified and pharmacologically evaluated in several tissues and cells, including those of brain, cardiovascular, endothelial, and smooth muscle origin.
Based on pharmacological properties, these receptors are divided into at least two subtypes, denoted CGRPi and CGRP2. Human oc-CGRP-(8-37), a fragment of CGRP that lacks seven N-terminal amino acid residues, is a selective antagonist of CGRP l, whereas the linear analogue of CGRP, diacetoamido methyl cysteine CGRP ([Cys(ACM)2,7]CGRP), is a selective agonist of CGRP2. CGRP is a potent neuromodulator that has been implicated in the pathology of cerebrovascular disorders such as migraine and cluster headache. In clinical studies, elevated levels of CGRP in the jugular vein were found to occur during migraine attacks (Goadsby et al., Ann. Neurol., 1990, 28, 183-187), salivary levels of CGRP are elevated in migraine subjects between attacks (Bellamy et al., Headache, 2006, 46, 24-33), and CGRP itself has been shown to trigger migrainous headache (Lassen et al., Cephalalgia, 2002, 22, 54-61). In clinical trials, the CGRP antagonist BIBN4096BS has been shown to be effective in treating acute attacks of migraine (Olesen et al., New Engl. J. Med., 2004, 350, 1104-1110) and was able to prevent headache induced by CGRP infusion in a control group (Petersen et al., Clin. Pharmacol. Ther., 2005, 77, 202-213).
CGRP-mediated activation of the trigeminovascular system may play a key role in migraine pathogenesis. Additionally, CGRP activates receptors on the smooth muscle of intracranial vessels, leading to increased vasodilation, which is thought to contribute to headache pain during migraine attacks (Lance, Headache Pathogenesis: Monoamines, Neuropeptides, Purines and Nitric Oxide, Lippincott-Raven Publishers, 1997, 3-9). The middle meningeal artery, the principle artery in the dura mater, is innervated by sensory fibers from the trigeminal ganglion which contain several neuropeptides, including CGRP. Trigeminal ganglion stimulation in the cat resulted in increased levels of CGRP, and in humans, activation of the trigeminal system caused facial flushing and increased levels of CGRP in the external jugular vein (Goadsby et al, Ann. Neurol., 1988, 23, 193-196). Electrical stimulation of the dura mater in rats increased the diameter of the middle meningeal artery, an effect that was blocked by prior administration of CGRP(8-37), a peptide CGRP antagonist (Williamson et al., Cephalalgia, 1997, 17, 525-531). Trigeminal ganglion stimulation increased facial blood flow in the rat, which was inhibited by CGRP(8-37) (Escott et al., Brain Res. 1995, 669, 93-99). Electrical stimulation of the trigeminal ganglion in marmoset produced an increase in facial blood flow that could be blocked by the non-peptide CGRP antagonist BIBN4096BS (Doods et al., Br. J.Pharmacol., 2000, 129, 420-423). Thus the vascular effects of CGRP may be attenuated, prevented or reversed by a CGRP antagonist.
CGRP-mediated vasodilation of rat middle meningeal artery was shown to sensitize neurons of the trigeminal nucleus caudalis (Williamson et al., The CGRP Family: Calcitonin Gene-Related Peptide (CGRP), Amylin, and Adrenomedullin, Landes Bioscience, 2000, 245-247). Similarly, distention of dural blood vessels during migraine headache may sensitize trigeminal neurons. Some of the associated symptoms of migraine, including extracranial pain and facial allodynia, may be the result of sensitized trigeminal neurons (Burstein et al., Ann. Neurol. 2000, 47, 614-624). A CGRP antagonist may be beneficial in attenuating, preventing or reversing the effects of neuronal sensitization.
The ability of the compounds to act as CGRP antagonists makes them useful pharmacological agents for disorders that involve CGRP in humans and animals, but particularly in humans. Such disorders include migraine and cluster headache (Doods, Curr Opin Inves Drugs, 2001, 2 (9), 1261-1268; Edvinsson et al., Cephalalgia, 1994, 14, 320-327); chronic tension type headache (Ashina et al., Neurology, 2000, 14, 1335-1340); pain (Yu et al., Eur. J. Pharm., 1998, 347, 275-282); chronic pain (Hulsebosch et al., Pain, 2000, 86, 163-175);neurogenic inflammation and inflammatory pain (Holzer, Neurosci., 1988, 24, 739-768; Delay-Goyet et al., Acta Physiol. Scanda. 1992, 146, 537-538; Salmon et al., Nature Neurosci., 2001, 4(4), 357-358); eye pain (May et al. Cephalalgia, 2002, 22, 195-196), tooth pain (Awawdeh et al., Int. Endocrin. J., 2002, 35, 30-36), non-insulin dependent diabetes mellitus (Molina et al., Diabetes, 1990, 39, 260-265); vascular disorders; inflammation (Zhang et al, Pain, 2001, 89, 265), arthritis, bronchial hyperreactivity, asthma, (Foster et al., Ann. NY Acad. Sci., 1992, 657, 397-404; Schini et al., Am. J. Physiol., 1994, 267, H2483-H2490; Zheng et al., J. Virol., 1993, 67, 5786-5791); shock, sepsis (Beer et al., Crit. Care Med., 2002, 30 (8), 1794-1798); opiate withdrawal syndrome (Salmon et al., Nature Neurosci., 2001, 4(4), 357-358); morphine tolerance (Menard et al., J. Neurosci., 1996, 16 (7), 2342-2351); hot flashes in men and women (Chen et al., Lancet, 1993, 342, 49; Spetz et al., J. Urology, 2001, 166, 1720-1723); allergic dermatitis (Wallengren, Contact Dermatitis, 2000, 43 (3), 137-143); psoriasis; encephalitis, brain trauma, ischaemia, stroke, epilepsy, and neurodegenerative diseases (Rohrenbeck et al., Neurobiol. of Disease 1999, 6, 15-34); skin diseases (Geppetti and Holzer, Eds., Neurogenic Inflammation, 1996, CRC Press, Boca Raton, FL), neurogenic cutaneous redness, skin rosaceousness and erythema; tinnitus (Herzog et al., J. Membrane Biology, 2002, 189(3), 225); inflammatory bowel disease, irritable bowel syndrome, (Hoffman et al. Scandinavian Journal of Gastroenterology,2002, 37(4) 414-422) and cystitis. Of particular importance is the acute or prophylactic treatment of headache, including migraine and cluster headache.
Ubrogepant (MK-1602), an oral calcitonin gene-related peptide (CGRP) antagonist, is in phase III clinical development at Allergan for the acute treatment of migraine attacks.
In August 2015, the product was licensed to Allergan by Merck, for the development and marketing worldwide for the treatment of migraine.
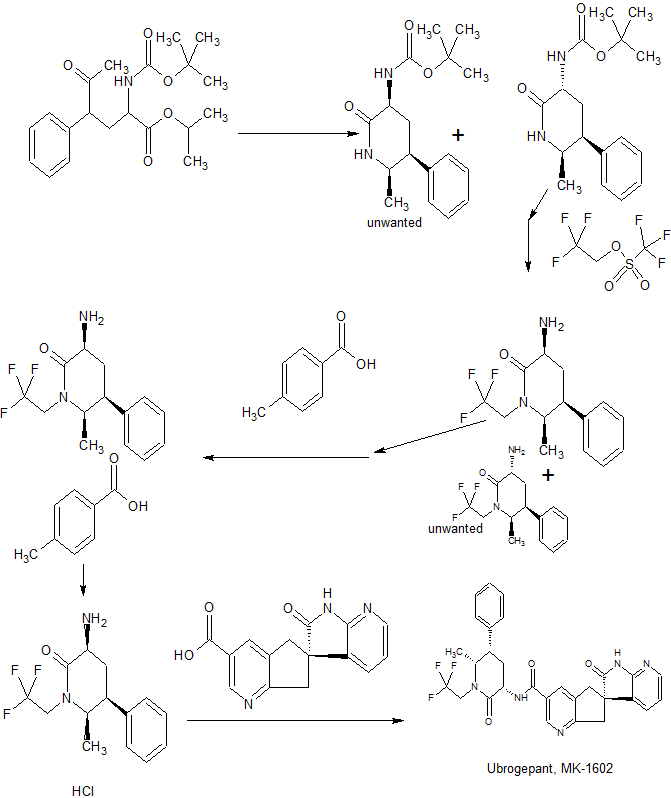
Synthesis
WO 2013138418

CONTD………..

CONTD……….

Ian Bell


Mark Fraley

Steven Gallicchio
EXAMPLE 1

(65yN-[(3£5£ )-6-Methyl-2-oxo-5-pheny
i’,2′,5 J-tetrahvdrospiro[cyclopenta|“^lpyridine-6,3′-pyrroloj“2,3-¾lpyridine1-3-carboxamide (Benzotriazol- 1 -yloxy)tr/i,(dimethylamino)phosphonium hexafluorophosphate (1.89 g, 4.28 mmol) was added to a solution of (6S -2′-oxo- ,2,,5,7- tetrahydrospiro[cyclopenta[&]pyridine-6,3′-pyrrolo[2,3-&]pyridine]-3-carboxylic acid (described in Intermediate 1) (1.10 g, 3.92 mmol), (3JS’,55′,6J?)-3-amino-6-methyl~5~phenyl-l-(2,2,2- trifluoroethyl)piperidin-2-one hydrochloride (described in Intermediate 4) (1.15 g, 3.56 mmol), and NjiV-diisopropylethylamine (3.1 1 m.L, 17.8 mmol) in DMF (40 mL), and the resulting mixture was stirred at 23 °C for 3 h. The reaction mixture was then partitioned between saturated aqueous sodium bicarbonate solution (200 mL) and ethyl actetate (3 χ 200 mL). The combined organic layers were washed with brine, dried over sodium sulfate, and concentrated. The residue was purified by flash column chromatography on silica gel, eluting with hexanes initially, then grading to 100% EtOAc before stepping to 5% MeOH in EtOAc to afford the title compound as an amorphous solid, which was further purified by the following crystallization procedure. A solution of the amorphous product in a minimal amount of methanol required for dissolution was diluted with 10 volumes water, and the resulting slurry was seeded with crystalline product and stirred at 23 °C for 4 h. The solids were filtered, washed with water, and dried under a stream of nitrogen to give the title compound as a crystalline solid. HRMS: m/z = 550.2068, calculated m/z – 550.2061 for C29H27F3N503. lH NMR (500 MHz, CDC13) δ 8.91 (s, 1H), 8.70 (s, 1H), 8.17 (dd, 1H, J- 5.4, 1.5 Hz), 8.04 (s5 1H), 7.37 (m, 3H), 7.29 (t, 1H, J= 7.3 Hz), 7.21 (d, 2H, J= 7.3 Hz), 7.13 (dd, 1H, J = 7.3, 1.2 Hz), 6.89 (dd, 1H, J = 7.3, 5.4 Hz), 4.99- 4.90 (m, 1H), 4.53 (dt, 1H, J= 10.7, 6.6 Hz), 3.94 (p, 1H, J = 5.9 Hz), 3.78 (d, 1H, J = 17.1 Hz), 3.67 (d, 1H, J- 16.4 Hz), 3.65 (m, 1H), 3.34-3.26 (m, 1H), 3.28 (d, 1H, J- 17.1 Hz), 3.17 (d, 1H, J = 16.6 Hz), 2.79 (m, 1H), 2.58 (q, 1H, J – 12.7 Hz), 1.07 (d, 3H, J= 6.6 Hz).
PATENT
(5)-N-((3^,5^,6i?)-6-Methyl-2-oxo-5-phenyl 2,2,2-trifluoroethyl)piperidine-3-yl)-2*-oxo- l\2 5,7-tetrahydrospiro[cyclopenta[¾]pyridine-6,3′-pyrrolo[2,3-¾]pyridine]-3-carboxam trihydrate (15)

To a suspension of 11 (465 g, 96% wt, 0.99 mol) in iPAc (4.6 L) was added 5% aqueous K3PO4 (4.6 L). The mixture was stirred for 5 min. The organic layer was separated and washed with 5%> aqueous K3PO4 (4.6 L) twice and concentrated in vacuo and dissolved in acetonitrile (1.8 L).
To another flask was added 14 (303 g, 91.4 wt%>), acetonitrile (1.8 L) and water (1.8 L) followed by 10 N NaOH (99 mL). The resulting solution was stirred for 5 min at room temperature and the chiral amine solution made above was charged to the mixture and the container was rinsed with acetonitrile (900 mL). HOBT hydrate (164 g) was charged followed by EDC hydrochloride (283 g). The mixture was agitated at room temperature for 2.5 h. To the mixture was added iPAc (4.6 L) and organic layer was separated, washed with 5%> aqueous NaHC03 (2.3 L) followed by a mixture of 15%> aqueous citric acid (3.2 L) and saturated aqueous NaCl (1.2 L). The resulting organic layer was finally washed with 5%> aqueous NaHC03 (2.3 L). The organic solution was concentrated below 50 °C and dissolved in methanol (2.3 L). The solution was slowly added to a mixture of water (6 L) and methanol (600 mL) with ~ 2 g of seed crystal. And the resulting suspension was stirred overnight at room temperature. Crystals were filtered, rinsed with water/methanol (4 L, 10 : 1), and dried under nitrogen flow at room temperature to provide 15 (576 g, 97 % yield) as trihydrate.
Ή NMR (500 MHz, CDCI3): δ 10.15 (br s, 1 H), 8.91 (br s, 1 H), 8.21 (d, J= 6.0 Hz, 1 H), 8.16 (dd, J= 5.3, 1.5 Hz, 1 H), 8.01 (br s, 1 H), 7.39-7.33 (m, 2 H), 7.31-7.25 (m, 1 H), 7.22-7.20 (m, 2 H), 7.17 (dd, J= 7.4, 1.6 Hz, 1 H), 6.88 (dd, J= 7.4, 5.3 Hz, 1 H), 4.94 (dq, J= 9.3, 7.6 Hz, 1 H), 4.45-4.37 (m, 1 H), 3.94-3.87 (m, 1 H), 3.72 (d, J= 17.2 Hz, 1 H), 3.63-3.56 (m, 2 H), 3.38-3.26 (m, 1 H), 3.24 (d, J= 17.3 Hz, 1 H), 3.13 (d, J= 16.5 Hz, 1 H), 2.78 (q, J= 12.5 Hz, 1 H), 2.62-2.56 (m, 1 H), 1.11 (d, J= 6.5 Hz, 3 H); 13C NMR (126 MHz, CD3CN): δ 181.42, 170.63, 166.73, 166.63, 156.90, 148.55, 148.08, 141.74, 135.77, 132.08, 131.09, 130.08, 129.66, 129.56, 128.78, 128.07, 126.25 (q, J= 280.1 Hz), 119.41, 60.14, 53.07, 52.00, 46.41 (q, J= 33.3 Hz), 45.18, 42.80, 41.72, 27.79, 13.46; HRMS m/z: calcd for C29H26F3N503 550.2061 (M+H): found 550.2059.
Alternative procedure for 15:

13
To a suspension of 13 (10 g, 98 wt%, 23.2 mmol) in MTBE (70 mL) was added 0.6 N HCI (42 mL). The organic layer was separated and extracted with another 0.6 N HCI (8 mL). The combined aqueous solution was washed with MTBE (10 mL x3). To the resulting aqueous solution was added acetonitrile (35 mL) and 14 (6.66 g, 99 wt%). To the resulting suspension was neutralized with 29 % NaOH solution to pH 6. HOPO (0.26 g) was added followed by EDC hydrochloride (5.34 g). The mixture was stirred at room temperature for 6-12 h until the conversion was complete (>99%). Ethanol (30 ml) was added and the mixture was heated to 35 °C. The resulting solution was added over 2 h to another three neck flask containing ethanol (10 mL), water (30 mL) and 15 seeds (0.4 g). Simultaneously, water (70 mL) was also added to the mixture. The suspension was then cooled to 5 °C over 30 min and filtered. The cake was washed with a mixture of ethanol/water (1 :3, 40 mL). The cake was dried in a vacuum oven at 40 °C to give 15 trihydrate (13.7 g, 95%) as crystals.
PATENT
PATENT
US 9174989
CLIP
Practical Asymmetric Synthesis of a Calcitonin Gene-Related Peptide (CGRP) Receptor Antagonist Ubrogepant
 , Ed Cleator*‡, Birgit Kosjek†, Jianguo Yin†, Bangping Xiang†, Frank Chen†, Shen-Chun Kuo†, Kevin Belyk†, Peter R. Mullens‡, Adrian Goodyear‡, John S. Edwards‡, Brian Bishop‡, Scott Ceglia§, Justin Belardi§, Lushi Tan†, Zhiguo J. Song†, Lisa DiMichele†, Robert Reamer†, Fabien L. Cabirol∥, Weng Lin Tang∥, and Guiquan Liu⊥
, Ed Cleator*‡, Birgit Kosjek†, Jianguo Yin†, Bangping Xiang†, Frank Chen†, Shen-Chun Kuo†, Kevin Belyk†, Peter R. Mullens‡, Adrian Goodyear‡, John S. Edwards‡, Brian Bishop‡, Scott Ceglia§, Justin Belardi§, Lushi Tan†, Zhiguo J. Song†, Lisa DiMichele†, Robert Reamer†, Fabien L. Cabirol∥, Weng Lin Tang∥, and Guiquan Liu⊥Abstract

The development of a scalable asymmetric route to a new calcitonin gene-related peptide (CGRP) receptor antagonist is described. The synthesis of the two key fragments was redefined, and the intermediates were accessed through novel chemistry. Chiral lactam 2 was prepared by an enzyme mediated dynamic kinetic transamination which simultaneously set two stereocenters. Enzyme evolution resulted in an optimized transaminase providing the desired configuration in >60:1 syn/anti. The final chiral center was set via a crystallization induced diastereomeric transformation. The asymmetric spirocyclization to form the second fragment, chiral spiro acid intermediate 3, was catalyzed by a novel doubly quaternized phase transfer catalyst and provided optically pure material on isolation. With the two fragments in hand, development of their final union by amide bond formation and subsequent direct isolation is described. The described chemistry has been used to deliver over 100 kg of our desired target, ubrogepant.
(S)-N-((3S,5S,6R)-6-Methyl-2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)piperidin-3-yl)-2′-oxo-1′,2′,5,7-tetrahydrospiro[cyclopenta[b]pyridine-6,3′-pyrrolo[2,3-b]pyridine]-3-carboxamide Trihydrate (1)


REFERENCES
1: Voss T, Lipton RB, Dodick DW, Dupre N, Ge JY, Bachman R, Assaid C, Aurora SK, Michelson D. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016 Aug;36(9):887-98. doi: 10.1177/0333102416653233. PubMed PMID: 27269043.
/////////////ubrogepant, MK-1602, Phase III, Migraine
O=C(C1=CN=C2C(C[C@@]3(C4=CC=CN=C4NC3=O)C2)=C1)N[C@@H]5C(N(CC(F)(F)F)[C@H](C)[C@H](C6=CC=CC=C6)C5)=O

