PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
To treat platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer after one to three prior systemic treatment regimens, at least one of which included bevacizumab
Mechanism of ActionGlucocorticoid receptor antagonists
Orphan Drug StatusYes – Pancreatic cancer; Cushing syndrome
Phase IIICushing syndrome; Ovarian cancer; Pancreatic cancer
Phase IIFallopian tube cancer; Peritoneal cancer; Prostate cancer
Phase I/IISolid tumours
Phase IAdrenocortical carcinoma
Most Recent Events
09 Sep 2022Subgroup analysis efficacy data from a phase-II trial in Ovarian cancer presented at the 47th European Society for Medical Oncology Congress (ESMO-2022)
29 Jun 2022Phase-III clinical trials in Ovarian cancer (Combination therapy, Recurrent, Second-line therapy or greater) in USA (PO)
06 Jun 2022Corcept Therapeutics announces intentions to submit a NDA for Ovarian cancer
The drug was approved by the USFDA in 2026 for the treatment of platinum-resistant ovarian cancer.[3]
Relacorilant is an orally available antagonist of the glucocorticoid receptor (GR), with potential antineoplastic activity. Upon administration, relacorilant competitively binds to and blocks GRs. This inhibits the activity of GRs, and prevents both the translocation of the ligand-GR complexes to the nucleus and gene expression of GR-associated genes. This decreases the negative effects that result from excess levels of endogenous glucocorticoids, like those seen when tumors overproduce glucocorticoids. In addition, by binding to GRs and preventing their activity, inhibition with CORT125134 also inhibits the proliferation of GR-overexpressing cancer cells. GRs are overexpressed in certain tumor cell types and promote tumor cell proliferation.
OriginatorCorcept Therapeutics
DeveloperCorcept Therapeutics; University of Chicago
Mechanism of ActionGlucocorticoid receptor antagonists
Orphan Drug StatusYes – Pancreatic cancer; Ovarian cancer; Cushing syndrome
RegisteredFallopian tube cancer; Ovarian cancer; Peritoneal cancer
PreregistrationCushing syndrome
Phase IIIAdenocarcinoma
Phase IIProstate cancer
DiscontinuedAdrenocortical carcinoma
27 Mar 2026Discontinued – Phase-I for Adrenocortical carcinoma (Inoperable/Unresectable, Late-stage disease, Metastatic disease, Combination therapy) in USA (PO), before March 2026 (Corcept Therapeutics pipeline, March 2026)
27 Mar 2026Corcept Therapeutics plans the phase II STELLA trial for Cervical cancer (Combination therapy, Second-line therapy or greater) in first quarter of 2026
25 Mar 2026Registered for Fallopian tube cancer (Combination therapy, Second-line therapy or greater) in USA (PO) – First global approval
Relacorilant (CORT125134)118) is being developed by Corcept Therapeutics, Inc. It is an orally active, high-affinity, selective antagonist of the glucocorticoid receptor that may benefit from the modulation of cortisol activity. In structural optimization, the introduction of a trifluoromethyl group to the 4-position on the pyridyl moiety was found to increase HepG2 tyrosine amino transferase assay potency by a factor of four. Relacorilant is currently being evaluated in a phase II clinical study in patients with Cushing’s syndrome.119)
2-Bromo-4-(trifluoromethyl)pyridine (17) prepared from (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one is employed as a key intermediate for the preparation of relacorilant as shown in Scheme 31.120)
118) H. Hunt, T. Johnson, N. Ray and I. Walters (Corcept Therapeutics, Inc.): PCT Int. Appl. WO2013/177559 (2013).
119) H. J. Hunt, J. K. Belanoff, I. Walters, B. Gourdet, J. Thomas, N. Barton, J. Unitt, T. Phillips, D. Swift and E. Eaton: Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexahydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methanone (CORT125134): A Selective Glucocorticoid Receptor (GR) Antagonist. J. Med. Chem. 60, 3405–3421 (2017). [Abstract] [Google Scholar]
120) B. Lehnemann, J. Jung and A. Meudt (Archimica GmbH): PCT Int. Appl. WO 2007/000249 (2007).
The nonselective glucocorticoid receptor (GR) antagonist mifepristone has been approved in the U.S. for the treatment of selected patients with Cushing’s syndrome. While this drug is highly effective, lack of selectivity for GR leads to unwanted side effects in some patients. Optimization of the previously described fused azadecalin series of selective GR antagonists led to the identification of CORT125134, which is currently being evaluated in a phase 2 clinical study in patients with Cushing’s syndrome.
Cushing’s syndrome (CS) is a metabolic disorder caused by chronic hypercortisolism. CS is associated with cardiovascular, metabolic, skeletal and psychological dysfunctions and can be fatal if left untreated. The first-line treatment for all forms of CS is a surgery. However, medical therapy has to be chosen if surgical resection is not an option or is deemed ineffective. Currently available therapeutics are either not selective and have side effects or are only available as an injection (pasireotide).
Arimoclomol maleate is in a phase III clinical trials by Orphazyme for the treatment of Niemann-Pick disease type C (NP-C). It is also in phase II clinical studies for the treatment of amyotrophic lateral sclerosis (ALS).
Arimoclomol (INN; originally codenamed BRX-345, which is a citrate salt formulation of BRX-220) is an experimental drug developed by CytRx Corporation, a biopharmaceutical company based in Los Angeles, California. In 2011 the worldwide rights to arimoclomol were bought by Danish biotech company Orphazyme ApS.[1] The European Medicines Agency (EMA) and U.S. Food & Drug Administration (FDA) granted orphan drug designation to arimoclomol as a potential treatment for Niemann-Pick type C in 2014 and 2015 respectively.[2][3]
Fig. 1 Structures of (±)-bimoclomol (1) and (R)-(+)-arimoclomol (2).
The present disclosure provides an optimized four-step process for preparing an ultra-pure composition comprising arimoclomol citrate, i.e. N-{[(2R)-2-hydroxy-3-piperidin-l-ylpropyl]oxy}pyridine-3-carboximidoyl chloride 1-oxide citrate. The optimized process comprises a plurality of optimized sub-steps, each contributing to an overall improved process, providing the ultra-pure composition comprising arimoclomol citrate. The ultra-pure composition comprising arimoclomol citrate meets the medicines agencies’ high regulatory requirements. An overview of the four-steps process is outlined below:
Step 1: Overview of process for preparing ORZY-01
Step 2: Overview of process for preparing ORZY-03
Step 4: Overview of process for preparing BRX-345 (ORZY-05)
The previously reported two-step synthesis of ORZY-01 as shown below includes a 2 hour reflux in step 1A, followed by purification of intermediate compound (V) to increase the batch quality.
(R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide1 – (R)-(+)-Arimoclomol – 2 A solution of (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carbamimidoyl)pyridine-1-oxide 12 (205 mg, 0.70 mmol) in conc. hydrochloric acid (1.1 mL, 13.9 mmol) and water (3 mL) was cooled to -5 °C for 15 minutes. Sodium nitrite (63 mg, 0.91 mmol) in water (0.5 mL) was then added dropwise to the reaction mixture and the reaction was stirred at -5 °C for 2.5 hours. The reaction mixture was made alkaline with NaOH (7 M, 3 mL). An additional 10 mL of water was added followed by DCM (30 mL) containing EtOAc (5 mL) and the organics were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on Biotage Isolera using Biotage SNAP 10 g Si cartridge eluting with gradient elution 0-30% MeOH:DCM both containing 0.1% Et3N to afford the title compound (160 mg, 73% yield) as a colourless semi-solid. Analytical data was consistent with literature values. See ESI section SFC traces for specific enantiomeric ratios of 2 synthesised under the various methodologies quoted in the text. Optical rotation was not determined as it was determined in the ultimate product of this 2·citrate and comparative run times on SFC. 1H NMR (600 MHz, CDCl3) δ: 8.63 (t, J = 1.4 Hz, 1H), 8.16 (ddd, J = 6.4, 1.6, 0.9 Hz, 1H), 7.66 – 7.62 (m, 1H), 7.25 (dd, J = 8.0, 6.6 Hz, 1H), 4.26 (qd, J = 11.3, 5.2 Hz, 2H), 4.07 (dd, J = 9.2, 4.7 Hz, 1H), 2.62 (s, 2H), 2.47 – 2.31 (m, 4H), 1.65 – 1.51 (m, 4H), 1.42 (s, 2H); 13C NMR (151 MHz, CDCl3) δ: 140.3, 137.7, 133.1, 132.5, 125.7, 123.9, 78.7, 64.9, 60.9, 54.8, 25.8, 24.0.
(R)-(+)- Arimoclomol citrate – 2·citrate (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide (159 mg, 0.51 mmol) was dissolved in acetone (3 mL) and citric acid (97 mg, 0.51 mmol) was added. The reaction mixture was left to stir at room temperature for 18 hours. After this time the mixture was sonicated and the precipitate was filtered, rinsed with cold acetone (1 mL) and dried under vacuum to afford the title compound (165 mg, 64% yield) as a white amorphous solid. Analytical data was consistent with literature values. m.p. 161-162 °C, Acetone (lit. 163-165 °C, EtOH); [α]D 20 +8.0 (c=1, H2O); IR νmax (neat): 3423, 3228, 2949, 2868, 1722, 1589, 1483, 1433, 1307, 1128, 972, 829 cm-1; 1H NMR (600 MHz, d6-DMSO) δ: 8.54 (t, J = 1.5 Hz, 1H), 8.39 – 8.35 (m, 1H), 7.72 – 7.68 (m, 1H), 7.55 (dd, J = 8.0, 6.5 Hz, 1H), 4.28 (ddd, J = 17.6, 13.3, 7.4 Hz, 3H), 3.35 (br. s, 2H), 3.13 – 2.74 (m, 6H), 2.59 (d, J = 15.2 Hz, 2H), 2.56 – 2.51 (m, 2H), 1.77 – 1.61 (m, 4H), 1.48 (s, 2H); 13C NMR (151 MHz, d6-DMSO) δ: 176.6, 171.3, 140.5, 136.4, 132.7, 131.5, 126.8, 123.3, 77.8, 71.4, 63.8, 58.7, 53.1, 44.0, 30.7, 23.0, 21.9; HRMS (m/z TOF MS ES+) for C14H20ClN3O3 [M+H]+ calc. 314.1271, observed 314.1263; SFC er purity R:S >99:1
Procedure for the conversion of (R)-(+)-Bimoclomol 1 into (R)-(+)-Arimoclomol 2 To a solution of (R)-(+)-bimoclomol (61 mg, 0.21 mmol) in acetone (2 mL) was added benzenesulfonic acid (33 mg, 0.21 mmol). The reaction mixture was stirred at room temperature for 1.5 hours. The reaction mixture was concentrated in vacuo. Separately to a suspension of hydrogen peroxide-urea adduct (39 mg, 0.41 mmol) in acetonitrile (6 mL) at -5°C (ice-salt bath) was added trifluoroacetic anhydride (58 μL, 0.41 mmol) dropwise. A suspension of (R)-(+)-bimoclomol, 1, benzenesulfonic acid salt, as made above, in acetonitrile (3 mL) was then added dropwise to this solution. The reaction mixture was stirred for 18 hours, whilst slowly warming to room temperature. Aqueous Na2S2O5 solution (0.5 M, 1 mL) was added and the reaction mixture stirred for 1 hour. The reaction mixture was made alkaline with NaOH (7 M) and extracted with DCM (2 x 30 mL). The combined organics were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on a Biotage Isolera using Biotage SNAP 10g Si cartridge eluting with gradient elution 0-35% MeOH in DCM to afford the title compound (35 mg, 55% yield) as a colourless semi-solid. Analytical data of the products was consistent with literature and/or previous samples synthesised above.
Arimoclomol is believed to function by stimulating a normal cellular protein repair pathway through the activation of molecular chaperones. Since damaged proteins, called aggregates, are thought to play a role in many diseases, CytRx believes that arimoclomol could treat a broad range of diseases.
Arimoclomol has been shown to extend life in an animal model of ALS[11] and was well tolerated in healthy human volunteers in a Phase I study. CytRx is currently conducting a Phase II clinical trial.[12]
Arimoclomol also has been shown to be an effective treatment in an animal model of Spinal Bulbar Muscular Atrophy (SBMA, also known as Kennedy’s Disease).[13]
Arimoclomol was discovered by Hungarian researchers, as a drug candidate to treat insulin resistance[14][15] and diabetic complications such as retinopathy, neuropathy and nephropathy. Later, the compound, along with other small molecules, was screened for further development by Hungarian firm Biorex, which was sold to CytRx Corporation, who developed it toward a different direction from 2003.
^ Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (April 2004). “Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice”. Nat. Med. 10 (4): 402–5. doi:10.1038/nm1021. PMID15034571. S2CID2311751.
^ Kalmar B, Greensmith L, Malcangio M, McMahon SB, Csermely P, Burnstock G (December 2003). “The effect of treatment with BRX-220, a co-inducer of heat shock proteins, on sensory fibers of the rat following peripheral nerve injury”. Exp. Neurol. 184 (2): 636–47. doi:10.1016/S0014-4886(03)00343-1. PMID14769355. S2CID5316222.
^ Rakonczay Z, Iványi B, Varga I, et al. (June 2002). “Nontoxic heat shock protein coinducer BRX-220 protects against acute pancreatitis in rats”. Free Radic. Biol. Med. 32 (12): 1283–92. doi:10.1016/S0891-5849(02)00833-X. PMID12057766.
^ Kalmar B, Burnstock G, Vrbová G, Urbanics R, Csermely P, Greensmith L (July 2002). “Upregulation of heat shock proteins rescues motoneurones from axotomy-induced cell death in neonatal rats”. Exp. Neurol. 176 (1): 87–97. doi:10.1006/exnr.2002.7945. PMID12093085. S2CID16071543.
Madrigal Pharmaceuticals , following the merger between Synta and Madrigal Pharmaceuticals (pre-merger) (following the acquisition of VIA Pharmaceuticals ‘ assets (originally under license from Roche )), is developing resmetirom (MGL-3196, VIA-3196), the lead from oral capsule formulation thyroid hormone receptor (THR) beta agonists, cholesterol and triglyceride modulators, for the use in the treatment of metabolic disorders including hypercholesterolemia and other dyslipidemias, and non-alcoholic steatohepatitis.
MGL-3196 is a first-in-class, orally administered, small-molecule, liver-directed, THR β-selective agonist. Preclinical, toxicology and Phase 1 clinical data suggest MGL-3196 has an attractive, differentiated profile as a potential treatment for non-alcoholic steatohepatitis (NASH) and dyslipidemias. THR-β selectivity also enhances the safety profile of MGL-3196, compared to non-selective agents. MGL-3196 has shown no suppression of the central thyroid axis, no THR-α effects on heart rate or bone, and no elevation of liver enzymes. These characteristics make MGL-3196 among the most promising molecules in development in this therapeutic area. MGL-3196 is in a Phase 2 clinical trial for the treatment of non-alcoholic steatohepatitis (NASH).
PATENT
WO-2020010068
Novel crystalline salt of resmetirom as thyroid hormone receptor agonists useful for treating obesity, hyperlipidemia, hypercholesterolemia and diabetes. Appears to be the first filing from the assignee and the inventors on this compound,
Thyroid hormones are critical for normal growth and development and for maintaining metabolic homeostasis (Paul M. Yen, Physiological reviews, Vol. 81(3): pp. 1097-1126 (2001)). Circulating levels of thyroid hormones are tightly regulated by feedback mechanisms in the hypothalamus/pituitary/thyroid (HPT) axis. Thyroid dysfunction leading to hypothyroidism or hyperthyroidism clearly demonstrates that thyroid hormones exert profound effects on cardiac function, body weight, metabolism, metabolic rate, body temperature, cholesterol, bone, muscle and behavior.
[0005] The biological activity of thyroid hormones is mediated by thyroid hormone receptors (TRs or THRs) (M. A. Lazar, Endocrine Reviews, Vol. 14: pp. 348-399 (1993)). TRs belong to the superfamily known as nuclear receptors. TRs form heterodimers with the retinoid receptor that act as ligand-inducible transcription factors. TRs have a ligand binding domain, a DNA binding domain, and an amino terminal domain, and regulate gene expression through interactions with DNA response elements and with various nuclear co-activators and co repressors. The thyroid hormone receptors are derived from two separate genes, a and b. These distinct gene products produce multiple forms of their respective receptors through differential RNA processing. The major thyroid receptor isoforms are aΐ, a2, bΐ, and b2. Thyroid hormone receptors aΐ, bΐ, and b2 bind thyroid hormone. It has been shown that the thyroid hormone receptor subtypes can differ in their contribution to particular biological responses. Recent studies suggest that TIIb 1 plays an important role in regulating TRH (thyrotropin releasing hormone) and on regulating thyroid hormone actions in the liver. T11b2 plays an important role in the regulation of TSH (thyroid stimulating hormone) (Abel et. al, J. Clin. Invest., Vol 104: pp. 291-300 (1999)). TIIb 1 plays an important role in regulating heart rate (B. Gloss et. al. Endocrinology, Vol. 142: pp. 544-550 (2001); C. Johansson et. al, Am. J. Physiol., Vol. 275: pp. R640-R646 (1998)).
[0006] Efforts have been made to synthesize thyroid hormone analogs which exhibit increased thyroid hormone receptor beta selectivity and/or tissue selective action. Such thyroid hormone mimetics may yield desirable reductions in body weight, lipids, cholesterol, and lipoproteins, with reduced impact on cardiovascular function or normal function of the hypothalamus/pituitary/thyroid axis (see, e.g., Joharapurkar et al, J. Med. Chem, 2012, 55 (12), pp 5649-5675). The development of thyroid hormone analogs which avoid the undesirable effects of hyperthyroidism and hypothyroidism while maintaining the beneficial effects of thyroid hormones would open new avenues of treatment for patients with metabolic disease such as obesity, hyperlipidemia, hypercholesterolemia, diabetes and other disorders and diseases such as liver steatosis and NASH, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer, thyroid diseases, a resistance to thyroid hormone (RTH) syndrome, and related disorders and diseases.
Example 3: Preparation of (Z)-ethyl (2-cyano-2-(2-(3,5-dichloro-4-((5-isopropyl-6- oxo- l,6-dihydropyridazin-3-yl)oxy)phenyl)hydrazono)acetyl)carbamate (Int. 8)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet was charged with Int. 7 (75.0 g, 0.239 mol, 1 wt), acetic acid (600 mL, 8 vol), water (150 mL, 2 vol), and concentrated HC1 (71.3 mL, 0.95 vol). The resulting thin slurry was cooled to 6 °C and a solution of NaN02 (16.8 g, 0.243 mol, 1.02 equiv) in water (37.5 mL, 0.5 vol) was added over a period of 10 min while maintaining a batch temperature below 10 °C. After an additional 10 min of agitation between 5-10 °C, HPLC analysis showed complete conversion of Int. 7 to the diazonium intermediate. A solution of NaOAc (54.5 g, 0.664 mol, 2.78 equiv) in water (225 mL, 3 vol) was added over a period of 6 min while maintaining a batch temperature below 10 °C. N-cyanoacetylurethane (37.9 g, 0.243 mol, 1.02 equiv) was immediately added, the cooling was removed, and the batch naturally warmed to 8 °C over 35 min. HPLC analysis showed complete consumption of the diazonium intermediate and the reaction was deemed complete. The batch warmed naturally to 21 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (375 mL, 5 vol) twice. The collected orange solid was dried in a 35 °C vacuum oven for 64 h to provide crude Int. 8 (104.8 g, 91%).
A I L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, and N2 inlet/outlet was charged with crude Int. 8 (104.4 g, 1 wt) and acetic acid (522 mL, 5 vol). The resulting slurry was heated to 50 °C and held at that temperature for 1.5 h. The batch cooled naturally to 25 °C over 2 h and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (522 mL, 5 vol) and the cake conditioned under vacuum for 1.75 h. The light orange solid was dried to constant weight in a 40 °C vacuum oven to provide 89.9 g (78% from Int. 7) of the desired product. 1H NMR (DMSO) was consistent with the assigned structure.
Example 4: Preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, N2 inlet/outlet, and reflux condenser was charged with Int. 8 (89.3 g, 0.185 mol, 1 wt), DMAC (446 mL, 5 vol), and KOAc (20.0 g, 0.204 mol, 1.1 equiv). The mixture was heated to 120 °C and held at that temperature for 2 h. HPLC analysis showed complete conversion to Compound A. The batch temperature was adjusted to 18 °C over 1 h, and acetic acid (22.3 mL, 0.25 vol) was added. The batch temperature was adjusted to 8 °C, and water (714 mL, 8 vol) was added over 1 h; an orange slurry formed. The batch was filtered through Sharkskin filter paper and the cake was allowed to condition overnight under N2 without vacuum for convenience. A premixed solution of 1 : 1 acetone/water (445 mL, 5 vol) was charged to the flask and added to the cake as a rinse with vacuum applied. After 2 h of conditioning the cake under vacuum, it was transferred to a clean 1 L, three-neck, round- bottom flask equipped with overhead stirring, a thermocouple, and N2inlet/outlet. Ethanol (357 mL, 4 vol) and acetone (357 mL, 4 vol) were charged and the resulting slurry was heated to 60 °C; dissolution occurred. Water (890 mL, 10 vol) was added over a period of 90 min while maintaining a batch temperature between 55-60 °C. The resulting slurry was allowed to cool to 25 °C and filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with a solution of 1:1 EtOH/water (446 mL, 5 vol). The cake was conditioned overnight under N2 without vacuum for convenience. The cracks in the cake were smoothed and vacuum applied. The cake was washed with water (179 mL, 2 vol) and dried in a 45 °C vacuum oven to a constant weight of 70.5 g (87%, crude Compound A). HPLC analysis showed a purity of 94.8%.
A 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser was charged with crude Compound A (70.0 g) and MIBK (350 mL, 5 vol). The orange slurry was heated to 50 °C and held at that temperature for 2 h. The batch cooled naturally to 23 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with MIBK (35 mL, 0.5 vol) twice. The collected solids were dried in a 45 °C vacuum oven to a constant weight of 58.5 g (84%). This solid was charged to a 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser. Ethanol (290 mL, 5 vol) was added and the slurry was heated to reflux. After 3.5 h at reflux, XRPD showed the solid was consistent with Form I, and heating was removed. Upon reaching 25 °C, the batch was filtered through filter paper, and the reactor and cake were washed sequentially with EtOH (174 mL, 3 vol). The tan solid Compound A was dried in a 40 °C vacuum oven to a constant weight of 50.4 g (87%, 64% from Int. 8). HPLC analysis showed a purity of 99.1%. 1H NMR (DMSO) was consistent with the assigned structure.
Example 5: Scaled up preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A larger scale batch of Compound A was synthesized according to the scheme below. The conditions in the scheme below are similar to those described in Examples 1-4 above.
6A
Compound A
Synthesis of 4: A 50 L jacketed glass vessel (purged with N2) was charged with 3,6- dichloropyridazine (2.00 kg), 4-amino-2,6-dichlorophenol (2.44 kg) and N,N- dimethylacetamide (10.0 L). The batch was vacuum (26 inHg) / nitrogen (1 PSIG) purged 3 times. Cesium carbonate (5.03 kg) was added and the batch temperature was adjusted from 22.3 °C to 65.0 °C over 3.5 hours. The batch was held at 65.0 °C for 20 hours. At this point,
NMR analysis indicated 3.34% 3.6-dichloropyridazine relative to 2. The batch temperature was adjusted to 21.5 °C and ethyl acetate (4.00 L) was added to the batch. The batch was agitated for 10 minutes and then filtered through a 18″ Nutsche filter equipped with polypropylene filter cloth. The filtration took 15 minutes. Ethyl acetate (5.34 L) was charged to the vessel and transferred to the filter as a rinse. The batch was then manually re- suspended in the filter before re-applying vacuum. This process was repeated 2 more times and the filter cake was conditioned for 10 minutes. The filtrate was charged to a 100-L vessel that contained (16.0 L) of a previously prepared 15% sodium chloride in H20. The batch was agitated for 5 minutes and then allowed to separate for 35 minutes. The interface was not visible, so the calculated 23 L of the lower aqueous phase was removed. 16.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 6 minutes and then allowed to separate for 7 minutes. The interface was visible at -19 L and the lower aqueous phase was removed. 17.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 7 minutes and then allowed to separate for 11 minutes. The lower aqueous phase was removed. The vessel was set up for vacuum distillation and the batch was concentrated from 17.0 L to 8.0 L over 2 hours 20 minutes with the batch temperature kept around 21 °C. Benzoic anhydride (3.19 kg) and acetic acid (18.0 L) were charged to the vessel. The vessel was set up for vacuum distillation and the batch was concentrated from 28.0 L to 12.0 L over 2 days (overnight hold at 20 °C) with the batch temperature kept between 20 and 55 °C. At this point, JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.015. Acetic acid (4.0 L) was charged to the batch and the batch was distilled to 12 L. JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.0036. Acetic acid (20.0 L) was charged to the batch and the batch temperature was adjusted to 70.0 °C. The batch was sampled for HPLC analysis and 2 was 0.16%. Sodium acetate (2,20 kg) was added to the batch and the batch temperature was adjusted from 72.4 °C to 110.0 °C. After 18.5 hours, HPLC analysis indicated no Int. B detected. The batch temperature was adjusted from 111.3 to 74.7 °C and DI water (30.0 L) was added to the batch over 2 hours. The batch temperature was adjusted to 20 .5 °C and then filtered using a 24″ Haselloy Nutsche filter equipped with polypropylene filter cloth. A previously prepared solution of 1:1 acetic acid in DI H20 (10.0 L) was charged to the vessel and agitated for 5 minutes. The wash was transferred to the filter and the batch was then manually re- suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged to the vessel and then transferred to the filter. The batch was manually re-suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged directly to the filter and the batch was then manually re-suspended in the filter before re-applying vacuum. The filter cake was allowed to condition for 18 hours to give 14.4 kg of 4. HPLC analysis indicated a purity of 93.7%. This wet cake was carried forward into the purification. A 100 L jacketed glass vessel (purged with N2) was charged with crude 4 (wet cake 14.42 kg), acetic acid (48.8 L) and the agitator was started. DI H20 (1.74 L) was charged. The batch (a slurry) temperature was adjusted from 18.1 to 100.1 °C over 4.25 hours. The batch was held at 100.1 to 106.1 °C for 1 hour and then adjusted to 73.1 °C. DI H20 (28.0 L) was added to the batch over 1 hour keeping the batch temperature between 73.1 and 70.3 °C. The batch temperature was adjusted further from 70.3 °C to 25.0 °C overnight. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with polypropylene filter cloth. The filtration took 13 minutes. A solution of DI H20 (9.00 L) and acetic acid (11.0 L) was prepared and added to the 100 L vessel. The mixture was agitated for 5 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 6 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 9 minutes and then transferred to the filter cake. The batch was allowed to condition for 3 days and then transferred to drying trays for vacuum oven drying. After 3 days at 50 °C and 28’7Hg, the batch gave a 74% yield (3.7 kg) of4 as an off-white solid. The JH NMR spectrum was consistent with the assigned structure, HPLC analysis indicated a purity of 98.87% and KF analysis indicated 0.14% H20. Synthesis of Int. 7: A 100-L jacketed glass vessel (purged with N2) was charged with tetrahydrofuran (44.4 L). The agitator was started (125 RPM) and 4 (3.67 kg) was charged followed by lithium chloride (1.26 kg). The batch temperature was observed to be 26.7 ° C and was an amber solution. Isopropenylmagnesium bromide 1.64 molar solution in 2-methyl THF (21.29 kg) was added over 2 ½ hours keeping the batch between 24.3 and 33.6 °C. The batch was agitated at 24.5 °C for 17 hours at which point HPLC analysis indicated 9% 4. A 2nd 100-L jacketed glass vessel (purged with N2) was charged with 3N hydrogen chloride (18.3 L). The batch was transferred to the vessel containing the 3N HC1 over 25 minutes keeping the batch temperature between 20 and 46 °C. A bi-phasic solution was observed. The quenched batch was transferred back to the 1st 100-L vessel to quench the small amount of residue left behind. THF (2.00 L) was used as a rinse. The batch temperature was observed to be 40.9 ° C and was agitated at 318 RPM for 45 minutes. The batch temperature was adjusted to 21.8 ° C and the layers were allowed to separate. The separation took 10 minutes. The lower aqueous phase was removed (-26.0 L). A solution of sodium chloride (1.56 kg) in DI water (14.0 L) was prepared and added to the batch. This was agitated at 318 RPM for 10 minutes and agitator was stopped. The separation took 3 minutes. The lower aqueous phase was removed (-16.0 L). The batch was vacuum distilled from 58.0 L to 18.4 L using ~24’7Hg and a jacket temperature of 50 to 55 °C. A solution of potassium hydroxide (2.30 kg) in DI water (20.7 L) was prepared in a 72-L round bottom flask. The vessel was set up for atmospheric distillation using 2 distillation heads and the batch was transferred to the 72-L vessel. THF (0.75 L) was used as a rinse. The batch volume was -41.0 L, the temperature was adjusted to 64.1 °C and distillation started with the aid of a N2 sweep. Heating was continued to drive the batch temperature to 85.4 °C while distilling at which point the 72-L vessel was set up for reflux (batch volume was about 28.0 L at the end of the distillation). The batch was held at 85 °C for 13 hours at which point HPLC analysis indicated 0.3% compound 6A. Heating was stopped and the batch was transferred to a 100-L jacketed glass vessel. Solids were observed. The batch temperature was adjusted from 70.6 °C to 56.7 °C. A previously prepared solution of sodium hydrogen carbonate (2.82 kg) in DI water (35.0 L) was added over 80 minutes keeping the batch temperature between 56.7 and 46.7 °C. The batch pH at the end of the addition was 9.8. The batch was held at
46.7 to 49.0 °C for 40 minutes and then cooled to 25.0 °C. The batch was filtered using a 18″ stainless steel Nutsche filter. DI water (18.4 L) was charged to the vessel and transferred to the filter. The filter cake was manually re-suspended in the filter and then the liquors were removed. This process was repeated once more and the filter cake was 3″ thick. The filter cake was conditioned on the filter for 3 days, was transferred to drying trays and dried in a vacuum oven at 45 °C to provide 2.93 kg Int. 7 (95% yield) with an HPLC purity of 87.6%.
Synthesis of Int. 8: A 100 L jacketed glass vessel (purged with N2 and plumbed to a caustic scrubber) was charged with acidic acid (13.0 L). Int. 7 (2.85 kg) was charged to the vessel and the agitator was started. N-Cyanoacetylurethane (1.56 kg) and DI water (5.70 L) were charged to the vessel. The batch temperature was adjusted from 17.0 °C to 5.5 °C and a thin slurry was observed. At this point 37% hydrogen chloride (2.70 L) was added over 10 minutes keeping the batch temperature between 4.8 °C and 8.8 °C. A previously prepared solution of sodium nitrite (638 g) in DI water (1.42 L) was added over 26 minutes keeping the batch temperature between 5.8 °C and 8.7 °C. A brown gas was observed in the vessel head space during the addition. HPLC analysis indicated no Int. 7 detected. At this point a previously prepared solution of sodium acetate (2.07 kg) in DI water (8.50 L) was added over 47 minutes keeping the batch temperature between 5.5 °C and 9.5 °C. After the addition, a thin layer of orange residue was observed on the vessel wall just above the level of the batch. The batch temperature was adjusted from 9.4 °C to 24.5 °C and held at 25 °C (+ 5 °C) for 12 hours. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with tight-weave polypropylene filter cloth. The filtration took 30 minutes. The vessel was rinsed with 14.3 L of a 1 : 1 acidic acid / DI water. The orange residue on the reactor washed away with the rinse. The rinse was transferred to the filter where the batch was manually re-suspended. Vacuum was re-applied to remove the wash. A 2nd 1 : 1 acidic acid / DI water wash was performed as above and the batch was conditioned on the filter for 26 hours. HPLC analysis of the wet filter cake indicated purity was 90.4%. The batch was dried to a constant weight of 3.97 kg (91% yield) in a vacuum oven at 45 °C and 287Hg. Preparation of Compound A DMAC Solvate
A 100 L, jacketed, glass vessel purged with N2 was charged with Int. 8 (3.90 kg) and potassium acetate (875 g). N,N-dimethylacetamide (DMAC, 18.3 L) was charged to the vessel and the agitator was started. The batch temperature was adjusted to 115 °C over 2 h. After 2 h at 115 °C, the batch was sampled and HPLC analysis indicated 0.27% Int. 8 remained. The batch temperature was adjusted to 25.0 °C overnight. Acetic acid (975 mL) was added to the batch and the batch was agitated further for 3 h. The batch was transferred to a carboy and the vessel was rinsed clean with 800 mL of DMAC. The batch was transferred back to the 100 L vessel using vacuum through a 10 μιη in-line filter and a DMAC rinse (1.15 L) was used. The filtration was fast at the beginning but slow at the end, plugging up the filter. The batch temperature was adjusted to 11.1 °C and DI water (35.1 L) was added over 2 h 20 min, keeping the batch temperature between 5-15 °C. The batch was held for 1 h and filtered, using an 18″ Nutsche filter equipped with tight-weave
polypropylene cloth. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel, cooled to 10 °C, and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg to give 89% yield (3.77 kg) of Compound A DMAC solvate as an orange/tan solid. The 1H NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated 0.49% H20. XRPD indicated the expected form, i.e., Compound A DMAC solvate. Thermogravimetric analysis (TGA) indicated 16% weight loss. HPLC analysis indicated a purity of 93.67%.
Preparation of Crude Compound A
A 100 L, jacketed, glass vessel purged with N2 was charged with Compound A
DMAC solvate (3.75 kg) and ethanol (15.0 L). The agitator was started and acetone (15.0 L) was added. The batch temperature was adjusted from 10.6 °C to 60.0 °C over 1 h. At this point, the batch was in solution. DI water was added to the batch over 1.5 h, keeping the batch temperature at 60 + 5 °C. The batch was held at 60 + 5 °C for 1 h and cooled to 23.5 °C. An 18″ Nutsche filter equipped with tight-weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg for five days to give a 94% yield (2.90 kg) of Compound A as a powdery tan solid. The NMR spectrum is consistent with the assigned structure and Karl Fischer analysis indicated 6.6% H20. XRPD indicated the expected form of dihydrate. TGA indicated 6.7% weight loss. HPLC analysis indicated a purity of 96.4% (AUC).
Purification of Crude Compound A
A 50 L, jacketed, glass vessel purged with N2 was charged with Compound A crude
(2.90 kg) and methyl isobutyl ketone (14.5 L). The agitator was started and the batch temperature was adjusted from 20.2 °C to 50.4 °C over 1.5 h. The batch was held at 50 °C (+ 5 °C) for 1 h and cooled to 20-25 °C. The batch was held at 20-25 °C for 2.5 h. An 18″ Nutsche filter equipped with tight- weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 20 min. Methyl isobutyl ketone (MIBK, 1.45 L) was charged to the vessel and transferred to the filter cake. The cake was manually resuspended and the liquors were pulled through with vacuum. Methyl isobutyl ketone (2.90 L) was charged to the filter cake and the cake was manually resuspended. The liquors were pulled through with vacuum and the cake was conditioned with vacuum and nitrogen for 15 h. The filter cake dried into a tan, hard 18″ x 1 ½” disc. This was manually broken up and run through coffee grinders to give a 76% yield (2.72 kg) of MGL-3196 MIBK solvate as a tan, powdery solid. No oven drying was necessary. The NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated <0.1 % H20. XRPD indicated the expected form MIBK solvate. TGA indicated 17.3% weight loss. HPLC analysis indicated a purity of 98.5%.
Example 6: Conversion of Compound A to Form I
Purified Compound A (4802 g) as a 1:1 MIBK solvate which was obtained from Int. 8 as described in Example 5 above was added into a jacketed, 100 L reactor along with 24 liters of ethanol. The resulting slurry was heated to 80 + 5 °C (reflux) over 1 h 25 min; the mixture was stirred at that temperature for 4 h 25 min. Analysis of the filtered solids at 2 h 55 min indicated that the form conversion was complete, with the XRPD spectra conforming to Form I. The mixture was cooled to 20 + 5 °C over 45 min and stirred at that temperature for 15 min. The slurry was filtered and the filter cake was washed twice with prefiltered ethanol (2 x 4.8 L). The wet cake (4.28 kg) was dried under vacuum at 40 + 5 °C for 118 h to afford 3390 g of Compound A form I.
PAPER
Journal of Medicinal Chemistry (2014), 57(10), 3912-3923
The beneficial effects of thyroid hormone (TH) on lipid levels are primarily due to its action at the thyroid hormone receptor β (THR-β) in the liver, while adverse effects, including cardiac effects, are mediated by thyroid hormone receptor α (THR-α). A pyridazinone series has been identified that is significantly more THR-β selective than earlier analogues. Optimization of this series by the addition of a cyanoazauracil substituent improved both the potency and selectivity and led to MGL-3196 (53), which is 28-fold selective for THR-β over THR-α in a functional assay. Compound 53 showed outstanding safety in a rat heart model and was efficacious in a preclinical model at doses that showed no impact on the central thyroid axis. In reported studies in healthy volunteers, 53 exhibited an excellent safety profile and decreased LDL cholesterol (LDL-C) and triglycerides (TG) at once daily oral doses of 50 mg or higher given for 2 weeks.
//////////////RESMETIROM , MGL-3196, VIA-3196, UNII-RE0V0T1ES0, Phase III
Hepatitis C virus (HCV), or hepatitis C virus infection, is a chronic blood-borne infection. Studies have shown that 40% of chronic liver diseases are associated with HCV infection, and an estimated 8,000-10,000 people die each year. HCV-related end-stage liver disease is the most common indication for liver transplantation in adults.
In the past ten years, antiviral therapy for chronic liver disease has developed rapidly, and significant improvement has been seen in the treatment effect. However, even with the combination therapy with pegylated IFN-α plus ribavirin, 40% to 50% of patients fail to treat, that is, they are non-responders or relapsers. These patients do not currently have an effective treatment alternative. Because the risk of HCV-related chronic liver disease is related to the duration of HCV infection, and the risk of cirrhosis increases in patients who have been infected for more than 20 years, chronic liver disease often progresses to advanced stages with cirrhosis, ascites, jaundice, and rupture of varicose veins. , Brain disease, and progressive liver failure, and the risk of liver cancer is also significantly increased.
HCV is a enveloped positive-strand RNA virus of the Flaviviridae family. The single-stranded HCV RNA genome is approximately 9500 nucleotides in length and has a single open reading frame (ORF) that encodes a single open reading frame (ORF) of approximately 3,000 amino acids. Mostly polyprotein. In infected cells, cellular and viral proteases cleave this polyprotein at multiple sites to produce viral structural and non-structural (NS) proteins. There are two viral proteases that affect the production of mature non-structural proteins (NS2, NS3, NS4, NS4A, NS4B, NS5A, and NS5B). The first viral protease is cleaved at the NS2-NS3 junction of the polyprotein; the second viral protease is A “NS3 protease” that mediates all subsequent cleavage events at a site downstream of the NS3 position relative to the polyprotein (ie, the site between the C-terminus of NS3 and the C-terminus of the polyprotein). The NS3 protease exhibits cis-activity at the NS3-NS4 cleavage site and, conversely, exhibits trans-activity at the remaining NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B sites. The NS4A protein is thought to provide multiple functions, such as acting as a cofactor for the NS3 protease, and may promote membrane localization of NS3 and other viral replicase components. The formation of a complex between NS3 and NS4A may be necessary for NS3-mediated processing events and improves the proteolytic efficiency at all sites recognized by NS3. NS3 protease may also exhibit nucleotide triphosphatase and RNA helicase activity. NS5B is an RNA-dependent RNA polymerase involved in HCV RNA replication. In addition, compounds that inhibit the effects of NS5A in viral replication may be useful for treating HCV.
Beijing Kawin Technology Share-Holding, in collaboration with Beijing Fu Rui Tiancheng Biotechnology and Ginkgo Pharma , is developing coblopasvir as an oral capsule formulation of dihydrochloride salt (KW-136), for treating hepatitis C virus infection. In June 2018, an NDA was filed in China by Beijing Kawin Technology and Sichuan Qingmu Pharmaceutical . In August 2018, the application was granted Priority Review in China . Also, Beijing Kawin is investigating a tablet formulation of coblopasvir dihydrochloride.
PATENT
WO2011075607 , claiming substituted heterocyclic derivatives as HCV replication inhibitors useful for treating HCV infection and liver fibrosis, assigned to Beijing Kawin Technology Share-Holding Co Ltd and InterMune Inc ,
Novel crystalline and amorphous forms of methyl carbamate compound, particularly coblopasvir dihydrochloride , (designated as Forms H) processes for their preparation, compositions and combinations comprising them are claimed. Also claim is an article or kit comprising a container and a package insert, wherein the container contains coblopasvir dihydrochloride.
Step 7
To a solution of compound 1-IXf (250 mg, 0.31 mmol) in toluene (10.0 mL) was added NH4OAc (4.0 g, 50 mmol) and the mixture was refluxed for 16 hours. The reaction mixture was diluted with ethyl acetate and washed with water and brine. The solvent was removed and the residue was purified by preparative HPLC to give Compound I (43.5 mg, yield 20%) as a white solid. MS (ESI) m / z (M + H) + 783.4.
Example 2 Preparation of a compound of formula II
Compound of formula (I) N-[(2S) -1-[(2S) -2- {4- [7- (4- {2-[(2S) -1-[(2S) -2-[(A Oxycarbonyl) amino] -3-methylbutanoyl] pyrrolidin-2-yl] -1H-imidazol-4-yl} phenyl) -2H-1,3-benzodioxo-4-yl] Preparation of -1H-imidazol-2-yl} pyrrolidin-1-yl] -3-methyl-1-oxobutane-2-yl] carbamate dihydrochloride
At room temperature, a solution of the pure product of structural formula I (800 g, 1.0 eq) and ethyl acetate (8 L) were sequentially added to a 20 L bottle and stirred. A 11.2% HCl / ethyl acetate solution (839 g) was added dropwise to the system, the temperature of the system was controlled at 15 ° C to 25 ° C, and the mixture was stirred for more than 3 hours to stop the reaction. The filter cake was filtered with ethyl acetate (2L). Wash the cake, bake the cake at a controlled temperature of 40-60 ° C, sample and test until the ethyl acetate residue is <0.5%, (about 73 hours of baking), to obtain the compound of formula II, off-white solid powder or granules, 774 g, HPLC Purity: 98.65%, yield: 88.5%, tested XRPD as amorphous.
///////////////Coblopasvir , KW-136, hepatitis C virus infection, CHINA, Beijing Kawin Technology, NDA, Phase III
PHASE 3, for the potential oral treatment of moderate-to-severe atopic dermatitis (AD)
Jak1 tyrosine kinase inhibitor
UPDATE…… JAPAN APPROVED, 2021, 2021/9/27, CIBINQO
ALSO
fda 2022, APPROVALS 2022, 1/14/2022
THE US
In February 2018, the FDA granted Breakthrough Therapy designation for the treatment of patients with moderate-to-severe AD
PHASEIII
In December 2017, a randomized, double-blind, placebo-controlled, parallel-group, phase III trial (NCT03349060; JADE Mono-1; JADE; B7451012; 2017-003651-29) of PF-04965842 began in patients aged 12 years and older (expected n = 375) with moderate-to-severe AD
PRODUCT PATENT
Pub. No.:
WO/2014/128591
International Application No.:
PCT/IB2014/058889
Publication Date:
28.08.2014
International Filing Date:
11.02.2014
EXPIRY Roughly 2034
form
powder
color
white to beige
solubility
DMSO: 10 mg/mL, clear
storage temp.
room temp
Biochem/physiol Actions
PF-04965842 is a Janus Kinase (JAK) inhibitor selective for JAK1 with an IC50value of 29 nM for JAK1 compared to 803 nM for JAK2, >10000 nM for JAK3 and 1250 nM for Tyk2. JAKs mediate cytokine signaling, and are involved in cell proliferation and differentiation. PF-04965842 has been investigated as a possible treatment for psoriasis.
Originator Pfizer
Class Skin disorder therapies; Small molecules
Mechanism of Action Janus kinase 1 inhibitors
Highest Development Phases
Phase IIIAtopic dermatitis
DiscontinuedLupus vulgaris; Plaque psoriasis
Most Recent Events
08 Mar 2018Phase-III clinical trials in Atopic dermatitis (In children, In adults, In adolescents) in USA (PO) (NCT03422822)
14 Feb 2018PF 4965842 receives Breakthrough Therapy status for Atopic dermatitis in USA
06 Feb 2018Pfizer plans the phase III JADE EXTEND trial for Atopic Dermatitis (In children, In adults, In adolescents) in March 2018 (PO) (NCT03422822)
This compound was developed by Pfizer for Kinase Phosphatase Biology research. To learn more about Sigma′s partnership with Pfizer and view other authentic, high-quality Pfizer compounds,
PF-04965842 is an oral Janus Kinase 1 inhibitor being investigated for treatment of plaque psoriasis.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified into tyrosine and serine/threonine kinases. Inappropriate kinase activity, arising from mutation, over-expression, or inappropriate regulation, dys-regulation or de-regulation, as well as over- or under-production of growth factors or cytokines has been i mplicated in many diseases, including but not limited to cancer, cardiovascular diseases, allergies, asthma and other respiratory diseases, autoimmune d iseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative disorders such as Alzheimer’s disease. Inappropriate kinase activity triggers a variety of biological cellular responses relating to cell growth, cell differentiation , survival, apoptosis, mitogenesis, cell cycle control, and cel l mobility implicated in the aforementioned and related diseases.
Thus, protein kinases have emerged as an important class of enzymes as targets for therapeutic intervention. In particular, the JAK family of cellular protein tyrosine kinases (JAK1, JAK2, JAK3, and Tyk2) play a central role in cytoki ne signaling (Kisseleva et al., Gene, 2002, 285 , 1; Yamaoka et al. Genome Biology 2004, 5, 253)). Upon binding to their receptors, cytokines activate JAK which then phosphorylate the cytokine receptor, thereby creating docking sites for signaling molecules, notably, members of the signal transducer and activator of transcription (STAT) family that ultimately lead to gene expression. Numerous cytokines are known to activate the JAK family. These cytokines include, the IFN family (IFN-alpha, IFN-beta, IFN-omega, Limitin, IFN-gamma, IL- 10, IL- 19, IL-20, IL-22), the gp 130 family (IL-6, IL- 11, OSM, LIF, CNTF, NNT- 1//SF-3, G-CSF, CT- 1, Leptin, IL- 12 , I L-23), gamma C family (IL-2 , I L-7, TSLP, IL-9, IL- 15 , IL-21, IL-4, I L- 13), IL-3 family (IL-3 , IL-5 , GM-CSF), single chain family (EPO, GH, PRL, TPO), receptor tyrosine kinases (EGF, PDGF, CSF- 1, HGF), and G-protein coupled receptors (ATI).
Abrocitinib is quickly absorbed from the gut and generally reaches highest blood plasma concentrations within one hour. Only 1.0 to 4.4% of the dose are found unmetabolized in the urine.[4]
History
April 2016: initiation of Phase 2b trial
December 2017: initiation of JADE Mono-1 Phase 3 trial[5]
May 2018: Results of Phase 2b trial posted
October 2019: Results of Phase 3 trial presented[6]
June 2020: Results of second Phase 3 trial published[7]
Society and culture
Legal status
In October 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Cibinqo, intended for the treatment of atopic dermatitis.[8] The applicant for this medicinal product is Pfizer Europe MA EEIG.[8] In December 2021, the European Commission approved abrocitinib for the treatment of atopic dermatitis.[2][9]
In January 2022, the United States Food and Drug Administration (FDA) approved abrocitinib for adults with moderate-to-severe atopic dermatitis.[10]
Introduction The finished product is presented as immediate release film-coated tablets containing 50 mg, 100 mg or 200 mg of abrocitinib as active substance. Other ingredients are: Tablet core: microcrystalline cellulose (E460i), anhydrous dibasic calcium phosphate (E341ii), sodium starch glycolate and magnesium stearate (E470b). Film-coat: hypromellose (E464), titanium dioxide (E171), lactose monohydrate, macrogol (E1521), triacetin (E1518) and red iron oxide (E172). The product is available in high-density polyethylene (HDPE) bottles with polypropylene closure or polyvinylidene chloride (PVDC) blisters with aluminium foil lidding film, as described in section 6.5 of the SmPC.
The chemical name of abrocitinib is N-((1S,3S)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4- yl)amino)cyclobutyl)propane-1-sulfonamide corresponding to the molecular formula C14H21N5O2S. It has a relative molecular mass of 323.42 Daltons and the following structure depicted in Figure 1:
The chemical structure of abrocitinib was elucidated by a combination of UV/VIS and IR spectroscopy, mass spectrometry, NMR spectroscopy and X-ray diffraction. The active substance is a white to pale-purple or pale pink crystalline powder. It is non-hygroscopic and its solubility is pH dependent. Abrocitinib is classified as BCS Class II. The impact of particle size on finished product uniformity of dosage units and dissolution has been studied (see finished product section). Based on the abrocitinib finished product biopharmaceutics performance, stability, and manufacturing experience, the active substance particle size specification was established. Abrocitinib is an achiral molecule, but with 2 stereocentres. Only one crystalline anhydrous form (Form 1) of abrocitinib has been identified. This form has been the only form used in all toxicology and clinical studies. Extensive polymorph and hydrate screening have been conducted to investigate if additional solid forms of abrocitinib could be discovered. Abrocitinib, Form 1 was the only anhydrous crystalline form identified from these studies. No new anhydrous polymorphs, hydrates or amorphous solids of abrocitinib were isolated from these screens. Experiments with 1,4 dioxane and dimethyl sulfoxide yielded solvated forms of abrocitinib. When these solvated structures were subjected to high temperature, these materials desolvated and converted to Form 1, free base anhydrous form of abrocitinib. However, these are not relevant since the commercial crystallisation step does not utilise either of these solvent systems. It has been confirmed that the manufacturing process consistently yields polymorphic form I. This form is physically and chemically stable under normal manufacturing and storage conditions as well as under accelerated conditions. Hence the absence of control of form I is justified.
FDA
U.S. FDA Approves Pfizer’s CIBINQO® (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis
CIBINQO is a once-daily oral treatment with proven efficacy to manage symptoms for adults who have not yet found relief with current options
NEW YORK–(BUSINESS WIRE)– Pfizer Inc. (NYSE: PFE) announced today that the United States (U.S.) Food and Drug Administration (FDA) approved CIBINQO® (abrocitinib), an oral, once-daily, Janus kinase 1 (JAK1) inhibitor, for the treatment of adults living with refractory, moderate-to-severe atopic dermatitis (AD) whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
CIBINQO is approved at the recommended doses of 100 mg and 200 mg, with the 200 mg dose being recommended for patients who are not responding to the 100 mg dose. Additionally, a 50 mg dose was approved to treat moderate-to-severe AD specifically in patients with moderate renal impairment (kidney failure), certain patients receiving treatment with inhibitors of cytochrome P450 (CYP) 2C19, or patients who are known or suspected to be poor metabolizers of CYP2C19. For patients with moderate renal impairment who are not responding to 50 mg once daily, 100 mg once daily may also be prescribed.
“The reality for patients living with chronic inflammatory skin disease such as moderate-to-severe atopic dermatitis is that many experience debilitating symptoms that are not managed by current treatment options. Today’s approval of CIBINQO will provide an important new oral option that could help those who have yet to find relief,” said Jonathan Silverberg, MD, PhD, MPH, Department of Dermatology, The George Washington University School of Medicine and Health Sciences. “In multiple large-scale clinical trials, CIBINQO demonstrated strong efficacy at clearing skin, improving itch, and managing the extent and severity of eczema, offering a benefit-risk profile that supports the use of this treatment in the FDA-approved patient population.”
The FDA approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The safety and efficacy of CIBINQO was evaluated in three randomized, placebo-controlled, Phase 3 trials. Additionally, safety was evaluated through a randomized, placebo-controlled, dose-ranging trial and an ongoing long-term open-label extension trial. Across the trials, CIBINQO demonstrated a consistent safety profile and profound improvements in skin clearance, extent of disease, and severity, as well as rapid improvement in itch after two weeks, for some people living with AD versus placebo. In addition, a higher proportion of subjects treated with CIBINQO in two monotherapy trials achieved improvement in itching at week 12 compared to placebo.
“The FDA’s approval offers hope to the millions of patients across the U.S. who are suffering daily with an immuno-inflammatory condition that can cause intense and persistent itching, pain, discomfort, and distress if left uncontrolled,” said Mike Gladstone, Global President of Pfizer Inflammation & Immunology. “CIBINQO, an efficacious once-daily pill, is a medical breakthrough made possible by Pfizer researchers and the people living with moderate-to-severe atopic dermatitis who participated in our clinical trials.”
“Atopic dermatitis is so much more than just a rash, and it goes beyond the surface of the skin. It’s a chronic condition that can both significantly disrupt patients’ daily lives and negatively impact their emotional well-being,” said Julie Block, President and CEO, National Eczema Association. “We appreciate Pfizer’s commitment to this resilient patient community and eagerly await the positive impact CIBINQO could have on the treatment landscape for moderate-to-severe atopic dermatitis.”
The most common adverse events reported in ≥5% of patients with CIBINQO included nasopharyngitis (12.4% with CIBINQO 100 mg, 8.7% with CIBINQO 200 mg, and 7.9%, with placebo), nausea (6%, 14.5%, and 2.1%, respectively), and headache (6%, 7.8%, and 3.5%, respectively).
The full prescribing information for CIBINQO can be found here. CIBINQO will be made available in the coming weeks.
Additional Details on the CIBINQO Clinical Trial Program
Five clinical trials in the CIBINQO JAK1 Atopic Dermatitis Efficacy and Safety (JADE) global development program were included in the New Drug Application (NDA) to support the FDA approval.
The safety and efficacy of CIBINQO was evaluated in three Phase 3, randomized, placebo-controlled clinical trials. The trials evaluated measures of improvements in skin clearance, itch, disease extent, and severity, including the Investigator Global Assessment (IGA), Eczema Area and Severity Index (EASI), and Peak Pruritus Numerical Ratings Scale (PP-NRS). In each of the trials, over 40% of patients had prior exposure to a systemic therapy:
JADE MONO-1 and JADE MONO-2: A pair of randomized, double-blind, placebo-controlled trials designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO monotherapy in 778 patients 12 years of age and older with moderate-to-severe AD. The trials assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
JADE COMPARE: A randomized, double-blind, placebo-controlled trial designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO in 837 adult patients with moderate-to-severe AD on background topical medicated therapy. The trial also included an active control arm with dupilumab, a biologic treatment administered by subcutaneous injection, compared with placebo. The trial assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
Select findings for CIBINQO 100 mg, 200 mg, and placebo follow (*p<0.01 or **p<0.001):
JADE MONO-1:
IGA Response Rate (Week 12): 24%*, 44%**, and 8%, respectively
EASI-75 Response Rate (Week 12): 40%**, 62%**, and 12%, respectively
JADE MONO-2
IGA Response Rate (Week 12): 28%**, 38%**, and 9%, respectively
EASI-75 Response Rate (Week 12): 44%**, 61%**, and 10%, respectively
JADE COMPARE
IGA Response Rate (Week 12): 36%**, 47%**, and 14%, respectively
EASI-75 Response Rate (Week 12): 58%**, 68%**, and 27%, respectively
Safety was additionally evaluated through a randomized dose-ranging trial and a long-term, open-label, extension trial (JADE EXTEND).
U.S. IMPORTANT SAFETY INFORMATION
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, AND THROMBOSIS
Serious Infections
Patients treated with CIBINQO may be at increased risk for developing serious infections that may lead to hospitalization or death. The most frequent serious infections reported with CIBINQO were herpes simplex, herpes zoster, and pneumonia.
If a serious or opportunistic infection develops, discontinue CIBINQO and control the infection.
Reported infections from Janus kinase (JAK) inhibitors used to treat inflammatory conditions:
Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Test for latent TB before and during therapy; treat latent TB prior to use. Monitor all patients for active TB during treatment, even patients with initial negative, latent TB test.
Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
Bacterial, viral (including herpes zoster), and other infections due to opportunistic pathogens.
Avoid use of CIBINQO in patients with an active, serious infection, including localized infections. The risks and benefits of treatment with CIBINQO should be carefully considered prior to initiating therapy in patients with chronic or recurrent infections or those who have resided or traveled in areas of endemic tuberculosis or endemic mycoses.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with CIBINQO, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
Consider yearly screening for patients in highly endemic areas for TB. CIBINQO is not recommended for use in patients with active TB. For patients with a new diagnosis of latent TB or prior untreated latent TB, or for patients with a negative test for latent TB but who are at high risk for TB infection, start preventive therapy for latent TB prior to initiation of CIBINQO.
Viral reactivation, including herpes virus reactivation (eg, herpes zoster, herpes simplex), was reported in clinical studies with CIBINQO. If a patient develops herpes zoster, consider interrupting CIBINQO until the episode resolves. Hepatitis B virus reactivation has been reported in patients receiving JAK inhibitors. Perform viral hepatitis screening and monitoring for reactivation in accordance with clinical guidelines before starting therapy and during therapy with CIBINQO. CIBINQO is not recommended for use in patients with active hepatitis B or hepatitis C.
Mortality
In a large, randomized postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another JAK inhibitor to TNF blocker treatment, a higher rate of all-cause mortality (including sudden cardiovascular death) was observed with the JAK inhibitor. CIBINQO is not approved for use in RA patients.
Malignancies
Malignancies, including non-melanoma skin cancer (NMSC), were reported in patients treated with CIBINQO. Lymphoma and other malignancies have been observed in patients receiving JAK inhibitors used to treat inflammatory conditions. Perform periodic skin examination for patients who are at increased risk for skin cancer. Exposure to sunlight and UV light should be limited by wearing protective clothing and using broad-spectrum sunscreen.
In a large, randomized postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. CIBINQO is not approved for use in RA patients. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Major Adverse Cardiovascular Events
Major adverse cardiovascular events were reported in patients treated with CIBINQO. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients. Patients who are current or past smokers are at additional increased risk. Discontinue CIBINQO in patients that have experienced a myocardial infarction or stroke.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur.
Thrombosis
Deep vein thrombosis (DVT) and pulmonary embolism (PE) have been reported in patients treated with CIBINQO. Thrombosis, including PE, DVT, and arterial thrombosis have been reported in patients receiving JAK inhibitors used to treat inflammatory conditions. Many of these adverse reactions were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of overall thrombosis, DVT, and PE were observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients.
Avoid CIBINQO in patients that may be at increased risk of thrombosis. If symptoms of thrombosis occur, discontinue CIBINQO and treat patients appropriately.
Contraindication
CIBINQO is contraindicated in patients taking antiplatelet therapies, except for low-dose aspirin (≤81 mg daily), during the first 3 months of treatment.
Laboratory Abnormalities
Hematologic Abnormalities: Treatment with CIBINQO was associated with an increased incidence of thrombocytopenia and lymphopenia. Prior to CIBINQO initiation, perform a complete blood count (CBC). CBC evaluations are recommended at 4 weeks after initiation and 4 weeks after dose increase of CIBINQO. Discontinuation of CIBINQO therapy is required for certain laboratory abnormalities.
Lipid Elevations: Dose-dependent increase in blood lipid parameters were reported in patients treated with CIBINQO. Lipid parameters should be assessed approximately 4 weeks following initiation of CIBINQO therapy, and thereafter patients should be managed according to clinical guidelines for hyperlipidemia. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Immunizations
Prior to initiating CIBINQO, complete all age-appropriate vaccinations as recommended by current immunization guidelines, including prophylactic herpes zoster vaccinations. Avoid vaccination with live vaccines immediately prior to, during, and immediately after CIBINQO therapy.
Renal Impairment
Avoid use in patients with severe renal impairment or end stage renal disease, including those on renal replacement therapy.
Hepatic Impairment
Avoid use in patients with severe hepatic impairment.
Adverse Reactions
Most common adverse reactions (≥1%) in subjects receiving 100 mg and 200 mg include: nasopharyngitis, nausea, headache, herpes simplex, increased blood creatinine phosphokinase, dizziness, urinary tract infection, fatigue, acne, vomiting, oropharyngeal pain, influenza, gastroenteritis.
Most common adverse reactions (≥1%) in subjects receiving either 100 mg or 200 mg also include: impetigo, hypertension, contact dermatitis, upper abdominal pain, abdominal discomfort, herpes zoster, and thrombocytopenia.
Use in Pregnancy
Available data from pregnancies reported in clinical trials with CIBINQO are not sufficient to establish a drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Advise females of reproductive potential that CIBINQO may impair fertility.
There will be a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to CIBINQO during pregnancy. Pregnant women exposed to CIBINQO and health care providers are encouraged to call 1-877-311-3770.
Lactation
Advise women not to breastfeed during treatment with CIBINQO and for one day after the last dose.
Indication
CIBINQO is indicated for the treatment of adults with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
Limitations of Use: CIBINQO is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
About CIBINQO® (abrocitinib)
CIBINQO is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD, including interleukin IL-4, IL-13, IL-31, IL-22, and thymic stromal lymphopoietin (TSLP).
In addition to receiving regulatory approval in the U.S., CIBINQO has received marketing authorization in the European Union, Great Britain, Japan, Korea, the United Arab Emirates, Norway, Iceland, and Singapore.
About Atopic Dermatitis
AD is a chronic skin disease characterized by inflammation of the skin and skin barrier defects.i,ii Most people know AD is a skin condition. But many don’t realize it can be caused in part by an abnormal immune response beneath the skin. This dysregulated immune response is thought to contribute to inflammation within the skin and the signs of AD on the surface. Lesions of AD are characterized by erythema (red/pink or discolored skin patches, depending on normal skin color), itching, lichenification (thick/leathery skin), induration (hardening)/papulation (formulation of papules), and oozing/crusting.i,ii
AD is one of the most common inflammatory skin diseases, affecting approximately 5-10% of adults in the U.S.iii,iv Approximately 1 in 3 adults with AD have moderate-to-severe disease.v,vi
About Pfizer Inflammation & Immunology
At Pfizer Inflammation & Immunology, we strive to deliver breakthroughs that enable freedom from day-to-day suffering for people living with autoimmune and chronic inflammatory diseases, which can be debilitating, disfiguring and distressing, dramatically affecting what they can do. With a focus on immuno-inflammatory conditions in Rheumatology, Gastroenterology and Medical Dermatology, our current portfolio of approved medicines and investigational molecules spans multiple action and delivery mechanisms, from topicals to small molecules, biologics and biosimilars. The root cause of many immunological diseases is immuno-inflammation, which requires specifically designed agents. Our differentiated R&D approach resulted in one of the broadest pipelines in the industry, where we purposefully match molecules to diseases where we believe they can make the biggest difference. Building on our decades-long commitment and pioneering science, we continue to advance the standard of care for patients living with immuno-inflammatory diseases and are working hand-in-hand with patients, caregivers and the broader healthcare community on healthcare solutions for the many challenges of managing chronic inflammatory diseases, allowing patients to live their best lives.
Pfizer Inc.: Breakthroughs that Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety, and value in the discovery, development, and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments, and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments, and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
There remains a need for new compounds that effectively and selectively inhibit specific JAK enzymes, and JAK1 in particular, vs. JAK2. JAK1 is a member of the Janus family of protein kinases composed of JAK1, JAK2, JAK3 and TYK2. JAK1 is expressed to various levels in all tissues. Many cytokine receptors signal through pairs of JAK kinases in the following combinations: JAK1/JAK2, JAK1/JAK3, JAK1/TYK2 , JAK2/TYK2 or JAK2/JAK2. JAK1 is the most broadly
paired JAK kinase in this context and is required for signaling by γ-common (IL-2Rγ) cytokine receptors, IL—6 receptor family, Type I, II and III receptor families and IL- 10 receptor family. Animal studies have shown that JAK1 is required for the development, function and homeostasis of the immune system. Modulation of immune activity through inhibition of JAK1 kinase activity can prove useful in the treatment of various immune disorders (Murray, P.J.
J. Immunol., 178, 2623-2629 (2007); Kisseleva, T., et al., Gene, 285 , 1-24 (2002); O’Shea, J . J., et al., Ceil , 109, (suppl .) S121-S131 (2002)) while avoiding JAK2 dependent erythropoietin (EPO) and thrombopoietin (TPO) signaling (Neubauer H., et al., Cell, 93(3), 397-409 (1998);
Parganas E., et al., Cell, 93(3), 385-95 (1998)).
Tofacitinib (1), baricitinib (2), and ruxolitinib (3)
SYNTHESIS 5+1 =6 steps
Main synthesis
Journal of Medicinal Chemistry, 61(3), 1130-1152; 2018
INTERMEDIATE
CN 105732637
ONE STEP
CAS 479633-63-1, 7H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-7-[(4- methylphenyl)sulfonyl]-
Pfizer Receives Breakthrough Therapy Designation from FDA for PF-04965842, an oral JAK1 Inhibitor, for the Treatment of Patients with Moderate-to-Severe Atopic Dermatitis
Wednesday, February 14, 2018 8:30 am EST
Dateline:
NEW YORK
Public Company Information:
NYSE:
PFE
US7170811035
“We look forward to working closely with the FDA throughout our ongoing Phase 3 development program with the hope of ultimately bringing this important new treatment option to these patients.”
NEW YORK–(BUSINESS WIRE)–Pfizer Inc. (NYSE:PFE) today announced its once-daily oral Janus kinase 1 (JAK1) inhibitor PF-04965842 received Breakthrough Therapy designation from the U.S. Food and Drug Administration (FDA) for the treatment of patients with moderate-to-severe atopic dermatitis (AD). The Phase 3 program for PF-04965842 initiated in December and is the first trial in the JAK1 Atopic Dermatitis Efficacy and Safety (JADE) global development program.
“Achieving Breakthrough Therapy Designation is an important milestone not only for Pfizer but also for patients living with the often devastating impact of moderate-to-severe atopic dermatitis, their providers and caregivers,” said Michael Corbo, Chief Development Officer, Inflammation & Immunology, Pfizer Global Product Development. “We look forward to working closely with the FDA throughout our ongoing Phase 3 development program with the hope of ultimately bringing this important new treatment option to these patients.”
Breakthrough Therapy Designation was initiated as part of the Food and Drug Administration Safety and Innovation Act (FDASIA) signed in 2012. As defined by the FDA, a breakthrough therapy is a drug intended to be used alone or in combination with one or more other drugs to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If a drug is designated as a breakthrough therapy, the FDA will expedite the development and review of such drug.1
About PF-04965842 and Pfizer’s Kinase Inhibitor Leadership
PF-04965842 is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD including interleukin (IL)-4, IL-13, IL-31 and interferon gamma.
Pfizer has established a leading kinase research capability with multiple unique kinase inhibitor therapies in development. As a pioneer in JAK science, the Company is advancing several investigational programs with novel selectivity profiles, which, if successful, could potentially deliver transformative therapies for patients. Pfizer has three additional kinase inhibitors in Phase 2 development across multiple indications:
PF-06651600: A JAK3 inhibitor under investigation for the treatment of rheumatoid arthritis, ulcerative colitis and alopecia areata
PF-06700841: A tyrosine kinase 2 (TYK2)/JAK1 inhibitor under investigation for the treatment of psoriasis, ulcerative colitis and alopecia areata
PF-06650833: An interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor under investigation for the treatment of rheumatoid arthritis
Working together for a healthier world®
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products. Our global portfolio includes medicines and vaccines as well as many of the world’s best-known consumer health care products. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 150 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
DISCLOSURE NOTICE: The information contained in this release is as of February 14, 2018. Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments.
This release contains forward-looking information about PF-04965842 and Pfizer’s ongoing investigational programs in kinase inhibitor therapies, including their potential benefits, that involves substantial risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Risks and uncertainties include, among other things, the uncertainties inherent in research and development, including the ability to meet anticipated clinical trial commencement and completion dates and regulatory submission dates, as well as the possibility of unfavorable clinical trial results, including unfavorable new clinical data and additional analyses of existing data; risks associated with preliminary data; the risk that clinical trial data are subject to differing interpretations, and, even when we view data as sufficient to support the safety and/or effectiveness of a product candidate, regulatory authorities may not share our views and may require additional data or may deny approval altogether; whether regulatory authorities will be satisfied with the design of and results from our clinical studies; whether and when drug applications may be filed in any jurisdictions for any potential indication for PF-04965842 or any other investigational kinase inhibitor therapies; whether and when any such applications may be approved by regulatory authorities, which will depend on the assessment by such regulatory authorities of the benefit-risk profile suggested by the totality of the efficacy and safety information submitted, and, if approved, whether PF-04965842 or any such other investigational kinase inhibitor therapies will be commercially successful; decisions by regulatory authorities regarding labeling, safety and other matters that could affect the availability or commercial potential of PF-04965842 or any other investigational kinase inhibitor therapies; and competitive developments.
A further description of risks and uncertainties can be found in Pfizer’s Annual Report on Form 10-K for the fiscal year ended December 31, 2016 and in its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results”, as well as in its subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov and www.pfizer.com.
LC/MS m/z (M + H+) calcd for C14H22N5O2S: 324. Found: 324. Anal. Calcd for C14H21N5O2S: C, 51.99; H, 6.54; N, 21.65; O, 9.89; S, 9.91. Found: C, 52.06; H, 6.60; N, 21.48; O, 10.08; S, 9.97.
Schmieder, G.; Draelos, Z.; Pariser, D.; Banfield, C.; Cox, L.; Hodge, M.; Kieras, E.; Parsons-Rich, D.; Menon, S.; Salganik, M.; Page, K.; Peeva, E.Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled studyBr. J. Dermatol.2017, DOI: 10.1111/bjd.16004
Compound 25, N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide is available through MilliporeSigma (cat. no. PZ0304).
CLIP
REFERENCES
1: Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, Kieras E, Parsons-Rich D, Menon S, Salganik M, Page K, Peeva E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study. Br J Dermatol. 2017 Sep 26. doi: 10.1111/bjd.16004. [Epub ahead of print] PubMed PMID: 28949012
2 Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.
Originator Pfizer
Class Anti-inflammatories; Antipsoriatics; Pyrimidines; Pyrroles; Skin disorder therapies; Small molecules; Sulfonamides
Mechanism of Action Janus kinase 1 inhibitors
Phase III Atopic dermatitis
Discontinued Lupus vulgaris; Plaque psoriasis
21 May 2019Pfizer initiates enrolment in a phase I trial in Healthy volunteers in USA (PO) (NCT03937258)
09 May 2019 Pfizer plans a phase I pharmacokinetic and drug-drug interaction trial in healthy volunteers in May 2019 (NCT03937258)
30 Apr 2019 Pfizer completes a phase I trial (In volunteers) in USA (PO) (NCT03626415)
^ Jump up to:abcde“Cibinqo EPAR”. European Medicines Agency (EMA). 11 October 2021. Retrieved 17 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
“Abrocitinib”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
Clinical trial number NCT03575871 for “Study Evaluating Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-2)” at ClinicalTrials.gov
{{ClinicalTrialsGov|NCT03720470|Study Evaluating Efficacy and Safety of PF-04965842 and Dupilumab in Adult Subjects With Moderate to Severe Atopic Dermatitis on Background Topical Therapy (JADE Compare)}
CF 101 (known generically as IB-MECA) is an anti-inflammatory drug for rheumatoid arthritis patients. Its novel mechanism of action relies on antagonism of adenoside A3 receptors. CF101 is supplied as an oral drug and has an excellent safety profile. It is also being considered for the treatment of other autoimmune-inflammatory disorders, such as Crohn’s disease, psorasis and dry eye syndrome.
Originator Can-Fite BioPharma
Class Amides; Anti-inflammatories; Antineoplastics; Antipsoriatics; Antirheumatics; Eye disorder therapies; Iodobenzenes; Neuroprotectants; Purine nucleosides; Ribonucleosides; Small molecules
05 Feb 2019 Can-Fite BioPharma receives patent allowance for A3 adenosine receptor (A3AR) agonists in USA
05 Feb 2019 Can-Fite BioPharma receives patent allowance for A3 adenosine receptor (A3AR) agonists in North America, South America, Europe and Asia
21 Aug 2018 Phase-III clinical trials in Plaque psoriasis (Monotherapy) in Israel (PO)
Piclidenoson, also known as CF101, is a specific agonist to the A3 adenosine receptor, which inhibits the development of colon carcinoma growth in cell cultures and xenograft murine models. CF101 has been shown to downregulate PKB/Akt and NF-κB protein expression level. CF101 potentiates the cytotoxic effect of 5-FU, thus preventing drug resistance. The myeloprotective effect of CF101 suggests its development as an add-on treatment to 5-FU.
Piclidenoson is known to be a TNF-α synthesis inhibitor and a neuroprotectant. use as an A3 adenosine receptor agonist, useful for treating rheumatoid arthritis (RA), psoriasis, osteoarthritis and glaucoma.
Can-Fite BioPharma , under license from the National Institutes of Health (NIH), is developing a tablet formulation of CF-101, an adenosine A3 receptor-targeting, TNF alpha-suppressing low molecular weight molecule for the potential treatment of psoriasis, RA and liver cancer. The company is also investigating a capsule formulation of apoptosis-inducing namodenoson, the lead from a program of adenosine A3 receptor agonist, for treating liver diseases, including hepatocellular carcinoma (HCC). In January 2019, preclinical data for the treatment of obesity were reported. Also, see WO2019105217 , WO2019105359 and WO2019105082 , published alongside.
Novel crystalline forms of CF-101 (also known as piclidenoson; designated as Forms CS1, CS2 and CS3), processes for their preparation, compositions comprising them and their use as an A3 adenosine receptor agonist for treating rheumatoid arthritis, psoriasis, osteoarthritis and glaucoma are claimed
CF-101 was developed by Kan-Fete Biomedical Co., Ltd. By the end of 2018, CF-101 is in clinical phase III for the treatment of autoimmune diseases such as rheumatoid arthritis, osteoarthritis and psoriasis, as well as glaucoma. CF-101 is an A3 adenosine receptor (A3AR) agonist, and adenosine plays an important role in limiting inflammation through its receptor. Adenosine can produce anti-inflammatory effects by inhibiting TNF-a, interleukin-1, and interleukin-6. Studies have shown that A3AR agonists are in different experimental autoimmune models, such as rheumatoid arthritis, Crohn’s disease, and silver swarf. In the disease, it acts as an anti-inflammatory agent by improving the inflammatory process.
The chemical name of CF-101 is: 1-deoxy-I-(6-{[(3-iodophenyl)methyl]amino}-9H-fluoren-9-yl)-N-methyl-bD-ribofuranose Carbonamide (hereinafter referred to as “Compound I”) has the following structural formula:
A crystal form is a solid in which a compound molecule is orderedly arranged in a microstructure to form a crystal lattice, and a drug polymorphism phenomenon means that two or more different crystal forms of a drug exist.
Due to different physical and chemical properties, different crystal forms of drugs may have different dissolution and absorption in the body, which may affect the clinical efficacy and safety of the drug to a certain extent; especially for poorly soluble solid drugs, the crystal form will have greater influence. Therefore, the drug crystal form is inevitably an important part of drug research and an important part of drug quality control. Most importantly, the study of crystal forms is beneficial to find a crystal form that is clinically therapeutically meaningful and has stable and physicochemical properties.
There are no reports of CF-101 related crystal forms so far. Amorphous is generally not suitable as a medicinal form, and the molecules in the amorphous material are disorderly arranged, so they are in a thermodynamically unstable state. Amorphous solids are in a high-energy state, and generally have poor stability. During the production and storage process, amorphous drugs are prone to crystal transformation, which leads to the loss of consistency in drug bioavailability, dissolution rate, etc., resulting in changes in the clinical efficacy of the drug. In addition, the amorphous preparation is usually a rapid kinetic solid precipitation process, which easily leads to excessive residual solvents, and its particle properties are difficult to control by the process, making it a challenge in the practical application of the drug.
Therefore, there is a need to develop a crystalline form of CF-101 that provides a usable solid form for drug development. The inventors of the present application have unexpectedly discovered the crystalline forms CS1, CS2 and CS3 of Compound I, which have melting point, solubility, wettability, purification, stability, adhesion, compressibility, fluidity, dissolution in vitro and in vivo, and biological effectiveness. There is an advantage in at least one of the properties and formulation processing properties. Crystalline CS1 has advantages in physical and chemical properties, especially physical and chemical stability, low wettability, good solubility and good mechanical stability. It provides a new and better choice for the development of drugs containing CF-101, which is very important. The meaning.
Figure 7 is a 1 H NMR spectrum of the crystalline form CS3 obtained according to Example 7 of the present invention
The nuclear magnetic data of the crystalline form CS3 obtained in Example 7 was: { 1 H NMR (400 MHz, DMSO) δ 8.82 – 8.93 (m, 1H), 8.53 – 8.67 (m, 1H), 8.45 (s, 1H), 8.31 ( s, 1H), 7.73 (s, 1H), 7.59 (d, J = 7.7 Hz, 1H), 7.36 (d, J = 7.7 Hz, 1H), 7.11 (t, J = 7.8 Hz, 1H), 5.98 ( d, J = 7.4 Hz, 1H), 5.74 (s, 1H), 5.56 (s, 1H), 4.64 (d, J = 29.3 Hz, 3H), 4.32 (s, 1H), 4.15 (s, 1H), 2.71 (d, J = 4.6 Hz, 3H), 1.91 (s, 3H).}. Form CS3 has a single peak at 1.91, corresponding to the hydrogen chemical shift of the acetic acid molecule. According to the nuclear magnetic data, the molar ratio of acetic acid molecule to CF-101 is 1:1, and its 1 H NMR is shown in FIG.7
PAPER
Journal of medicinal chemistry (1994), 37(5), 636-46
Preparation Example 1: Synthesis of Compound (5) (S) -2 – ((R) -1- (2-Chloro-6- (3-iodobenzylamino) -9H- purin- Hydroxyethoxy) -3-hydroxy-N-methylpropanamide)
Scheme 1
Step 1: A solution of (2R, 3S, 4S, 5R) -2- (benzoyloxymethyl) -5- (2,6- dichloro-9H- purin-9- yl) tetrahydrofuran- Preparation of benzoate (7)
Starting material A mixture of (2R, 3R, 4S, 5R) -2-acetoxy-5- (benzoyloxymethyl) tetrahydrofuran-3,4-diyldibenzoate (7.5 g, 14.9 mmol) (3.09 g, 16.4 mmol) was dissolved in acetonitrile (50 mL), and a solution of N, O-bis (trimethylsilyl) acetamid (8.9 mL, 36.4 mmol) was slowly added dropwise for 10-15 minutes Then, the mixture is stirred at 60 DEG C for 30 minutes. After cooling the reaction solution to -30 ° C, TiCl 4 (60 mL, 1 M methylene chloride solution, 59.5 mmol) is added dropwise, and the mixture is stirred at 60-65 ° C for 20 minutes. After confirming the completion of the reaction, methylene chloride (500 mL) and saturated sodium hydrogencarbonate solution (500 mL) are added. The reaction solution was stirred at 0 ° C for 30 minutes, and the organic layer was extracted and dried over anhydrous magnesium sulfate. After concentration under reduced pressure, the obtained residue was separated by column chromatography to obtain the intermediate compound (2R, 3S, 4S, 5R) -2- (benzoyloxymethyl) -5- (2,6- dichloro- Yl) tetrahydrofuran-3,4-diyl dibenzoate (9.3 g, 98.8%).
The analytical data of the obtained compound are as follows.
The intermediate compound (204 mg, 0.32 mmol) and 3-iodobenzylamine hydrochloride (113 mg, 0.41 mmol) prepared in the above step 1 were dissolved in anhydrous ethanol (5 mL) under a nitrogen atmosphere, triethylamine (0.13 mL, 0.96 mmol) is stirred at room temperature for 24 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure, and the obtained residue was separated by column chromatography to obtain the intermediate compound (2R, 3S, 4S, 5R) -2- (benzoyloxymethyl) (3-iodobenzylamino) -9H-purin-9-yl) tetrahydrofuran-3,4-diyldibenzoate (230 mg, 86.14%).
The analytical data of the obtained compound are as follows.
The intermediate compound (20 g, 24.09 mmol) prepared in the above step 2 was dissolved in methanolic ammonia (1 L) and stirred at room temperature for 3 days. The reaction mixture was concentrated under reduced pressure to obtain a triol intermediate. The triol intermediate thus obtained (20 g, 38.63 mmol) was dissolved in anhydrous acetone (400 mL), and 2,2-dimethoxypropane (23.68 mL, 193.15 mmol) and p-toluenesulfonic acid monohydrate (7.34 g, 38.63 mmol) was added dropwise thereto, followed by stirring at room temperature for 12 hours. After confirming the completion of the reaction, saturated sodium hydrogencarbonate solution (400 mL) was added thereto. The reaction mixture was concentrated under reduced pressure. The organic layer was extracted with chloroform (4 x 250 mL), washed with a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. The reaction mixture was concentrated under reduced pressure, and the obtained residue was then separated by column chromatography to obtain the intermediate compound ((3aR, 4R, 6R, 6aR) -6- (2-Chloro-6- (3-iodobenzylamino) Yl] -2,2-dimethyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl) methanol (12 g, 89.35%).
The analytical data of the obtained compound are as follows.
The intermediate compound (15 g, 26.89 mmol) prepared in step 3 was dissolved in a solution of acetonitrile-water (130 mL, 1: 1) and then (diacetoxy iodo) -benzene (19 g, 59.16 mmol) 2,2,6,6-Tetramethyl 1-piperidinyloxyl (840 mg, 5.37 mmol) was added dropwise, followed by stirring at room temperature for 4 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain an acid intermediate without purification. The obtained intermediate (15 g, 26.23 mmol) was dissolved in anhydrous ethanol (500 mL) under a nitrogen stream, cooled to 0 ° C, thionyl chloride (9.52 mL, 131.17 mmol) was slowly added dropwise and the mixture was stirred at room temperature for 12 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure to obtain an ethyl ester intermediate without purification. Methylamine (750 mL, 2 N THF solution) was added dropwise to the resulting ethyl ester intermediate (15.5 g, 25.84 mmol) and the mixture was stirred at room temperature for 12 hours. After confirming the completion of the reaction, the reaction mixture was concentrated under reduced pressure. The obtained residue was purified by column chromatography to obtain the intermediate compound (2S, 5R) -5- (2-Chloro-6- (3- -9-yl) -3,4-dihydroxy-N-methyltetrahydrofuran-2-carboxamide (5 g, 31.80%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 )? Ppm 2.72-2.73 (d, J = 3.91 Hz, 3H), 4.17 (brs, 1H), 4.33 (s, 1H), 4.55-4.56 (D, J = 6.35 Hz, 1H, exchangeable with D 2 O, 2′-OH), 5.71-5.72 d, J = 3.91 Hz, 1H, exchangeable with d 2 O, 3′-OH), 5.92-5.93 (d, J = 7.33 Hz, 1H), 7.11-7.14 (t, J = 7.82 Hz, 1H), 7.35 (D, J = 6.84 Hz, 1H), 7.59-7.61 (d, J = 7.82 Hz, 1H), 7.75 (s, 1H), 8.27-8.28 exchangeable with D 2 O, NH), 8.48 (s, 1 H), 8.98-8.99 (br. t, J = 5.86 Hz, 1H, exchangeable with D 2 O, N 6 H); 13 C NMR (125 MHz; DMSO-d 6) [delta] 26.07, 43.02, 72.79, 73.42, 84.95, 88.10, 95.12, 119.46, 127.31, 130.99, 136.06, 136.51, 141.57, 142.23, 150.00, 153.44, 155.31, 170.14; mp = 207-209 [deg.] C.
The intermediate compound (2.0 g, 3.67 mmol) prepared in step 4 was dissolved in water / methanol (210 mL, 1: 2), cooled to 0 ° C and then sodium per iodate (1.57 g, 7.34 mmol) And then stirred at the same temperature for 2 hours. After completion of the reaction was confirmed, sodium borohydride (694 mg, 18.35 mmol) was added and stirred for 1 hour. After confirming the completion of the reaction, the reaction mixture was concentrated under reduced pressure, and the residue was concentrated under reduced pressure using toluene (3 x 50 mL). The residue was separated by column chromatography to obtain the title compound (1.58 g, 79%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 ) δ ppm 2.40 (.. Br s, 3H), 3.53-3.60 (m, 1H), 3.71-3.73 (d, J = 10.27 Hz, 1H), 3.85 (br . s., 1H), 3.95 (br. s., 2H), 4.60 (br. s., 2H), 4.98-5.00 (t, J = 5.38 Hz, 1H, exchangeable with D 2 O, OH), 5.19 -5.20 (t, J = 5.86 Hz, 1H, exchangeable with D 2 O, OH), 5.78-5.80 (t, J = 5.38 Hz, 1H), 7.10-7.13 (t, J = 7.82Hz, 1H), 7.35 -7.36 (d, J = 6.84Hz, 1H), 7.59-7.60 (d, J = 5.86 Hz, 2H, exchangeable with D 2 O, NH), 7.73 (br. s., 1H), 8.30 (s, 1H ), 8.85 (br s, 1H, exchangeable with D 2 O, NH); 13 C NMR (125 MHz; DMSO-d 6) [delta] 25.59, 42.98, 62.02, 62.25, 80.43, 84.90, 95.11, 118.57, 127.27, 130.96, 136.03, 136.45, 140.78, 142.34, 150.69, 153.53, 155.17, 169.31; HRMS (FAB) m / z calcd for C 18 H20 ClIN 6 O 4 [M + Na] + 546.0279, found 569.0162; mp = 226-229 [deg.] C.
Example 2
Preparation Example 2: Synthesis of Compound (11) ((R) -2- (1- (2-Chloro-6- (3-iodobenzylamino) -9H- purin-9-yl) -2- hydroxyethoxy) Propane-1,3-diol)
Scheme 2
The intermediate compound (230 mg, 0.27 mmol) prepared in Step 2 of Example 1 was dissolved in methanolic ammonia (25 mL) and stirred at room temperature for 3 days. The reaction mixture was concentrated under reduced pressure to obtain a triol intermediate. The obtained triol intermediate (248 mg, 0.47 mmol) was treated in the same manner as in Step 5 of Example 1 to obtain the desired compound (109 mg, 75.69%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 )? Ppm 3.13-3.17 (m, 1H), 3.22-3.26 (m, 1H), 3.43-3.47 (m, 2H), 3.54-3.56 (Br s, 2H), 4.41-4.42 (br t, J = 5.38 Hz, 1H, exchangeable with D 2 O, OH), 4.60 , J = 5.38 Hz, 1H, exchangeable with D 2 O, OH), 5.13 (br. s., 1H, exchangeable with D 2 O, OH), 5.80-5.82 (t, J = 4.89 Hz, 1H), 7.11 (D, J = 7.33 Hz, 1H), 7.36-7.37 (d, J = 7.33 Hz, 1H), 7.59-7.60 s, 1 H), 8.82 (br s, 1H, exchangeable with D 2 O, NH); 13 C NMR (125 MHz; DMSO-d 6) [delta] 43.01, 61.12, 61.23, 62.64, 80.90, 84.53, 95.12, 118.56, 127.36, 130.99, 136.04, 136.58, 140.68, 142.44, 150.73, 153.40, 155.12; HRMS (FAB) m / z calcd for C 17 H 19 ClIN 5 O 4 [M + Na] + 519.0170, found 542.0054; mp = 170-172 [deg.] C.
(Hydroxymethyl) tetrahydrofuran-3,4-diol (4.8 g, 16.74 mmol) and 2,2 & lt; RTI ID = 0.0 & -Dimethoxypropane (10.26 mL, 83.71 mmol) was dissolved in anhydrous acetone (120 mL) under a nitrogen stream, p-toluenesulfonic acid monohydrate (3.18 g, 16.74 mmol) was added dropwise and the mixture was stirred at room temperature for 4 hours . After confirming the completion of the reaction, the reaction is terminated with a saturated sodium hydrogencarbonate solution. The reaction solution was concentrated under reduced pressure, and the organic layer was extracted with chloroform (4 x 20 mL), washed with a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. After concentration under reduced pressure, the obtained residue was separated by column chromatography to obtain the intermediate compound ((3aR, 4R, 6R, 6aR) -6- (6-Chloro-9H-purin-9- 3,4-d] [1,3] dioxol-4-yl) methanol (4.87 g, 89.03%).
The analytical data of the obtained compound are as follows.
The intermediate compound (2.8 g, 8.56 mmol) prepared in Step 1 was dissolved in anhydrous methylene chloride (100 mL), and then cooled to 0 ° C. Triethylamine (3.6 mL, 25.70 mmol) and dimethylaminopyridine (21 mg, 0.17 mmol). Benzoyl chloride (1.5 mL, 12.85 mmol) is slowly added dropwise at the same temperature and then stirred at room temperature for 2 hours. After confirming the completion of the reaction, the reaction is terminated with a saturated sodium hydrogencarbonate solution. The reaction solution was concentrated under reduced pressure, the organic layer was extracted with methylene chloride, washed with a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. After concentration under reduced pressure, the obtained residue was separated by column chromatography to obtain the intermediate compound ((3aR, 4R, 6R, 6aR) -6- (6-Chloro-9H-purin-9- 3,4-d] [1,3] dioxol-4-yl) methyl benzoate (3.68 g, 99.72%).
The analytical data of the obtained compound are as follows.
The intermediate compound (1.24 g, 2.87 mmol) prepared in the above step 2 was prepared in the same manner as in step 2 of Example 1 to give the intermediate compound ((3aR, 4R, 6R, 6aR) -6- (6- Yl) -2,2-dimethyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl) methyl benzoate (1.73 g, 96.11 %).
The analytical data of the obtained compound are as follows.
The intermediate compound (4.93 g, 7.85 mmol) prepared in the above step 3 was dissolved in 80% acetic acid (250 mL), and the mixture was refluxed at 100 ° C for 12 hours. After completion of the reaction was confirmed, the reaction solution was concentrated under reduced pressure, toluene (4 x 50 mL) was added, and the filtrate was concentrated under reduced pressure to obtain a diol intermediate without purification. The obtained diol intermediate (8.5 g, 14.47 mmol) was dissolved in anhydrous pyridine (250 mL), followed by addition of tetrabutyldimethylsilyl triflate (TBDMSOTf) (13.3 mL, 57.88 mmol) followed by stirring at 50 ° C for 5 hours. After confirming the completion of the reaction, the reaction solution was partitioned into methylene chloride / water. The organic layer was washed with water, saturated sodium hydrogencarbonate solution and saturated saturated sodium bicarbonate solution, and then dried over anhydrous magnesium sulfate. After concentration under reduced pressure, the obtained residue was separated by column chromatography to obtain the intermediate compound ((2R, 3R, 4R, 5R) -3,4-bis (tert-butyldimethylsilyloxy) -9H-purin-9-yl) tetrahydrofuran-2-yl) methylbenzoate (4.47 g, 69.73%).
The analytical data of the obtained compound are as follows.
The intermediate compound (1.28 g, 1.56 mmol) prepared in step 4 was dissolved in anhydrous methanol (100 mL), 25% sodium methoxide / methanol (15 mL) was added, and the mixture was stirred at room temperature for 12 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure, and the obtained residue was separated by column chromatography to obtain the intermediate compound ((2R, 3R, 4R, 5R) -3,4- bis (tert- butyldimethylsilyloxy) Yl) tetrahydrofuran-2-yl) methanol (960 mg, 86.48%) was obtained as a pale-yellow amorphous solid.
The analytical data of the obtained compound are as follows.
The intermediate compound (450 mg, 0.63 mmol) prepared in Step 5 and pyridinium dichromate (5.47 g, 14.54 mmol) were dissolved in DMF (50 mL) under a nitrogen stream, followed by stirring at room temperature for 12 hours. After confirming completion of the reaction, the resulting solid was washed with water to obtain an acid intermediate. The obtained intermediate (450 mg, 0.62 mmol) was dissolved in anhydrous ethanol (10 mL) under a nitrogen stream, cooled to 0 ° C, thionyl chloride (0.25 mL, 3.10 mmol) was slowly added dropwise and the mixture was stirred at room temperature for 5 hours Lt; / RTI & gt; After completion of the reaction was confirmed, the reaction solution was concentrated under reduced pressure, and the residue was partitioned into ethyl acetate / water. The organic layer was washed with water and saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. After concentration under reduced pressure, an intermediate ethyl ester was obtained. The ethyl ester thus obtained is added with a methylamine / 2N-THF solution under a nitrogen stream, followed by stirring at room temperature for 12 hours. After confirming the completion of the reaction, the reaction mixture was concentrated under reduced pressure and the residue was purified by column chromatography to obtain the intermediate compound (2S, 5R) -3,4-bis (tert-butyldimethylsilyloxy) -5- -9H-purin-9-yl) -N-methyltetrahydrofuran-2-carboxamide (400 mg, 85.65%).
The analytical data of the obtained compound are as follows.
The intermediate compound (65 mg, 0.08 mmol) prepared in Step 6 was dissolved in anhydrous THF under a nitrogen stream, and then tetrabutylammonium fluoride (TBAF) (0.44 mL, 0.43 mmol, (1 M solution THF) Stir at room temperature for 1 hour. After confirming the completion of the reaction, the reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by column chromatography to obtain the intermediate compound (2S, 5R) -3,4-dihydroxy-5- (6- (3-iodobenzylamino) -9H-purin-9-yl) -N-methyltetrahydrofuran-2-carboxamide (52 mg, 95.45%).
The analytical data of the obtained compound are as follows.
Step 8: (S) -3-Hydroxy-2 – ((R) -2-hydroxy-1- (6- (3-iodobenzylamino) -9H- purin- Preparation of N-methylpropanamide (12)
The intermediate compound (52 mg, 0.10 mmol) prepared in the above Step 7 was treated in the same manner as in Step 5 of Example 1 to obtain the title compound (35 mg, 83.33%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 .) Δ ppm 2.35 (s, 3H), 3.5-3.6 (m, 1H), 3.72-3.74 (d, J = 9.29 Hz, 1H), 3.86 (br s. 2H), 4.99 (br s, 1H, exchangeable with D2O, OH), 5.21 (br. S., 1H), 4.00 (d, J = (d, J = 6.84 Hz, 1H), 7.56 d, J = 6.35 Hz, 2H, exchangeable with D 2 O, NH), 7.71 (s, 1H), 8.22 (s, 1H), 8.30 (s, 1H), 8.37 (br. s., 1H, exchangeable with D2O, NH); 13 C NMR (125 MHz; DMSO-d 6 ) δ 25.53, 42.81, 61.98, 62.26, 80.30, 84.62, 95.09, 119.43, 127.11, 130.91, 135.81, 136.16, 140.19, 143.31, 149.79, 152.98, 154.66, 169.42; HRMS (FAB) m / z calcd for C 18H 21 IN 6 O 4 [M + Na] +512.0669, found 535.0578; mp = 176-182 [deg.] C.
Example 4
Production Example 4: Synthesis of Compound (21) ((R) -2- (2-hydroxy-1- (6- (3-iodobenzylamino) -9H- purin- 3-diol)
The intermediate compound (1.73 g, 2.75 mmol) prepared in the step 2 of Example 1 was treated in the same manner as in the step 5 of Example 3 with (3aR, 4R, 6R, 6aR) -6- L, 3-dioxol-4-yl) methanol (1.04 g, 93.69 & lt; RTI ID = 0.0 & gt; %).
The analytical data of the obtained compound are as follows.
The intermediate compound (250 mg, 0.47 mmol) prepared in the above step 1 was dissolved in 80% acetic acid (250 mL), and the mixture was heated under reflux at 100 ° C for 12 hours. After confirming the completion of the reaction, the reaction solution was concentrated under reduced pressure, toluene (4 x 50 mL) was added, and the mixture was concentrated under reduced pressure. The obtained residue was purified by column chromatography to obtain the intermediate (2R, 3S, 4R, Methyl) -5- (6- (3-iodobenzylamino) -9H-purin-9-yl) tetrahydrofuran-3,4-diol (177 mg, 76.95%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; DMSO-d 6 )? Ppm 3.56 (br s, 1H), 3.67-3.69 (d, J = 10.27 Hz, 1H), 3.97 ), 4.61-4.67 (m, 3H), 5.17 (d, J = 2.93 Hz, 1H, exchangeable with D 2 O, OH), 5.35-5.36 (t, J = 5.38 Hz, 1H, exchangeable with D 2 O, OH), 5.43-5.44 (d, J = 5.38 Hz, 1H, exchangeable with d 2O, OH), 5.89-5.90 (d, J = 4.89 Hz, 1H), 7.09-7.11 (t, J = 7.33 Hz, 1H), 7.35-7.36 (d, J = 6.84 Hz, 1H), 7.56-7.58 (d, J = 7.33 Hz, 1H), 7.72 ), 8.45 (br s, 1H, exchangeable with D 2 O, NH); 13 C NMR (125 MHz; DMSO-d 6) [delta] 42.69, 62.08, 71.06, 73.97, 86.32, 88.42, 95.09, 120.24, 127.08, 130.91, 135.81, 136.16, 140.47, 143.22, 149.00, 152.76, 154.79; mp = 174-178 [deg.] C.
The intermediate compound (77 mg, 0.15 mmol) prepared in the above Step 2 was treated in the same manner as in Step 5 of Example 1 to obtain the desired compound (62 mg, 80.51%).
The analytical data of the obtained compound are as follows.
1 H NMR (500 MHz; CD 3 OD)? Ppm 3.42 – 3.44 (d, J = 5.38 Hz, 2H), 3.54-3.58 (m, 1H), 3.65-3.68 ), 3.75-3.78 (dd, J = 11.73,4.44 Hz, 1H), 4.01-4.02 (d, J = 5.38 Hz, 2H), 4.53 (s, 2H), 6.04-6.06 J = 7.82 Hz, 1H), 7.76 (s, 1H), 7.07-7.10 (t, J = 7.82 Hz, 1H), 7.38-7.40 (d, J = 7.82 Hz, 1H), 7.59-7.60 ), 8.27 (s, 1 H), 8.29 (s, 1 H); 13 C NMR (125 MHz; CD 3 OD)? 42.88, 60.80, 61.25, 62.76, 80.26, 84.02, 93.52, 119.00, 126.44, 129.93, 135.90, 136.10, 139.65, 141.72, 148.97, 152.52, 154.53; HRMS (FAB) m / z calcd for C 17 H 20 IN 5 O 4 [M + H] +485.0560, found 486.0625; mp = 72-76 [deg.] C.
PATENT
WO 2008111082
REFERENCES
1: Avni I, Garzozi HJ, Barequet IS, Segev F, Varssano D, Sartani G, Chetrit N, Bakshi E, Zadok D, Tomkins O, Litvin G, Jacobson KA, Fishman S, Harpaz Z, Farbstein M, Yehuda SB, Silverman MH, Kerns WD, Bristol DR, Cohn I, Fishman P. Treatment of Dry Eye Syndrome with Orally Administered CF101 Data from a Phase 2 Clinical Trial. Ophthalmology. 2010 Mar 19. [Epub ahead of print] PubMed PMID: 20304499.
2: Bar-Yehuda S, Rath-Wolfson L, Del Valle L, Ochaion A, Cohen S, Patoka R, Zozulya G, Barer F, Atar E, Piña-Oviedo S, Perez-Liz G, Castel D, Fishman P. Induction of an antiinflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum. 2009 Oct;60(10):3061-71. PubMed PMID: 19790055.
3: Borea PA, Gessi S, Bar-Yehuda S, Fishman P. A3 adenosine receptor: pharmacology and role in disease. Handb Exp Pharmacol. 2009;(193):297-327. Review. PubMed PMID: 19639286.
4: Moral MA, Tomillero A. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2008 Mar;30(2):149-71. PubMed PMID: 18560631.
5: Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J Rheumatol. 2008 Jan;35(1):41-8. Epub 2007 Nov 15. PubMed PMID: 18050382
Class Antihyperglycaemics; Antihypertensives; Pyrroles; Small molecules; Sulfones
Mechanism of Action Mineralocorticoid receptor antagonists
Registered Hypertension
Phase III Diabetic nephropathies
No development reported Cardiovascular disorders; Heart failure
09 Jan 2019 Registered for Hypertension in Japan (PO) – First global approval
27 Nov 2018 Daiichi Sankyo completes a phase III trial in Diabetic nephropathies in Japan (PO) (JapicCTI-173696)
08 Jun 2018 Efficacy and adverse events data from the phase III ESAX-HTN trial in Essential hypertension presented 28th European Meeting on Hypertension and Cardiovascular Protection (ESH-2018)
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease similar to olmesartan , losartan, candesartan , valsartan, irbesartan, telmisartan, eprosartan,
Cas name 1H-Pyrrole-3-carboxamide, 1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-, (5S)-
Esaxerenone 1632006-28-0, FDA approved new drug will be a big potential drug. Original Route of Synthesis …
Esaxerenone, also known as CS-3150, XL-550, is a nonsteroidal antimineralocorticoid which was discovered by Exelixis and is now under development by Daiichi Sankyo Company for the treatment of hypertension, essential hypertension, hyperaldosteronism, and diabetic nephropathies. It acts as a highly selective silent antagonist of the mineralocorticoid receptor (MR), the receptor for aldosterone, with greater than 1,000-fold selectivity for this receptor over other steroid hormone receptors, and 4-fold and 76-fold higher affinity for the MR relative to the existing antimineralocorticoids spironolactone and eplerenone.
01 Jan 2015Daiichi Sankyo initiates a phase IIb trial for Diabetic nephropathies in Japan (NCT02345057)
01 Jan 2015Daiichi Sankyo initiates a phase IIb trial for Hypertension in Japan (NCT02345044)
01 May 2013Phase-II clinical trials in Diabetic nephropathies in Japan (PO)
Currently, angiotensin II receptor antagonists and calcium antagonists are widely used as a medicament for the treatment or prevention of such hypertension or heart disease.
Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
Renin – angiotensin II receptor antagonists are inhibitors of angiotensin system is particularly effective in renin-dependent hypertension, and show a protective effect against cardiovascular and renal failure. Also, the calcium antagonists, and by the function of the calcium channel antagonizes (inhibits), since it has a natriuretic action in addition to the vasodilating action, is effective for hypertension fluid retention properties (renin-independent) .
Therefore, the MR antagonist, when combined angiotensin II receptor antagonists or calcium antagonists, it is possible to suppress the genesis of multiple hypertension simultaneously, therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology is expected to exhibit.
Also, diuretics are widely used as a medicament for the treatment or prevention of such hypertension or heart disease. Diuretic agent is effective in the treatment of hypertension from its diuretic effect. Therefore, if used in combination MR antagonists and diuretics, the diuretic effect of diuretics, it is possible to suppress the genesis of multiple blood pressure at the same time, shows a therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology it is expected.
1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (hereinafter, compound ( I)) is, it is disclosed in Patent Documents 1 and 2, hypertension, for the treatment of such diabetic nephropathy are known to be useful.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Useful as a mineralocorticoid receptor (MR) antagonist, for treating hypertension, cardiac failure and diabetic nephropathy. It is likely to be CS-3150, a non-steroidal MR antagonist, being developed by Daiichi Sankyo (formerly Sankyo), under license from Exelixis, for treating hypertension and diabetic nephropathy (phase 2 clinical, as of March 2015). In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month (NCT02345057).
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Daiichi Sankyo (formerly Sankyo), under license from Exelixis, is developing CS-3150 (XL-550), a non-steroidal mineralocorticoid receptor (MR) antagonist, for the potential oral treatment of hypertension and diabetic nephropathy, microalbuminuria , By October 2012, phase II development had begun ; in May 2014, the drug was listed as being in phase IIb development . In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month. At that time, the trial was expected to complete in March 2017 .
Exelixis, following its acquisition of X-Ceptor Therapeutics in October 2004 , was investigating the agent for the potential treatment of metabolic disorders and cardiovascular diseases, such as hypertension and congestive heart failure . In September 2004, Exelixis expected to file an IND in 2006. However, it appears that the company had fully outlicensed the agent to Sankyo since March 2006 .
Description
Small molecule antagonist of the mineralocorticoid receptor (MR)
In January 2015, a multi-center, placebo-controlled, randomized, 5-parallel group, double-blind, phase II trial (JapicCTI-152774; NCT02345057; CS3150-B-J204) was planned to be initiated later that month in Japan, in patients with type 2 diabetes mellitus and microalbuminuria, to assess the efficacy and safety of different doses of CS-3150 compared to placebo. At that time, the trial was expected to complete in March 2017; later that month, the trial was initiated in the Japan
By October 2012, phase II development had begun in patients with essential hypertension
By January 2011, phase I trials had commenced in Japan
After methyl 4-methyl-5-[2-(trifluoromethyl) phenyl]-1H-pyrrole-3-carboxylate was obtained by the method described in Example 16 of WO 2006/012642 , the following reaction was performed using this compound as a raw material.
Methyl 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate (1.4 g, 4.9 mmol) was dissolved in methanol (12 mL), and a 5 M aqueous sodium hydroxide solution (10 mL) was added thereto, and the resulting mixture was heated under reflux for 3 hours. After the mixture was cooled to room temperature, formic acid (5 mL) was added thereto to stop the reaction. After the mixture was concentrated under reduced pressure, water (10 mL) was added thereto to suspend the resulting residue. The precipitated solid was collected by filtration and washed 3 times with water. The obtained solid was dried under reduced pressure, whereby 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylic acid (1.1 g, 83%) was obtained as a solid. The thus obtained solid was suspended in dichloromethane (10 mL), oxalyl chloride (0.86 mL, 10 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 2 hours. After the mixture was concentrated under reduced pressure, the residue was dissolved in tetrahydrofuran (10 mL), and 4-(methylsulfonyl)aniline hydrochloride (1.0 g, 4.9 mmol) and N,N-diisopropylethylamine (2.8 mL, 16 mmol) were sequentially added to the solution, and the resulting mixture was heated under reflux for 18 hours. After the mixture was cooled to room temperature, the solvent was distilled off under reduced pressure, and acetonitrile (10 mL) and 3 M hydrochloric acid (100 mL) were added to the residue. A precipitated solid was triturated, collected by filtration and washed with water, and then, dried under reduced pressure, whereby 4-methyl-N-[4-(methylsulfonyl) phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (1.4 g, 89%) was obtained as a solid. 1H-NMR (400 MHz, DMSO-d6) δ11.34 (1H, brs,), 9.89 (1H, s), 7.97 (2H, d, J = 6.6 Hz), 7.87-7.81 (3H, m), 7.73 (1H, t, J = 7.4 Hz), 7.65-7.61 (2H, m), 7.44 (1H, d, J = 7.8 Hz), 3.15 (3H, s), 2.01 (3H, s).
Sodium hydride (0.12 g, 3 mmol, 60% dispersion in mineral oil) was dissolved in N,N-dimethylformamide (1.5 mL), and 4-methyl -N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (0.47 g, 1.1 mmol) was added thereto, and then, the resulting mixture was stirred at room temperature for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (0.14 g, 1.2 mmol) was added thereto, and the resulting mixture was stirred at room temperature. After 1 hour, sodium hydride (40 mg, 1.0 mmol, oily, 60%) was added thereto again, and the resulting mixture was stirred for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (12 mg, 0.11 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, methanol (5 mL) was added to the residue and insoluble substances were removed by filtration, and the filtrate was concentrated again. To the residue, tetrahydrofuran (2 mL) and 6 M hydrochloric acid (2 mL) were added, and the resulting mixture was stirred at 60°C for 16 hours. The reaction was cooled to room temperature, and then dissolved in ethyl acetate, and washed with water and saturated saline. The organic layer was dried over anhydrous sodium sulfate and filtered. Then, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate), whereby the objective compound (0.25 g, 48%) was obtained. 1H-NMR (400 MHz, CDCl3) δ: 7.89-7.79 (m, 6H), 7.66-7.58 (m, 2H), 7.49 (s, 1H), 7.36 (d, 1H, J = 7.4Hz), 3.81-3.63 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1246.
Anal. calcd for C22H21F3N2O4S: C, 56.65; H, 4.54; N, 6.01; F, 12.22; S, 6.87. found: C, 56.39; H, 4.58; N, 5.99; F, 12.72; S, 6.92.
(Example 4)
Optical Resolution of Compound of Example 3
Resolution was performed 4 times in the same manner as in Example 2, whereby 74 mg of Isomer C was obtained as a solid from a fraction containing Isomer C (tR = 10 min), and 71 mg of Isomer D was obtained as a solid from a fraction containing Isomer D (tR = 11 min).
1- [2- (trifluoromethyl) phenyl] propan-1-one 75 g (370 mmol) in t- butyl methyl ether (750 mL), and I was added bromine 1.18 g (7.4 mmol). After confirming that the stirred bromine color about 30 minutes at 15 ~ 30 ℃ disappears, cooled to 0 ~ 5 ℃, was stirred with bromine 59.13 g (370 mmol) while keeping the 0 ~ 10 ℃. After stirring for about 2.5 hours, was added while maintaining 10 w / v% aqueous potassium carbonate solution (300 mL) to 0 ~ 25 ℃, was further added sodium sulfite (7.5 g), was heated to 20 ~ 30 ℃. The solution was separated, washed in the resulting organic layer was added water (225 mL), to give t- butyl methyl ether solution of the title compound and the organic layer was concentrated under reduced pressure (225 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.91 (3H, D, J = 4.0 Hz), 4.97 (1H, Q, J = 6.7 Hz), 7.60 ~ 7.74 (4H, M).
2-bromo-1- [2- (trifluoromethyl) phenyl] propan-1 / t- butyl methyl ether solution (220 mL) in dimethylacetamide (367 mL), ethyl cyanoacetate obtained in Example 1 53.39 g (472 mmol), potassium carbonate 60.26 g (436 mmol) were sequentially added, and the mixture was stirred and heated to 45 ~ 55 ℃. After stirring for about 2 hours, 20 is cooled to ~ 30 ℃, water (734 mL) and then extracted by addition of toluene (367 mL), washed by adding water (513 mL) was carried out in the organic layer (2 times implementation). The resulting organic layer was concentrated under reduced pressure to obtain a toluene solution of the title compound (220 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.33 ~ 1.38 (6H, M), 3.80 ~ 3.93 (2H, M), 4.28 ~ 4.33 (2H, M), 7.58 ~ 7.79 (4H, M).
The 20 ~ 30 ℃ 2-cyano-3-methyl-4-oxo-4 was obtained [2- (trifluoromethyl) phenyl] butanoate in toluene (217 mL) by the method of Example 2 ethyl acetate (362 mL) Te, after the addition of thionyl chloride 42.59 g (358 mmol), cooled to -10 ~ 5 ℃, was blown hydrochloric acid gas 52.21 g (1432 mmol), further concentrated sulfuric acid 17.83 g (179 mmol) was added, and the mixture was stirred with hot 15 ~ 30 ℃. After stirring for about 20 hours, added ethyl acetate (1086 mL), warmed to 30 ~ 40 ℃, after the addition of water (362 mL), and the layers were separated. after it separated organic layer water (362 mL) was added for liquid separation, and further 5w / v% was added for liquid separation aqueous sodium hydrogen carbonate solution (362 mL).
Subsequently the organic layer was concentrated under reduced pressure, the mixture was concentrated under reduced pressure further added toluene (579 mL), was added toluene (72 mL), and cooled to 0 ~ 5 ℃. After stirring for about 2 hours, the precipitated crystals were filtered, and washed the crystals with toluene which was cooled to 0 ~ 5 ℃ (217 mL). The resulting wet goods crystals were dried under reduced pressure at 40 ℃, the title compound was obtained (97.55 g, 82.1% yield).
Example obtained by the production method of the three 2-chloro-4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylate 97.32 g (293 mmol) in ethanol (662 mL), tetrahydrofuran (117 mL), water (49 mL), sodium formate 25.91 g (381 mmol) and 5% palladium – carbon catalyst (water content 52.1%, 10.16 g) was added at room temperature, heated to 55 ~ 65 ℃ the mixture was stirred. After stirring for about 1 hour, cooled to 40 ℃ less, tetrahydrofuran (97 mL) and filter aid (KC- flock, Nippon Paper Industries) 4.87 g was added, the catalyst was filtered and the residue using ethanol (389 mL) was washed. The combined ethanol solution was used for washing the filtrate after concentration under reduced pressure, and with the addition of water (778 mL) was stirred for 0.5 hours at 20 ~ 30 ℃. The precipitated crystals were filtered, and washed the crystals with ethanol / water = 7/8 solution was mixed with (292 mL). The resulting wet goods crystals were dried under reduced pressure at 40 ℃, the title compound was obtained (86.23 g, 98.9% yield).
N to the fourth embodiment of the manufacturing method by the resulting 4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylate 65.15 g (219 mmol), N- dimethylacetamide ( 261 mL), ethylene carbonate 28.95 g (328.7 mmol), 4- dimethylaminopyridine 2.68 g (21.9 mmol) were sequentially added at room temperature, and heated to 105 ~ 120 ℃, and the mixture was stirred. After stirring for about 10 hours, toluene was cooled to 20 ~ 30 ℃ (1303 mL), and the organic layer was extracted by adding water (326 mL). Subsequently, was washed by adding water (326 mL) to the organic layer (three times). The resulting organic layer was concentrated under reduced pressure, ethanol (652 mL) was added, and was further concentrated under reduced pressure, ethanol (130 mL) was added to obtain an ethanol solution of the title compound (326 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.35 (3H, t, J = 7.1 Hz), 1.84 (1H, Broad singlet), 2.00 (3H, s), 3.63 ~ 3.77 (4H, M), 4.27 (2H , m), 7.35 ~ 7.79 (5H, m).
Obtained by the method of Example 5 (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl / ethanol (321 mL) solution in water (128.6 mL), was added at room temperature sodium hydroxide 21.4 g (519 mmol), and stirred with heating to 65 ~ 78 ℃. After stirring for about 6 hours, cooled to 20 ~ 30 ℃, after the addition of water (193 mL), and was adjusted to pH 5.5 ~ 6.5, while maintaining the 20 ~ 30 ℃ using 6 N hydrochloric acid. was added as seed crystals to the pH adjustment by a liquid (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 6.4 mg , even I was added to water (193mL). Then cooled to 0 ~ 5 ℃, again, adjusted to pH 3 ~ 4 with concentrated hydrochloric acid and stirred for about 1 hour. Then, filtered crystals are precipitated, and washed the crystals with 20% ethanol water is cooled to 0 ~ 5 ℃ (93 mL). The resulting wet product crystals were dried under reduced pressure at 40 ℃, to give the title compound (64.32 g, 95.0% yield). 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.87 (3H, s), 3.38 ~ 3.68 (4H, M), 7.43 ~ 7.89 (5H, M).
(Example 7)
(S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
Quinine 31.05 g (96 mmol) in N, N- dimethylacetamide (25 mL), ethyl acetate (350 mL), was heated in water (15 mL) 65 ~ 70 ℃ was added, was added dropwise a solution 1. After about 1 hour stirring the mixture at 65 ~ 70 ℃, and slowly cooled to 0 ~ 5 ℃ (cooling rate standard: about 0.3 ℃ / min), and stirred at that temperature for about 0.5 hours. The crystals were filtered, 5 ℃ using ethyl acetate (100 mL) which was cooled to below are washed crystals, the resulting wet product crystals was obtained and dried under reduced pressure to give the title compound 43.66 g at 40 ℃ (Yield 42.9%). Furthermore, the diastereomeric excess of the obtained salt was 98.3% de. 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.30 ~ 2.20 (10H, M), 2.41 ~ 2.49 (2H, M), 2.85 ~ 3.49 (6H, M), 3.65 ~ 3.66 (1H, M), 3.88 (3H, s), 4.82 (1H, broad singlet), 4.92 ~ 5.00 (2H, m), 5.23 ~ 5.25 (1H, m), 5.60 (1H, br), 5.80 ~ 6.00 (1H, m), 7.36 ~ 7.92 (9H, M), 8.67 (1H, D, J = 4.6 Hz) (7-2) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3 diastereomeric excess of the carboxylic acid quinine salt HPLC measurements (% de) that the title compound of about 10 mg was collected, and the 10 mL was diluted with 50v / v% aqueous acetonitrile me was used as a sample solution.
Column: DAICEL CHIRALPAK IC-3 (4.6 mmI.D. × 250 mm, 3 μm)
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
[Table 1]
Detection: UV 237 nm
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
(Example 8)
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxamide (Compound (A))
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
(8-2) N – {[4- (methylsulfonyl) phenyl] amino} oxamic acid 2 – ((S) -3- methyl-4 – {[4- (methylsulfonyl) phenyl] carbamoyl} -2- [ 2- (trifluoromethyl) phenyl] -1H- pyrrol-1-yl) ethyl
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
After completion concentration under reduced pressure, acetonitrile (200 mL) was added and cooled to 10 ~ 15 ℃ (reaction 1).
Acetonitrile (240mL), pyridine 12.39 g (157 mmol), 4- were successively added (methylsulfonyl) aniline 26.85 g (157 mmol), the reaction solution 1 was added while maintaining the 10 ~ 15 ℃, the 20 ~ 25 ℃ and the mixture was stirred and heated to about 1 hour.
The resulting reaction solution in acetonitrile (40 mL), 2 N hydrochloric acid water (120 mL), was added sodium chloride (10.0 g) was stirred, and the layers were separated. Again, 2N aqueous hydrochloric acid to the organic layer (120 mL), was added sodium chloride (10.0 g) was stirred, and the layers were separated. After filtering the resulting organic layer was concentrated under reduced pressure (400 mL). Water (360 mL) was added to the concentrated liquid, after about 1 hour stirring, the crystals were filtered, washed with 50v / v% aqueous acetonitrile (120 mL), wet product of the title compound (undried product, 62.02 g) and obtained. 1 H NMR (500 MHz, DMSO-D 6 ) delta: 1.94 (s, 3H), 3.19 (s, 3H), 3.20 (s, 3H), 3.81 (t, 1H), 4.12 (t, 1H), 4.45 ( t, 2H, J = 5.81 Hz), 7.62 (t, 1H, J = 4.39 Hz), 7.74 (t, 2H, J = 3.68 Hz), 7.86 (dd, 3H), 7.92 (dd, 3H, J = 6.94 , 2.13 Hz), 7.97 (DD, 2H, J = 6.80, 1.98 Hz), 8.02 (DD, 2H), 10.03 (s, 1H), 11.19 (s, 1H)
(8-3) (S)-1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (Compound (A)) ( the resulting wet product crystals 8-2), t- butyl methyl ether (200 mL), acetonitrile (40 mL), 48w / w potassium hydroxide aqueous solution (16 g) and water (200 mL) was added, I was stirred for about 2 hours at 25 ~ 35 ℃. After stirring, and the mixture is separated, the resulting organic layer was concentrated under reduced pressure (120 mL), ethanol (240 mL) was added and further concentrated under reduced pressure (120 mL). After completion concentration under reduced pressure, ethanol (36 mL), and heated in water (12 mL) was added 35 ~ 45 ℃, while maintaining the 35 ~ 45 ℃ was added dropwise water (280 mL), and was crystallized crystals. After cooling the crystal exudates to room temperature, I was filtered crystal. Then washed with crystals 30v / v% aqueous ethanol solution (80 mL), where it was dried under reduced pressure at 40 ℃, the title compound was obtained in crystalline (26.26 g, 89.7% yield). Moreover, the enantiomers of the resulting crystals was 0.3%.
(8-4) (S)-1-(2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3- HPLC method for measuring the amount enantiomer carboxamide (%) and collected the title compound of about 10 mg is, what was the 10 mL was diluted with 50v / v% aqueous acetonitrile to obtain a sample solution.
(12-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H – pyrrole-3-carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
(12-2) N – {[4- (methylsulfonyl) phenyl] amino} oxamic acid 2 – ((S) -3- methyl-4 – {[4- (methylsulfonyl) phenyl] carbamoyl} -2- [ 2- (trifluoromethyl) phenyl] -1H- pyrrol-1-yl) ethyl
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
After concentration under reduced pressure end, is added acetonitrile (10 mL) and oxalyl chloride 0.10 g (1 mmol), and cooled to 0 ~ 5 ℃ (reaction 1).
Acetonitrile (30mL), pyridine 3.15 g (40 mmol), 4- were successively added (methylsulfonyl) aniline 6.71 g (39 mmol), the reaction solution 1 was added while maintaining the 10 ~ 15 ℃, the 20 ~ 25 ℃ and the mixture was stirred and heated to about 1 hour.
Insolubles from the resulting reaction solution was filtered, washed with acetonitrile (10 mL), and stirred for about 2 hours the addition of water (15 mL), followed by dropwise addition of water (75 mL) over about 1 hour . After about 1 hour stirring the suspension was filtered crystals were washed with 50v / v% aqueous acetonitrile (20 mL), wet product of the title compound (undried product, 15.78 g) to give a. 1 H NMR (500 MHz, DMSO-D 6 ) delta: 1.94 (s, 3H), 3.19 (s, 3H), 3.20 (s, 3H), 3.81 (t, 1H), 4.12 (t, 1H), 4.45 ( t, 2H, J = 5.81 Hz), 7.62 (t, 1H, J = 4.39 Hz), 7.74 (t, 2H, J = 3.68 Hz), 7.86 (dd, 3H), 7.92 (dd, 3H, J = 6.94 , 2.13 Hz), 7.97 (DD, 2H, J = 6.80, 1.98 Hz), 8.02 (DD, 2H), 10.03 (s, 1H), 11.19 (s, 1H)
(12-3) (S)-1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (Compound (A)) ( the resulting wet product crystals 12-2), t- butyl methyl ether (50 mL), acetonitrile (10 mL), 48w / w potassium hydroxide aqueous solution (4 g) and water (50 mL) was added, 15 I was about 2 hours of stirring at ~ 25 ℃. After stirring, and the mixture is separated, the resulting organic layer was concentrated under reduced pressure (30 mL), was added ethanol (60 mL), was further concentrated under reduced pressure (30 mL). After completion concentration under reduced pressure, ethanol (14 mL), after addition of water (20 mL), was added a seed crystal, and was crystallized crystals. After dropwise over about 1 hour water (50 mL), and about 1 hour stirring, and crystals were filtered off. Then washed with crystals 30v / v% aqueous ethanol solution (10 mL), where it was dried under reduced pressure at 40 ℃, the title compound was obtained in crystal (6.36 g, 87.0% yield). Moreover, the enantiomers of the resulting crystals was 0.05%. Enantiomers amount, I was measured by the method of (Example 8-4). 1 H NMR (400 MHz, CDCl 3 ) delta: 1.74 (1H, Broad singlet), 2.08 (3H, s), 3.04 (3H, s), 3.63 ~ 3.80 (4H, M), 7.36 (1H, D, J = 7.2 Hz), 7.48 (1H, s), 7.58 ~ 7.67 (2H, m), 7.77 ~ 7.90 (6H, m).
Angiotensin II receptor 桔抗 agent used as the component (A), olmesartan medoxomil, olmesartan cilexetil, losartan, candesartan cilexetil, valsartan, biphenyl tetrazole compounds such as irbesartan, biphenyl carboxylic acid compounds such as telmisartan, eprosartan, agile Sultan, and the like, preferably, a biphenyl tetrazole compound, more preferably, olmesartan medoxomil, is losartan, candesartan cilexetil, valsartan or irbesartan, particularly preferred are olmesartan medoxomil, losartan or candesartan cilexetil, Most preferably, it is olmesartan medoxomil.
Olmesartan medoxomil, JP-A-5-78328, US Patent No. 5,616,599
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
OLMESARTAN
Losartan (DUP-753) is, JP 63-23868, is described in US Patent No. 5,138,069 JP like, and its chemical name is 2-butyl-4-chloro-1- [2 ‘ – The (1H- tetrazol-5-yl) biphenyl-4-ylmethyl] -1H- is imidazol-5-methanol, application of losartan includes its pharmacologically acceptable salt (losartan potassium salt, etc.).
LOSARTAN
Candesartan cilexetil, JP-A-4-364171, EP-459136 JP, is described in US Patent No. 5,354,766 JP like, and its chemical name is 1- (cyclohexyloxycarbonyloxy) ethyl-2 ethoxy-1- [2 ‘one (1H- tetrazol-5-yl) -4-Bife~eniru ylmethyl] -1H- benzimidazole-7-carboxylate is a salt application of candesartan cilexetil, which is a pharmacologically acceptable encompasses.
Valsartan (CGP-48933), the JP-A-4-159718, are described in EP-433983 JP-like, and its chemical name, (S) -N- valeryl -N- [2 ‘- (1H- tetrazol – It is a 5-yl) biphenyl-4-ylmethyl) valine, valsartan of the present application includes its pharmacologically acceptable ester or a pharmacologically acceptable salt thereof.
Irbesartan (SR-47436), the Japanese Patent Publication No. Hei 4-506222, is described in JP WO91-14679 publication, etc., its chemical name, 2-N–butyl-4-spiro cyclopentane-1- [2′ The (tetrazol-5-yl) biphenyl-4-ylmethyl] -2-imidazoline-5-one, irbesartan of the present application includes its pharmacologically acceptable salts.
Eprosartan (SKB-108566) is described in US Patent No. 5,185,351 JP etc., the chemical name, 3- [1- (4-carboxyphenyl-methyl) -2-n- butyl – imidazol-5-yl] The 2-thienyl – methyl-2-propenoic acid, present in eprosartan, the carboxylic acid derivatives, pharmacologically acceptable ester or a pharmacologically acceptable salt of a carboxylic acid derivative (eprosartan mesylate, encompasses etc.).
Telmisartan (BIBR-277) is described in US Patent No. 5,591,762 JP like, and its chemical name is 4 ‘- [[4 Mechiru 6- (1-methyl-2-benzimidazolyl) -2 – is a propyl-1-benzimidazolyl] methyl] -2-biphenylcarboxylic acid, telmisartan of the present application includes its carboxylic acid derivative, a pharmacologically acceptable ester or a pharmacologically acceptable salt thereof of carboxylic acid derivatives .
Agile Sultan, is described in Patent Publication No. 05-271228 flat JP, US Patent No. 5,243,054 JP like, and its chemical name is 2-ethoxy-1 {[2 ‘- (5-oxo-4,5-dihydro 1,2,4-oxadiazole-3-yl) biphenyl-4-yl] methyl} -1H- benzo [d] imidazole-7-carboxylic acid (2-Ethoxy-1 {[2 ‘- (5- oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) biphenyl-4-yl] is a methyl} -1H-benzo [d] imidazole-7-carboxylic acid).
Phase III, AstraZeneca
Hutchison China MediTech (Chi-Med), Cancer, kidney (renal cell carcinoma, papillary)
A c-Met kinase inhibitor with antineoplastic activity.
NCI: volitinib. An orally bioavailable inhibitor of the c-Met receptor tyrosine kinase with potential antineoplastic activity. Volitinib selectively binds to and inhibits the activation of c-Met in an ATP-competitive manner, and disrupts c-Met signal transduction pathways. This may result in cell growth inhibition in tumors that overexpress the c-Met protein. C-Met encodes the hepatocyte growth factor receptor tyrosine kinase and plays an important role in tumor cell proliferation, survival, invasion, and metastasis, and tumor angiogenesis; this protein is overexpressed or mutated in a variety of cancers.(NCI Thesaurus)
Savolitinib is a first-in-class inhibitor of c-Met in phase III clinical development at at Hutchison China MediTech (Chi-Med) and AstraZeneca for the treatment of patients with MET-driven, unresectable and locally advanced or metastatic papillary renal cell carcinoma. Phase II trials are also under way for the oral treatment of locally advanced or metastatic pulmonary sarcomatoid carcinoma. AstraZeneca is conducting phase II clinical trials for the treatment of non-small cell lung cancer. Phase I/II trials are ongoing at Samsung Medical Center for the second-line treatment of advanced gastric adenocarcinoma patients with MET amplification.
In 2011, the drug was licensed to AstraZeneca by at Hutchison China MediTech (Chi-Med) for worldwide codevelopment and marketing rights for the treatment of cancer.
SYNTHESIS
PAPER
Journal of Organic Chemistry (2018),
A multidisciplinary approach covering synthetic, physical, and analytical chemistry, high-throughput experimentation and experimental design, process engineering, and solid-state chemistry is used to develop a large-scale (kilomole) Suzuki–Miyaura process. Working against clear criteria and targets, a full process investigation and optimization package is described highlighting how and why key decisions are made in the development of large-scale pharmaceutical processes.
Process Design and Optimization in the Pharmaceutical Industry: A Suzuki–Miyaura Procedure for the Synthesis of Savolitinib
Savolitinib (1) were added, and the resulting suspension was cooled to 0 °C over 8 h. After stirring for a further 4 h at 0 °C, the solid was collected via filtration, washed twice with cold s-BuOH (150 kg, 186 L), and dried in vacuo at 40 °C to give Savolitinib (1) as a white crystalline solid (105 kg, 304 mol, 76%): mp 205.9–208.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.83 (s, 1H), 8.64 (s, 1H), 8.31 (s, 1H), 8.01 (s, 1H), 7.62–7.55 (m, 2H), 7.42 (dd, J = 1.7, 9.4 Hz, 1H), 6.45 (q, J= 7.1 Hz, 1H), 3.98 (s, 3H), 2.22 (d, J = 7.1 Hz, 3H); 13C {1H} NMR (DMSO-d6, 101 MHz) δ 147.9, 147.2, 143.9, 141.9, 138.5, 137.4, 133.7, 131.6, 125.4, 124.3, 123.9, 119.4, 117.1, 113.8, 55.5, 40.1, 39.1, 19.6 ppm; HRMS (ESI/Q-ToF) m/z [M + H – N2]+ calcd for C17H16N7 318.1462, found 318.1486.
NMR Summary S6 1H‐NMR
S7 13C‐NMR
S8 HSQC‐DEPT‐NMR
S9 COSY‐NMR
S10 HMBC‐13C/1H‐NMR
S11 NOESY‐NMR
S12 HRMS
PAPER
Journal of Medicinal Chemistry (2014), 57(18), 7577-7589
HGF/c-Met signaling has been implicated in human cancers. Herein we describe the invention of a series of novel triazolopyrazine c-Met inhibitors. The structure–activity relationship of these compounds was investigated, leading to the identification of compound 28, which demonstrated favorable pharmacokinetic properties in mice and good antitumor activities in the human glioma xenograft model in athymic nude mice.
Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2,3]triazolo[4,5-b]pyrazine (Volitinib) as a Highly Potent and Selective Mesenchymal–Epithelial Transition Factor (c-Met) Inhibitor in Clinical Development for Treatment of Cancer
Preparation of (S)-2-(4-(1-(1-(pyrazolo[1,5-a]pyridin-5-yl)ethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol (30) and (R)-2-(4-(1-(1-(pyrazolo[1,5-a]pyridin-5-yl)ethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol (31)
The present invention provides a triazolopyrazine derivatives, the chemical name (S) -1- (l_ (imidazo [l, 2_a] pyrazin-6-yl) ethane-yl) -6-α _ -1H- pyrazol-4-yl-methyl) -1Η- [1,2,3] triazolo [4,5-b] pyrazine, of formula (I), the
[0005] This compound is an inhibitor of the activity c -Me t, may be used for treatment or prevention of inhibition of c -Me t sensitive cancers.In the Chinese patent CN 102906092A (W02011 / 079804), discloses the synthesis and use triazolopyrazine derivatives.Prepared by repeating the above patent, the compound powder obtained by detecting an amorphous state.As those skilled in the art, although amorphous higher solubility and dissolution rate than polymorph in most cases, but it is unstable, hygroscopic, readily converted to stable crystalline form.Thus, without the presence of processing stability and poor storage stability shaped, and in the production process, the smaller the bulk density of the particles of amorphous, high surface free energy, are likely to cause aggregation, poor flowability, and a series of powerful elastic deformation of the formulation problem seriously affecting the clinical value of amorphous Drug triazolopyrazine derivatives.
In some embodiments, the c-Met inhibitor is ARQ197 (Tivantinib). Tivantinib has the IUPAC name (3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)-2,5-pyrrolidinedione and the following chemical structure:
[0058] In some embodiments, the c-Met inhibitor is EMD1214063 (MSC2156119J; Tepotinib).
Tepotinib has the IUPAC name 3-(1-(3-(5-((1-methylpiperidin-4-yl)methoxy)pyrimidin-2-yl)benzyl)-1,6-dihydro-6-oxopyridazin-3-yl)benzonitrile and the following chemical structure:
[0059] In some embodiments, the c-Met inhibitor is GSK/1363089/XL880 (Foretinib). Foretinib has the IUPAC name N1’-[3-fluoro-4-[[6-methoxy-7-(3-morpholinopropoxy)-4-quinolyl]oxy]phenyl]-N1-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide and the following chemical structure:
[0060] In some embodiments, the c-Met inhibitor is XL184 (Cabozantinib). Cabozantinib has the IUPAC name N-(4-((6,7-Dimethoxyquinolin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide and the following chemical structure:
[0061] In some embodiments, the c-Met inhibitor is HMPL-504/AZD6094/volitinib (Savolitinib). Volitinib has the IUPAC name (S)-1-(1-(imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2,3]triazolo[4,5-b]pyrazine and the following chemical structure:
[0062] In some embodiments, the c-Met inhibitor is MSC2156119J (EMD 1214063, Tepotinib).
Tepotinib has the IUPAC name Benzonitrile, 3-[1,6-dihydro-1-[[3-[5-[(1-methyl-4-piperidinyl)methoxy]-2-pyrimidinyl]phenyl]methyl]-6-oxo-3-pyridazinyl]- and the following chemical structure:
[0063] In some embodiments, the c-Met inhibitor is LY2801653 (Merestinib). Merestinib has the IUPAC name N-(3-fluoro-4-{[1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5 yl]oxy}phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide and the following chemical structure:
[0064] In some embodiments, the c-Met inhibitor is AMG 337. AMG 337 has the IUPAC name 7-methoxy-N-((6-(3-methylisothiazol-5-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)methyl)-1,5-naphthyridin-4-amine and the following chemical structure:
[0065] In some embodiments, the c-Met inhibitor is INCB28060 (Capmatinib). Capmatinib has the IUPAC name 2-fluoro-N-methyl-4-[7-(quinolin-6-ylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide and the following chemical structure:
[0066] In some embodiments, the c-Met inhibitor is AMG 458. AMG 458 has the IUPAC name 1-(2-hydroxy-2-methylpropyl)-N-(5-((7-methoxyquinolin-4-yl)oxy)pyridin-2-yl)-5-methyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide and the following chemical structure:
[0067] In some embodiments, the c-Met inhibitor is PF-04217903. PF-04217903 has the IUPAC name 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol and the following chemical structure:
[0068] In some embodiments, the c-Met inhibitor is PF-02341066 (Crizotinib). Crizotinib has the IUPAC name (R)-3-(1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(1-(piperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine and the following chemical structure:
[0069] In some embodiments, the c-Met inhibitor is E7050 (Golvatinib). Golvatinib has the IUPAC name N-(2-fluoro-4-((2-(4-(4-methylpiperazin-1-yl)piperidine-1-carboxamido)pyridin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide and the following chemical structure:
[0070] In some embodiments, the c-Met inhibitor is MK-2461. MK-2461 has the IUPAC name N-((2R)-1,4-Dioxan-2-ylmethyl)-N-methyl-N’-[3-(1-methyl-1H-pyrazol-4-yl)-5-oxo-5H-benzo[4,5]cyclohepta[1,2-b]pyridin-7-yl]sulfamide and the following chemical structure:
[0071] In some embodiments, the c-Met inhibitor is BMS-777607. BMS-777607 has the IUPAC name N-(4-((2-amino-3-chloropyridin-4-yl)oxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide and the following chemical structure:
[0072] In some embodiments, the c-Met inhibitor is JNJ-38877605. JNJ-38877605 has the IUPAC name 6-(difluoro(6-(1-methyl-1H-pyrazol-3-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)methyl)quinoline and the following chemical structure:
Idorsia is developing nemorexant, a dual orexin receptor antagonist (DORA), for the oral treatment of insomnia and investigating the program for the treatment of COPD. In May 2018, a phase III study was initiated in subjects with insomnia disorder and in September 2018, a phase I trial was initiated in COPD.
SCHEME
SEE AT END OF PAGE
PATENT
WO2013182972 ,
PATENT
WO2015083094 ,
Patent
WO 2015083070
Synthesis of nemorexant, using 2-methyl-L-proline hydrochloride as the starting material
N-Protection of 2-methyl-L-proline hydrochloride with Boc2O gives N-Boc-2-methyl-L-proline,
Which upon condensation with 4-chloro-3-methylbenzene-1,2-diamine using HATU and DIEA in CH2Cl2 affords the corresponding amide.
Cyclization of diamine in the presence of AcOH at 100 °C provides imidazole derivative,
Whose Boc moiety is removed by means of HCl in dioxane to yield 5-chloro-4-methyl-2-[2(S)-methylpyrrolidin-2-yl]benzimidazole hydrochloride.
N-Acylation of pyrrolidine derivative with 5-methoxy-2-(1,2,3-triazol-2-yl)benzoic acid using HATU and DIEA in CH2Cl2 produces Nemorexant
5-methoxy-2-(1,2,3-triazol-2-yl)benzoic acid (prepared by the coupling of 2-iodo-5-methoxybenzoic acid with 1,2,3-triazole using CuI and Cs2CO3 in DMF)
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
Process for the preparation of a crystalline potassium salt of a 2-(2H-[1,2,3]triazol-2-yl)-benzoic acid derivatives is claimed. Compound is disclosed to be useful for the preparation of pharmaceuticals, especially certain orexin receptor antagonists such as nemorexant .
^ Jump up to:abEquihua-Benítez AC, Guzmán-Vásquez K, Drucker-Colín R (July 2017). “Understanding sleep-wake mechanisms and drug discovery”. Expert Opin Drug Discov. 12 (7): 643–657. doi:10.1080/17460441.2017.1329818. PMID28511597.
Muehlan, C.; Heuberger, J.; Juif, P.E.; Croft, M.; van Gerven, J.; Dingemanse, J. Accelerated development of the dual orexin receptor antagonist ACT-541468: Integration of a microtracer in a first-in-human study Clin Pharmacol Ther 2018, 104(5): 1022
A Study to Evaluate the Pharmacokinetics of ACT-541468 in Subjects With Mild, Moderate and Severe Hepatic Impairment (NCT03713242) ClinicalTrials.gov Web Site 2018, October 24
Boof, M.-.L.; Ufer, M.; Halabi, A.; Dingemanse, J. Impact of the dual orexin receptor antagonist ACT-541468 on the pharmacokinetics of the CYP3A4 probe drug midazolam and assessment of the effect of food on ACT-541468 119th Annu Meet Am Soc Clin Pharmacol Ther (ASCPT) (March 21-24, Orlando) 2018, Abst PI-043
Muehlan, C.; Brooks, S.; Zuiker, R.; van Gerven, J.; Dingemanse, J. Night-time administration of ACT-541468, a novel dual orexin receptor antagonist: Characterization of its pharmacokinetics, next-day residual effects, safety, and tolerability 32nd Annu Meet Assoc Sleep Soc (SLEEP) (June 2-6, Baltimore) 2018, Abst 0008
Proposed international nonproprietary names (Prop. INN): List 118 WHO Drug Inf 2017, 31(4): 635
CRYSTALLINE SALT FORM OF (S)-(2-(6-CHLORO-7-METHYL-1H-BENZO[D]IMIDAZOL-2-YL)-2-METHYLPYRROLIDIN-1-YL)(5-METHOXY-2-(2H-1, 2, 3-TRIAZOL-2-YL)PHENYL)METHANONE AS OREXIN RECEPTOR ANTAGONIST
CRYSTALLINE FORM OF (S)-(2-(6-CHLORO-7-METHYL-1H-BENZO[D]IMIDAZOL-2-YL)-2-METHYLPYRROLIDIN-1 -YL)(5-METHOXY-2-(2H-1, 2, 3-TRIAZOL-2-YL)PHENYL)METHANONE AND ITS USE AS OREXIN RECEPTOR ANTAGONISTS
Presented by: Yuji Koriyama, associate director at Shionogi & Co.
Target: β-site amyloid presursor protein cleaving enzyme 1 (BACE1), an enzyme whose buildup is implicated in Alzheimer’s disease
Disease: Alzheimer’s disease
Reporter’s notes: Presented by Koriyama, who told the audience he was attending the ACS National Meeting for the first time, JNJ-5486911 joins dozens of clinical candidates from many companies in Phase II and III trials to treat Alzheimer’s disease. Researchers started with a hit that inhibited BACE1 with approximately 2,600 nM affinity and advanced the program until finally reaching a compound with roughly 1 nM affinity. The compound is being jointly developed by Shionogi & Co. and Janssen Pharmaceuticals.
Originator Shionogi
Developer Janssen Research & Development
Class Antidementias; Small molecules
Mechanism of Action Amyloid precursor protein secretase inhibitors
Highest Development Phases
Phase II/III Alzheimer’s disease
Most Recent Events
16 Jul 2017 Pharmacodynamics data from preclinical trials in Alzheimer’s disease presented at the Alzheimer’s Association International Conference (AAIC-2017)
15 Dec 2016 Biomarkers information updated
01 Jun 2016 Janssen Research & Development completes a phase I pharmacokinetic interaction trial in Healthy volunteers in Germany (PO) (NCT02611518)
Compound 12 (3.0 g, 20.3 mmol) was dissolved in N-methylpyrrolidone (18 mL), and the solution was cooled to 5°C. Thionyl chloride (3.1 g, 26.1 mmol) was added to obtain a solution of Compound 13.
To a suspension of Compound 11 (5.0 g, 16.8 mmol) in ethyl acetate (50 mL) were added sodium bicarbonate (3.5 g, 42.0 mmol) and water (50 mL), and the mixture was stirred for 5 min at 20°C.
The layers were separated, and the organic layer was concentrated to 10 g under reduced pressure. N-Methylpyrrolidone (5 mL) and 35% hydrochloric acid (0.9 g) were added, and the mixture was cooled to 3°C. The solution of Compound 13 and N-methylpyrrolidone (1.5 mL) were added to obtain a solution of Compound 15.
The solution of Compound 15 was added to a mixture of water (15 mL) and ethyl acetate (10 mL). After stirring the mixture for 1 hour, triethylamine (14.8 g, 14.6 mmol), N-methylpyrrolidone (1.5 mL) and water (5 mL) were added and further stirred for 1 hour. Water (45 mL) was added, and the mixture was stirred for 1 hour, filtered and dried to obtain crystals of Compound 15 (Crystalline Form I, 5.71 g, 92.4%).
Example 1-5
To a suspension of Compound 11 (1831 g, 6.2 mol) in ethyl acetate (18L) were added sodium bicarbonate (1293 g, 15.4 mol) and water (18L), and the mixture was stirred for 5 min at 20°C. The layers were separated, and the organic layer was concentrated to 3.8 kg under reduced pressure to obtain a concentrated solution of Compound 14.
Compound 12 (912 g, 6.2 mol) was dissolved in N-methylpyrrolidone (64L), and the solution was cooled to 4°C. Thionyl chloride (951 g, 8.0 mol) was added, and the mixture was stirred for 30 min. The concentrated solution of Compound 14 was added to obtain a solution of Compound 15.
The solution of Compound 15 and N-methylpyrrolidone (1.6 L) were added to water (18 L), and the mixture was stirred for 40 min at 25°C. 24% sodium hydroxide in water (5 kg), sodium bicarbonate (259 g, 3.1 mmol) and water (2.7 L) were added to the mixture. The mixture was stirred for 1 hour, filtered and dried to obtain crystals (metastable Form II) of Compound 15 (1.93 kg, 85.4%).
Example 1-3
Preparation of Compound 11
[Chem. 30]
A suspension of Compound 9 (20.0 g, 29.0 mmol) in N,N-dimethylacetamide (30 mL) was cooled to 5°C. 1,8-diazabicyclo(5,4,0)-7-undecene (39.7 g, 260.8 mmol) was added, and the mixture was stirred for 22 hours. Water (70 mL) was added to afford a solution of Compound 10.
To a mixture of ethyl acetate (200 mL), water (40 mL) and 62% sulfuric acid (12.7 g) was added the solution of Compound 10, and the mixture was cooled to 10°C. 15% sulfuric acid (3.7 g) was added, and the mixture was warmed to 20°C. The layers were separated, and the organic layer was washed with 5% sodium chloride in water (95 g). The layers were separated, and the organic layer was concentrated in vacuo to 42 mL. Ethyl acetate (20 mL) and 50% potassium carbonate in water (20 g) were added, and the mixture was warmed to 40°C. 4-chlorobenzenethiol (6.29 g, 43.5 mmol) and ethyl acetate (11 mL) were added, and the mixture was stirred for 1 hour. After cooling to 20°C, ethyl acetate (100 mL), water (68 mL) and 15% hydrochloric acid (42.6 g) were added. The layers were separated, and ethyl acetate (149 mL) and 20% potassium carbonate in water (40.5 g) were added to the aqueous layer. The layers were separated, and the organic layer was washed with water (100 mL). The layers were separated, and the organic layer was concentrated to 20 mL. Acetic acid (1.7 g, 29.0 mmol) was added, and the mixture was cooled to 5°C and stirred for 90 min, filtered and dried to afford 7.19 g of crystals of Compound 11 (yield: 83.4%, optical purity of (S)-isomer: 100%).
The optical purity was determined as follows.
(Sample Preparation)
25 mg of Compound 11 was weighed and dissolved in a solvent to prepare a 50 mL sample solution.
(Method)
Using liquid chromatography, the peak area was determined by automatic integration method for each of (R)- and (S)-isomers of Compound 11.
(Conditions)
Detector: ultraviolet absorptiometer (wave length: 230 nm)
Column: CHIRALCEL OD-RH, φ4.6×150 mm, 5 μm, (Daicel Corporation)
Column Temp.: constant at around 40°C
Mobile Phase: water/acetonitrile (LC grade)/methanol (LC grade)/triethylamine (1320:340:340:1)
Flow Rate: 1.0 mL/min (retention time of Compound 11: about 8 min for (R)-isomer, about 9 min for (S)-isomer)
Time span of measurement: over 15 min from the sample injection
Injection Volume: 10 μL
Sample Cooler Temp.: constant at around 25°C
Autoinjector Rinse Solution: water/acetonitrile (1:1)


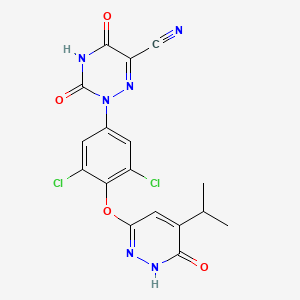
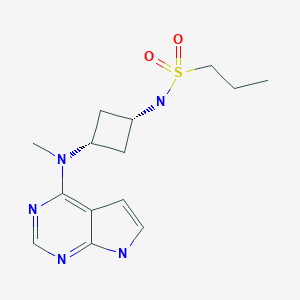
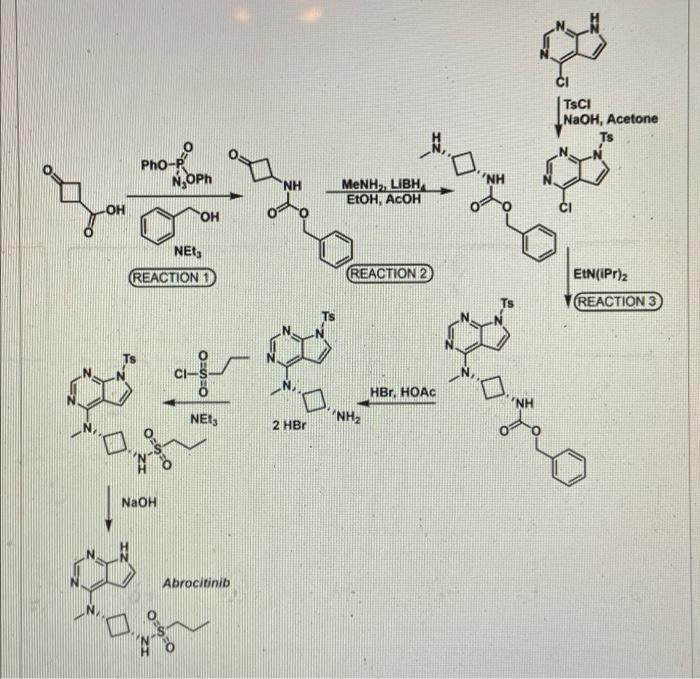
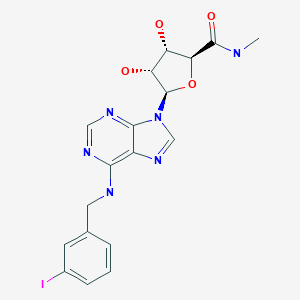
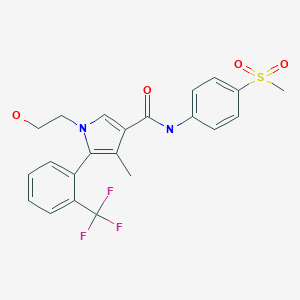

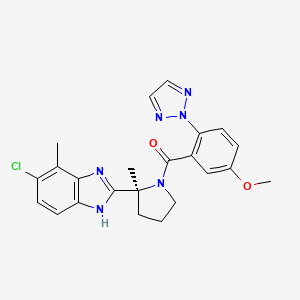
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader








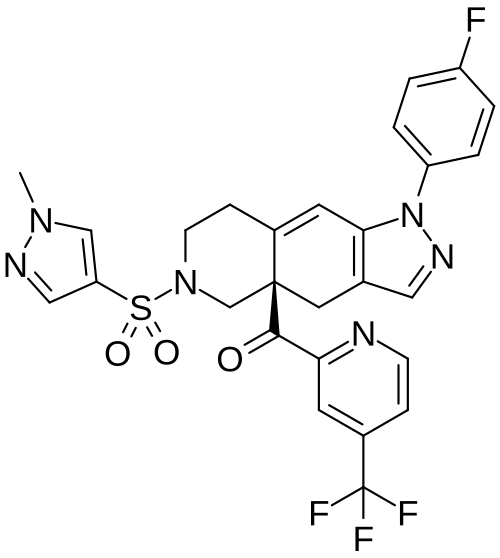
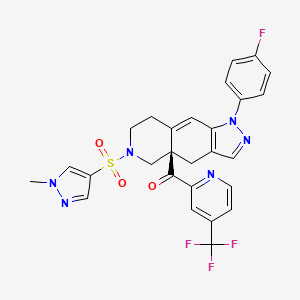












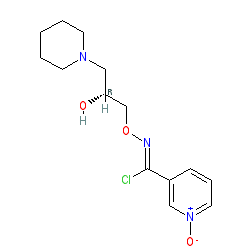
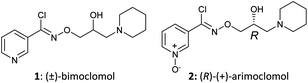




![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
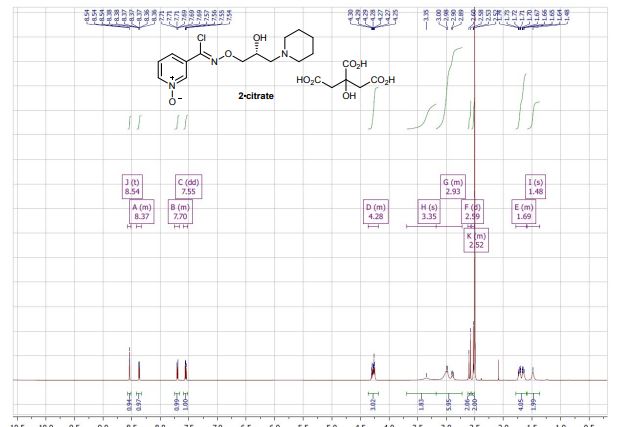





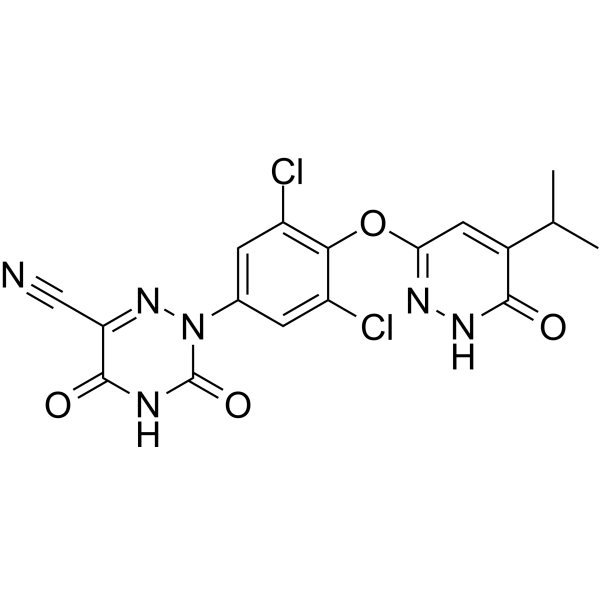







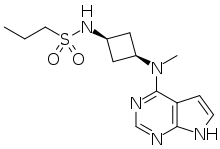
















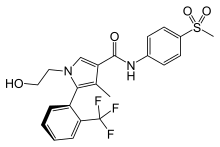
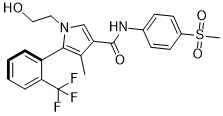












 OLMESARTAN
OLMESARTAN



 , Anna Jawor-Baczynska, Lucie Miller-Potucka, Michael Pilling, Katy Shepherd, Ross Tassone, Brian A. Taylor, and Aled Williams
, Anna Jawor-Baczynska, Lucie Miller-Potucka, Michael Pilling, Katy Shepherd, Ross Tassone, Brian A. Taylor, and Aled Williams













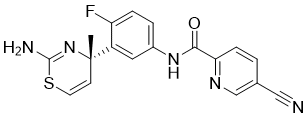















{kind=link}
{kind=link}
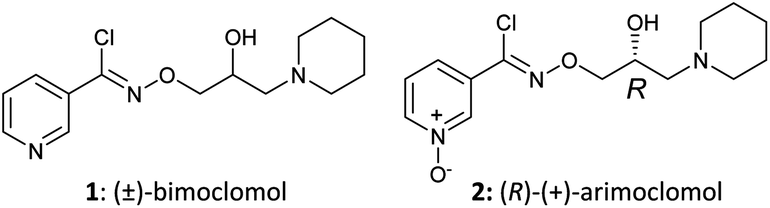{kind=link}
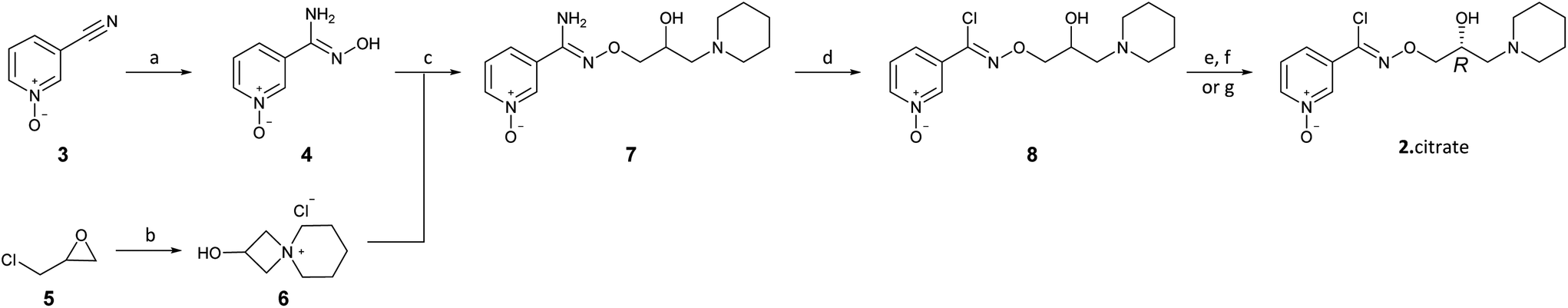{kind=link}
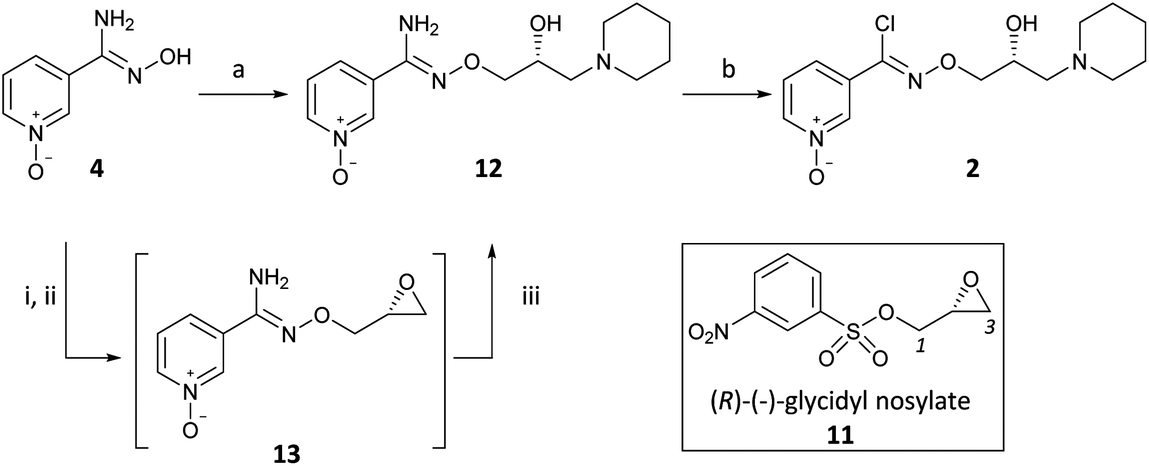{kind=link}
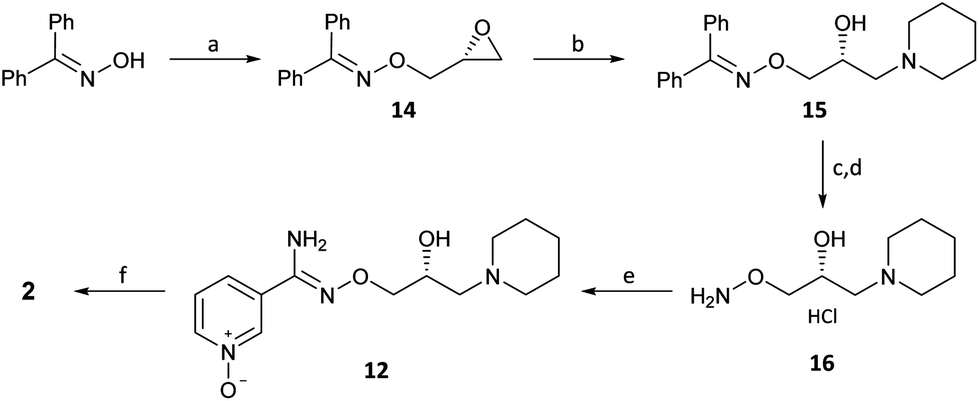{kind=link}
{kind=link}