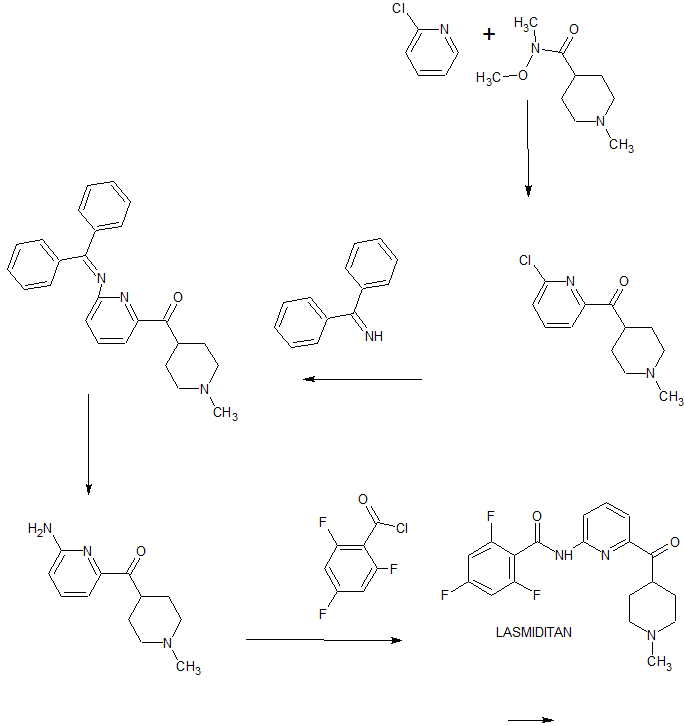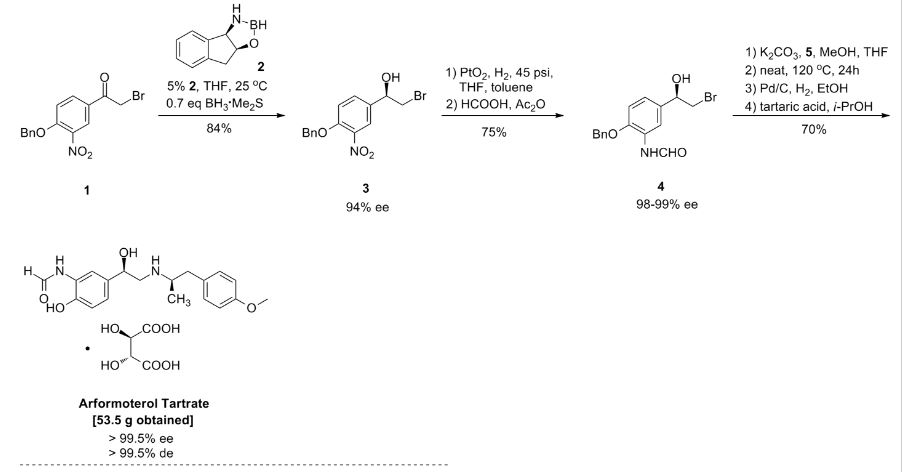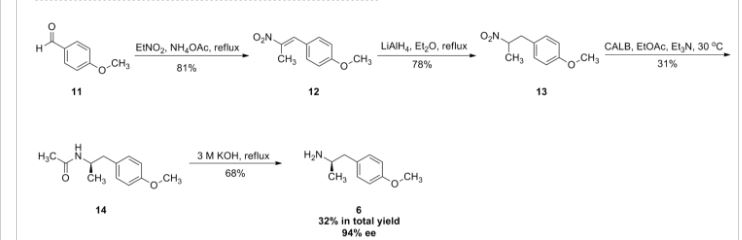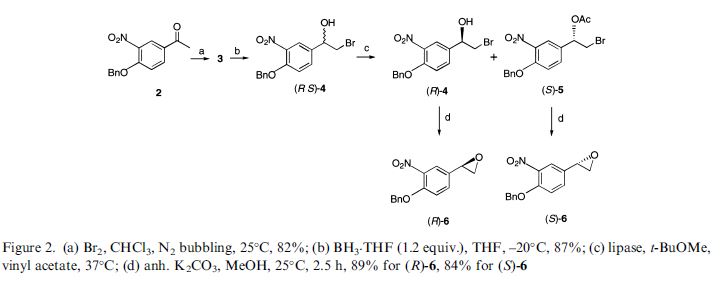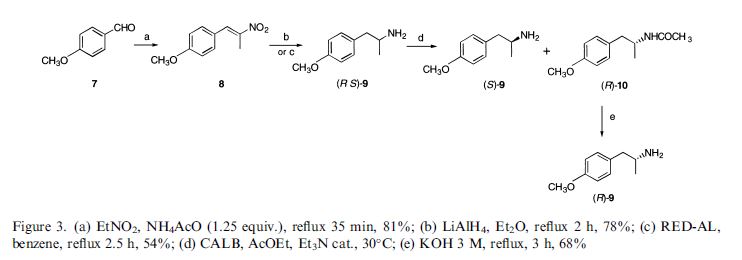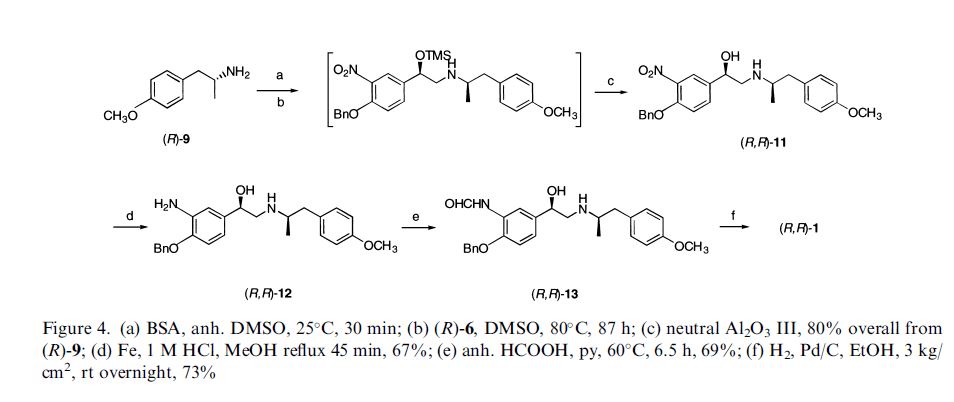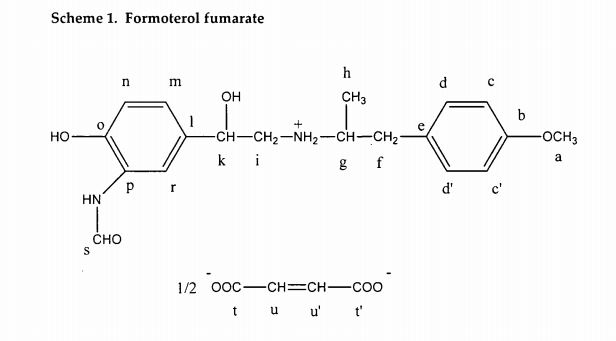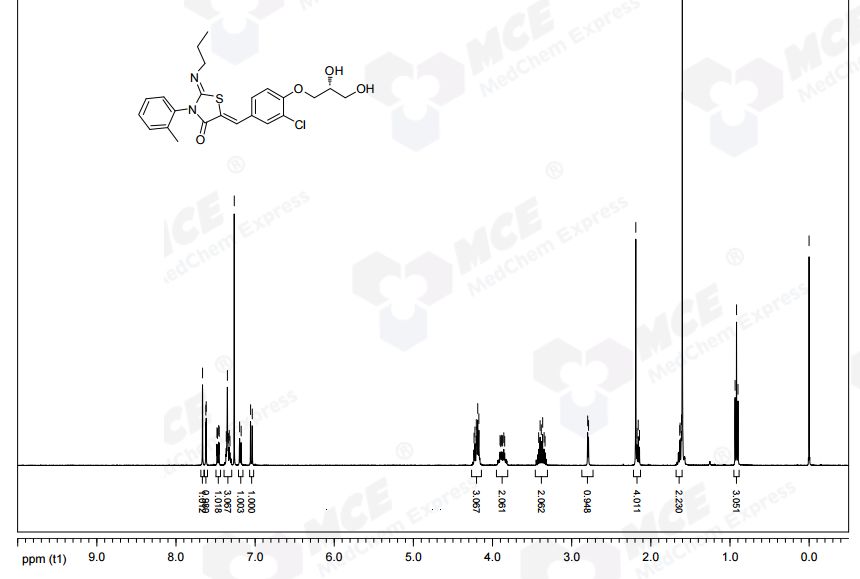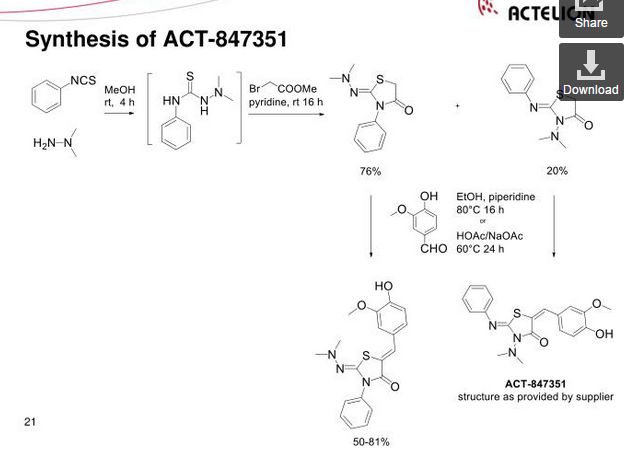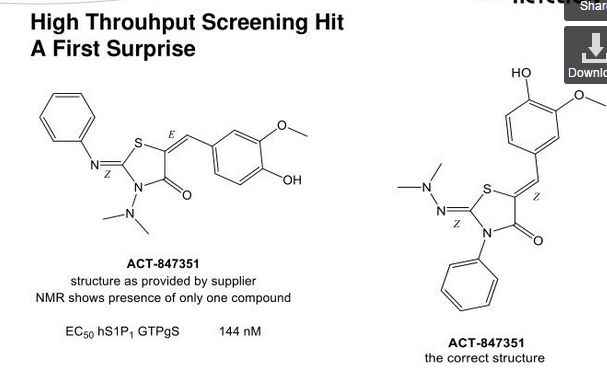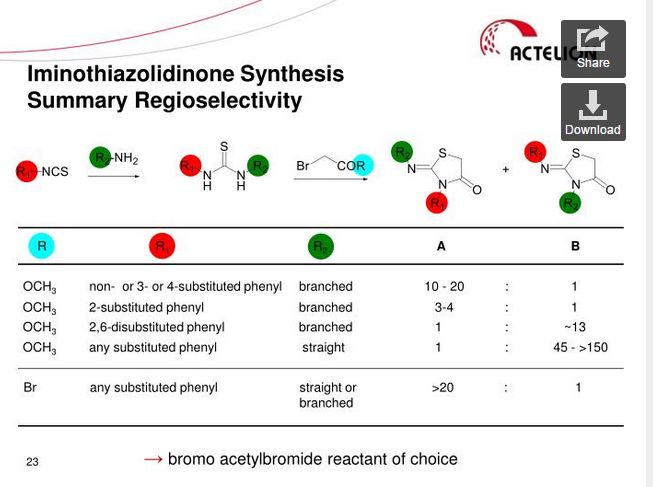

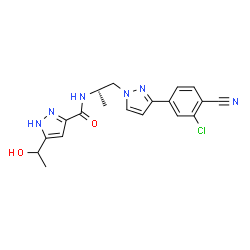
Darolutamide
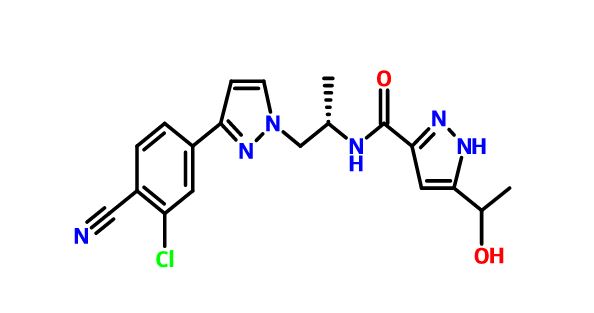
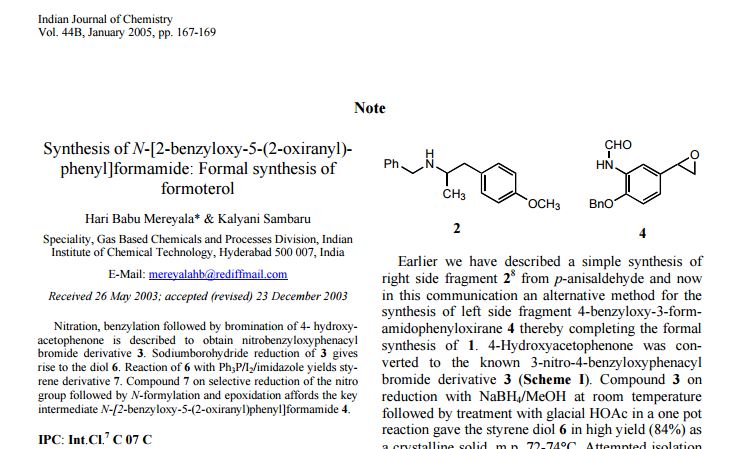
N-((S)-1-(3-(3-Chloro-4-cyanophenyl)-1H-pyrazol-1-yl)-propan-2-yl)-5-(1-hydroxyethyl)-1H-pyrazole-3-carboxamide
N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)-propan-2-yl)-5-(l-hydroxyethyl)-lH-pyrazole-3-carboxamide
- MF C19H19ClN6O2
- MW 398.846
BAY 1841788; ODM-201
даролутамид [Russian] [INN]
دارولوتاميد [Arabic] [INN]
ダロルタミド JAPANESE
ダロルタミド
Darolutamide

C19H19ClN6O2 : 398.85
[1297538-32-9] |

1H-Pyrazole-3-carboxamide, N-[(1S)-2-[3-(3-chloro-4-cyanophenyl)-1H-pyrazol-1-yl]-1-methylethyl]-5-(1-hydroxyethyl)-
BAY-1841788
N-{(2S)-1-[3-(3-Chlor-4-cyanphenyl)-1H-pyrazol-1-yl]-2-propanyl}-5-(1-hydroxyethyl)-1H-pyrazol-3-carboxamid
N-{(2S)-1-[3-(3-Chloro-4-cyanophenyl)-1H-pyrazol-1-yl]-2-propanyl}-5-(1-hydroxyethyl)-1H-pyrazole-3-carboxamide
N-{(2S)-1-[3-(3-Chloro-4-cyanophényl)-1H-pyrazol-1-yl]-2-propanyl}-5-(1-hydroxyéthyl)-1H-pyrazole-3-carboxamide
ODM-201
1297538-32-9 CAS
UNII:X05U0N2RCO
phase 3 for Hormone refractory prostate cancer; Hormone dependent prostate cancer
Orion and licensee Bayer are codeveloping darolutamide (ODM-201, BAY-1841788), an androgen receptor antagonist, for the potential treatment of castration-resistant prostate cancer (CRPC) and metastatic hormone-sensitive prostate cancer (HSPC) .
In September 2014, a phase III trial (ARAMIS) was initiated for non-metastatic CRPC; in April 2018, the trial was ongoing . In November 2016, a phase III trial in metatstic HSPC (ARASENS) was initiated .
PRODUCT PATENT
US-09657003 provides patent protection until May 2032.
Priority date 2009-10-27
InventorGerd WohlfahrtOlli TörmäkangasHarri SaloIisa HöglundArja KarjalainenPia KoivikkoPatrik HolmSirpa RaskuAnniina Vesalainen Current Assignee Orion Corp Original AssigneeOrion Corp
05-May-2011 WO-2011051540-A1, Priority date 2009-10-27
| Patent ID
|
Patent Title
|
Submitted Date
|
Granted Date
|
| US8921378 |
Androgen receptor modulating carboxamides |
2012-04-20
|
2014-12-30
|
| US8975254 |
ANDROGEN RECEPTOR MODULATING COMPOUNDS |
2010-10-27
|
2012-09-06
|
| US2017260206 |
ANDROGEN RECEPTOR MODULATING COMPOUNDS |
2017-04-13
|
|
| US9657003 |
ANDROGEN RECEPTOR MODULATING COMPOUNDS |
2015-01-16
|
2015-07-23
|
PHASE III
In September 2014, the double-blind, randomized, placebo-controlled, phase III trial (NCT02200614; ; ARAMIS) began to evaluate the safety and efficacy of darolutamide in patients (expected n = 1500, Taiwanese n = 20) in the US, Argentina, Australia, Brazil, Canada, Europe, Israel, Japan, Peru, South Korea, Russian Federation, South Africa, Taiwan and Turkey with non-metastatic CRPC. The primary endpoint was metastasis-free survival (MFS), defined as time between randomization and evidence of metastasis or death from any cause . In April 2018, the trial was expected to complete in September 2018

- Originator Orion
- Developer Bayer HealthCare; Orion
- Class Antineoplastics
- Mechanism of Action Androgen receptor antagonists
- Phase III Prostate cancer
-
Most Recent Events
- 03 Jun 2016 Bayer and Orion plan the phase III ARASENS trial for Prostate cancer
- 03 Jun 2016 Bayer and Orion expand the licensing agreement to include joint development of ODM 201 for Metastatic hormone-sensitive prostate cancer (mHSPC)
- 06 May 2016 Long-term combined adverse events data from the the ARADES (phase I/II) and the ARAFOR (phase I) trials in Prostate cancer presented at the 111th Annual Meeting of the American Urological Association (AUA -2016)
Darolutamide (INN) (developmental code names ODM-201, BAY-1841788) is a non-steroidal antiandrogen, specifically, a full and high-affinity antagonist of the androgen receptor (AR), that is under development by Orion and Bayer HealthCare[1] for the treatment of advanced, castration-resistant prostate cancer (CRPC).[2][3]
Orion and licensee Bayer are co-developing darolutamide, an androgen receptor antagonist, for treating castration-resistant prostate cancer and metastatic hormone-sensitive prostate cancer. In August 2016, darolutamide was reported to be in phase 3 clinical development. The drug appears to be first disclosed in WO2011051540, claiming novel heterocyclic derivatives as tissue-selective androgen receptor modulators, useful for the treatment of prostate cancer.

Mode of action
Relative to enzalutamide (MDV3100 or Xtandi) and apalutamide (ARN-509), two other recent non-steroidal antiandrogens, darolutamide shows some advantages.[3] Darolutamide appears to negligibly cross the blood-brain-barrier.[3] This is beneficial due to the reduced risk of seizures and other central side effects from off-target GABAA receptor inhibition that tends to occur in non-steroidal antiandrogens that are structurally similar to enzalutamide.[3] Moreover, in accordance with its lack of central penetration, darolutamide does not seem to increase testosterone levels in mice or humans, unlike other non-steroidal antiandrogens.[3] Another advantage is that darolutamide has been found to block the activity of all tested/well-known mutant ARs in prostate cancer, including the recently-identified clinically-relevant F876L mutation that produces resistance to enzalutamide and apalutamide.[3] Finally, darolutamide shows higher affinity and inhibitory efficacy at the AR (Ki = 11 nM relative to 86 nM for enzalutamide and 93 nM for apalutamide; IC50 = 26 nM relative to 219 nM for enzalutamide and 200 nM for apalutamide) and greater potency/efficaciousness in non-clinical models of prostate cancer.[3]
ORM-15341 is the main active metabolite of darolutamide.[3] It, similarly, is a full antagonist of the AR, with an affinity (Ki) of 8 nM and an IC50 of 38 nM.[3]

Clinical trials
Darolutamide has been studied in phase I and phase II clinical trials and has thus far been found to be effective and well-tolerated,[4] with the most commonly reported side effects including fatigue, nausea, and diarrhea.[5][6] No seizures have been observed.[6][7] As of July 2015, darolutamide is in phase III trials for CRPC.[3]

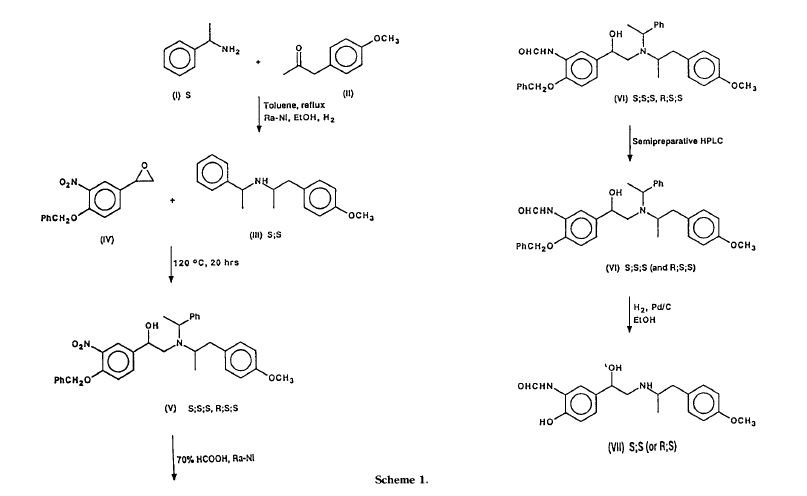
Representative binding affinities of ODM-201, ORM-15341, enzalutamide, and ARN-509 measured in competition with [3H]mibolerone using wtAR isolated from rat ventral prostates (C). All data points are means of quadruplicates ±SEM. Ki values are presented in parentheses. D. Antagonism to wtAR was determined using AR-HEK293 cells treated with ODM-201, ORM-15341, enzalutamide, or ARN-509 together with 0.45 nM testosterone in steroid-depleted medium for 24 hours before luciferase activity measurements. All data points are means of triplicates ±SEM. IC50 values are presented in parentheses.
WHIPPANY, N.J., Sept. 16, 2014 /PRNewswire/ — Bayer HealthCare and Orion Corporation, a pharmaceutical company based in Espoo, Finland, have begun to enroll patients in a Phase III trial with ODM-201, an investigational oral androgen receptor inhibitor in clinical development. The study, called ARAMIS, evaluates ODM-201 in men with castration-resistant prostate cancer who have rising Prostate Specific Antigen (PSA) levels and no detectable metastases. The trial is designed to determine the effects of the treatment on metastasis-free survival (MFS).
“The field of treatment options for prostate cancer patients is evolving rapidly. However, once prostate cancer becomes resistant to conventional anti-hormonal therapy, many patients will eventually develop metastatic disease,” said Dr. Joerg Moeller, Member of the Bayer HealthCare Executive Committee and Head of Global Development. “The initiation of a Phase III clinical trial for ODM-201 marks the starting point for a potential new treatment option for patients whose cancer has not yet spread. This is an important milestone for Bayer in our ongoing effort to meet the unmet needs of men affected by prostate cancer.”
Earlier this year, Bayer and Orion entered into a global agreement under which the companies will jointly develop ODM-201, with Bayer contributing a major share of the costs of future development. Bayer will commercialize ODM-201 globally, and Orion has the option to co-promote ODM-201 in Europe. Orion will be responsible for the manufacturing of the product.
About the ARAMIS Study
The ARAMIS trial is a randomized, Phase III, multicenter, double-blind, placebo-controlled trial evaluating the safety and efficacy of oral ODM-201 in patients with non-metastatic CRPC who are at high risk for developing metastatic disease. About 1,500 patients are planned to be randomized in a 2:1 ratio to receive 600 mg of ODM-201 twice a day or matching placebo. Randomisation will be stratified by PSA doubling time (PSADT less than or equal to 6 months vs. > 6 months) and use of osteoclast-targeted therapy (yes vs. no).
The primary endpoint of this study is metastasis-free survival (MFS), defined as time between randomization and evidence of metastasis or death from any cause. The secondary objectives of this study are overall survival (OS), time to first symptomatic skeletal event (SSE), time to initiation of first cytotoxic chemotherapy, time to pain progression, and characterization of the safety and tolerability of ODM-201.
About ODM-201
ODM-201 is an investigational androgen receptor (AR) inhibitor that is thought to block the growth of prostate cancer cells. ODM-201 binds to the AR and inhibits receptor function by blocking its cellular function.
About Oncology at Bayer
Bayer is committed to science for a better life by advancing a portfolio of innovative treatments. The oncology franchise at Bayer now includes three oncology products and several other compounds in various stages of clinical development. Together, these products reflect the company’s approach to research, which prioritizes targets and pathways with the potential to impact the way that cancer is treated.
About Bayer HealthCare Pharmaceuticals Inc.
Bayer HealthCare Pharmaceuticals Inc. is the U.S.-based pharmaceuticals business of Bayer HealthCare LLC, a subsidiary of Bayer AG. Bayer HealthCare is one of the world’s leading, innovative companies in the healthcare and medical products industry, and combines the activities of the Animal Health, Consumer Care, Medical Care, and Pharmaceuticals divisions. As a specialty pharmaceutical company, Bayer HealthCare provides products for General Medicine, Hematology, Neurology, Oncology and Women’s Healthcare. The company’s aim is to discover and manufacture products that will improve human health worldwide by diagnosing, preventing and treating diseases.
Bayer® and the Bayer Cross® are registered trademarks of Bayer.
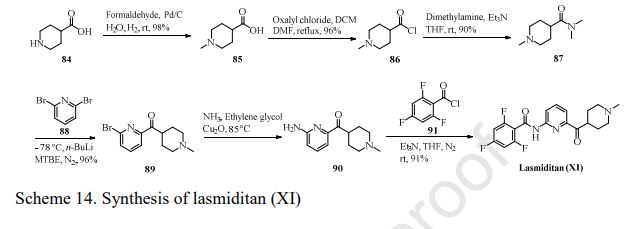
SYNTHESIS

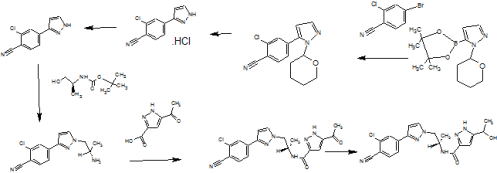
cas 1297538-32-9
Synthesis
WO 2016162604

POLYMORPH
CRYSTALLINE FORM I, I’, I” IN WO-2016120530
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016120530&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescriptionWO-2016120530


PATENTS
WO2011051540
https://www.google.com/patents/WO2011051540A1?cl=en
PATENT
US 2015203479
http://www.google.com/patents/WO2011051540A1?cl=en
PATENT
WO 2012143599
http://www.google.com/patents/US20140094474?cl=de
PATENT
IN 2011KO00570
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016120530&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
WO-2016120530
Compound of (I) (5 g) was dissolved in an acetonitrile and distilled water. The reaction mixture was heated at 75 °C and then slowly cooled down at RT and stirred at RT for 3 days. The solid obtained was filtered, washed twice with the acetonitrile: water and dried under vacuum at 40 °C and 60 °C to yield crystalline form of (I) (4.42 g) with 88% of yield (example 1, page 10).
Compound (I) can be synthetized using the procedures described in WO
201 1/051540.

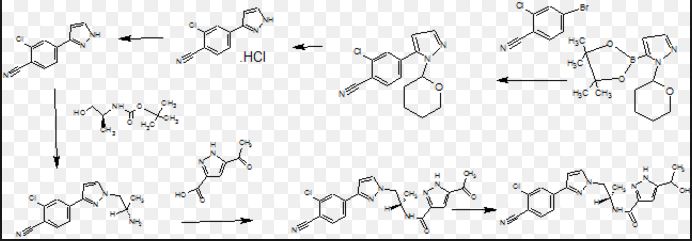
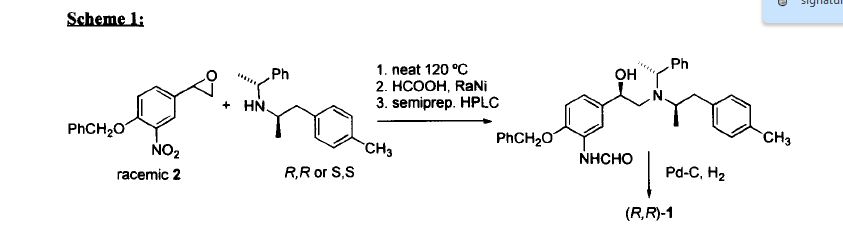
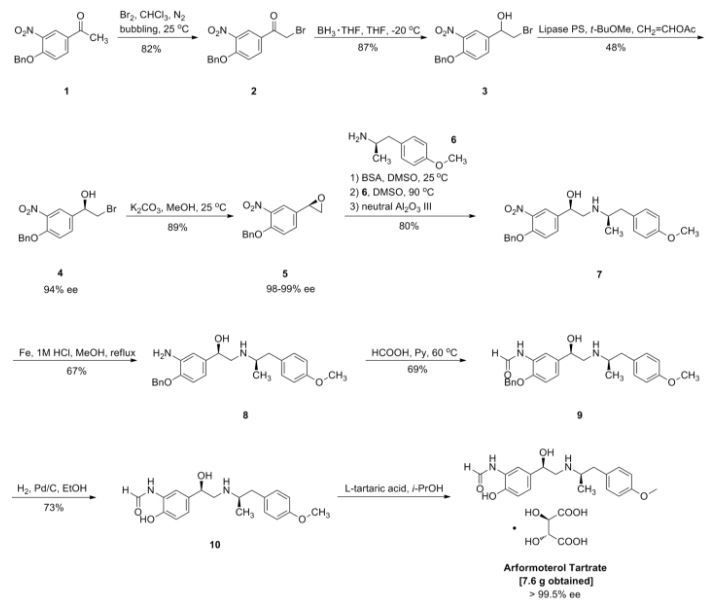
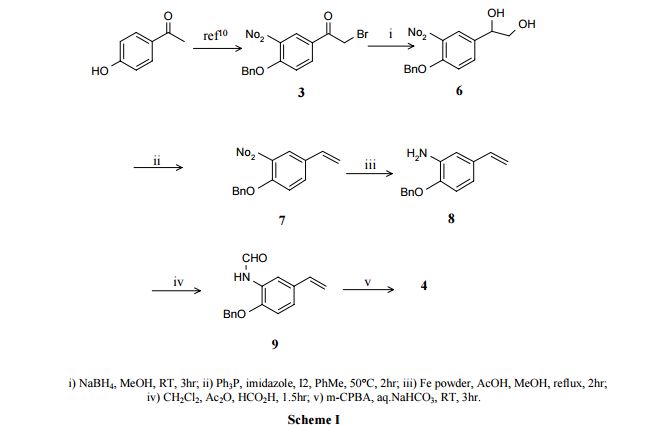
Pure diastereomers (la) and (lb) can be suitably synthetized, for example, using ketoreductase enzymes (KREDs) for both S- and R-selective reduction of compound 1 to compound 2 as shown in Scheme 1, wherein R is H or Ci_6 alkyl.


Scheme 1.
For example, Codexis KRED-130 and KRED -NADH-110 enzymes are useful for obtaining excellent stereoselectivity, even stereospecificity. In Scheme 1 the starting material 1 is preferably an ester (R= Ci_6 alkyl), for example ethyl ester (R=ethyl), such as to facilitate extraction of the product into the organic phase as the compound where R=H has a tendency to remain in the water phase. Intermediate 2 can be protected, preferably with silyl derivatives such as tert-butyldiphenylsilyl, in order to avoid esterification in amidation step. In the case of R=Ci_6 alkyl, ester hydrolysis is typically performed before amidation step, preferably in the presence of LiOH, NaOH or KOH. Amidation from compound 3 to compound 5_is suitably carried out using EDCI HBTU, DIPEA system but using other typical amidation methods is also possible. Deprotection of 5 give pure diastereomers (la) and (lb).
Pyrazole ring without NH substitution is known tautomerizable functionality and is described here only as single tautomer but every intermediate and end product here can exist in both tautomeric forms at the same time.
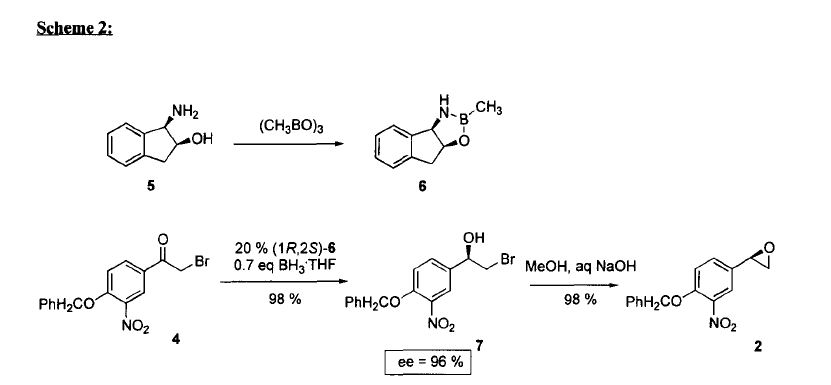
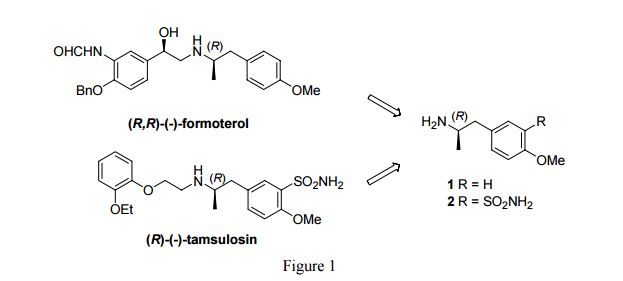
The stereochemistry of the compounds can be confirmed by using optically pure starting materials with known absolute configuration as demonstrated in Scheme 2, wherein R=H or Ci_6 alkyl, preferably alkyl, for example ethyl. The end products of Scheme 2 are typically obtained as a mixture of tautomers at +300K 1H-NMR analyses in DMSO.


Scheme 2. Synthesis pathway to stereoisomers by using starting materials with known absolute configuration
The crystalline forms I, Γ and Γ ‘ of compounds (I), (la) and (lb), respectively, can be prepared, for example, by dissolving the compound in question in an
acetonitrile: water mixture having volume ratio from about 85: 15 to about 99: 1, such as from about 90: 10 to about 98:2, for example about 95:5, under heating and slowly cooling the solution until the crystalline form precipitates from the solution. The concentration of the compound in the acetonitrile: water solvent mixture is suitably about 1 kg of the compound in 5-25 liters of acetonitrile: water solvent mixture, for example 1 kg of the compound in 10-20 liters of acetonitrile: water solvent mixture. The compound is suitably dissolved in the acetonitrile: water solvent mixture by heating the solution, for example near to the reflux temperature, for example to about 60-80 °C, for example to about 75 °C, under stirring and filtering if necessary. The solution is suitably then cooled to about 0-50 °C, for example to about 5-35 °C, for example to about RT, over about 5 to about 24 hours, for example over about 6 to 12 hours, and stirred at this temperature for about 3 to 72 hours, for example for about 5 to 12 hours. The obtained crystalline product can then be filtered, washed, and dried. The drying is suitably carried out in vacuum at about 40 to 60 °C, for example at 55 °C, for about 1 to 24 hours, such as for about 2 to 12 hours, for example 2 to 6 hours.
The crystalline forms I, Γ and I” of compounds (I), (la) and (lb), respectively, are useful as medicaments and can be formulated into pharmaceutical dosage forms, such as tablets and capsules for oral administration, by mixing with pharmaceutical excipients known in the art.
The disclosure is further illustrated by the following examples.
Example 1. Crystallization of N-((S)- 1 -(3 -(3 -chloro-4-cyanophenyl)- 1 H-pyrazol- 1 -yl)-propan-2-yl)-5 -( 1 -hydroxyethyl)- 1 H-pyrazole-3 -carboxamide (I)
N-((iS)- 1 -(3 -(3 -chloro-4-cyanophenyl)- 1 H-pyrazol- 1 -yl)-propan-2-yl)-5 -( 1 -hydroxyethyl)-! H-pyrazole-3 -carboxamide (I) (5 g), 71.25 ml of acetonitrile, and 3.75 ml of distilled water were charged to a flask, and the mixture was heated up to 75 °C. The mixture was slowly cooled down to RT and stirred at RT for 3 days. The solid obtained was filtered and washed twice with acetonitrile: water (9.5 ml:0.5 ml). The product was dried under vacuum at 40 °C and finally at 60°C to obtain 4.42 g of crystalline title compound (yield of 88 %) which was used in X-ray diffraction study.
Example 3. Synthesis of N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)-propan-2-yl)-5-((S)- 1 -hy droxy ethyl)- lH-pyrazole-3-carboxamide (la)
a) Ethyl-5 -((S) 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxylate


HO
MgS04 x7H20 (341 mg), NADP monosodium salt (596 mg), D(+)-glucose (9.26 g) and optimized enzyme CDX-901 lyophilized powder (142 mg) were added to 0.2 mM of KH2P04 buffer (pH 7.0, 709 ml) to prepare solution I. To this solution I was added solution II which contained ethyl-5 -acetyl- 1 H-pyrazole-3 -carboxylate (8.509 g; 46.70 mmol), EtOH (28 ml) and K ED-130 (NADPH ketoreductase, 474 mg). The mixture was agitated at 30-32°C for 5.5 h (monitoring by HPLC) and allowed to cool to RT. The mixture was evaporated to smaller volume and the residue was agitated with diatomaceous earth and filtered. The mother liquid was extracted with 3×210 ml of EtOAc and dried. The solution was filtered through silica (83 g) and evaporated to dryness to give 7.40 g of the title compound. The optical purity was 100 % ee.
b) Ethyl 5-((S)-l -((tert-butyldiphenylsilyl)oxy)ethyl)- 1 H-pyrazole-3 -carboxylate


Diphenyl-tert-butyl chlorosilane (7.48 g, 27.21 mmol) was added in 26 ml of DMF to a mixture of compound of Example 3(a) (5.00 g, 27.15 mmol) and imidazole (2.81 g, 41.27 mmol) in DMF (50 ml) at RT. The mixture was stirred at RT for 24 h.
Saturated aqueous NaHC03 (56 ml) and water (56 ml) were added and the mixture was stirred at RT for 20 min. The mixture was extracted with 2×100 ml of EtOAc. Combined organic phases were washed with water (1×100 ml, 1×50 ml), dried (Na2S04), filtered and concentrated to give 10.92 g of crude title compound.
c) 5-((S)-l -((tert-Butyldiphenylsilyl)oxy)ethyl)- 1 H-pyrazole-3 -carboxylic acid


2 M NaOH (aq) (38.8 ml; 77.5 mmol) was added to a solution of the compound of Example 3(b) (10.9 g, 25.8 mmol) in 66 ml of THF. The mixture was heated up to reflux temperature. Heating was continued for 2.5 h and THF was removed in vacuum. Water (40 ml) and EtOAc (110 ml) were added. Clear solution was obtained after addition of more water (10 ml). Layers were separated and aqueous phase was extracted with 100 ml of EtOAc. Combined organic phases were dried (Na2S04), filtered and concentrated to give 9.8 g of the title compound.
d) 5-((S)- 1 -((tert-Butyldiphenylsilyl)oxy)ethyl)-N-((S)- 1 -(3-(3-chloro-4-cyano-phenyl)- 1 H-pyrazol- 1 -yl)propan-2-yl)- 1 H-pyrazole-3 -carboxamide


Under nitrogen atmosphere HBTU (0.84 g; 2.22 mmol), EDCIxHCl (3.26 g; 17.02 mmol) and (S)-4-(l-(2-aminopropyl)-lH-pyrazol-3-yl)-2-chlorobenzonitrile (3.86 g; 14.80 mmol) were added to a mixture of crude compound of Example 3(c) (8.68g; purity 77.4 area-%) and DIPEA (2.20 g; 17.02 mmol) in DCM (50 ml). The mixture was stirred at RT for 46 h (6 ml of DCM was added after 20 h). The mixture was washed with 3×20 ml of water, dried (Na2S04), filtered and concentrated to give 13.7 g of crude title compound.
e) N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((S)- 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxamide (la)
TBAF hydrate (Bu4NF x 3H20; 2.34 g; 7.40 mmol) in 10 ml of THF was added to the solution of the compound of Example 3(d) (9.43 g; 14.79 mmol) in THF (94 ml) at 0 °C under nitrogen atmosphere. Stirring was continued at RT for 21.5 h and the mixture was concentrated. DCM (94 ml) was added to the residue and the solution was washed with 3×50 ml of water, dried (Na2S04), filtered and concentrated. Crude product was purified by flash chromatography (EtOAc/n-heptane) to give 2.1 g of the title compound. 1H-NMR (400MHz; d6-DMSO; 300K): Major tautomer (-85 %): δ 1.11 (d, 3H), 1.39 (d, 3H), 4.24-4.40 (m, 2H), 4.40-4.50 (m, 1H), 6.41(s, 1H), 6.93 (d, 1H), 7.77-7.82 (m, 1H), 7.88-8.01 (m, 2H), 8.08 (s, 1H), 8.19 (d, 1H), 13.02 (broad s, 1H). Minor tautomer (-15 %) δ 1.07-1.19 (m, 3H), 1.32-1.41 (m, 3H), 4.24-4.40 (m, 2H), 4.40-4.50 (m, 1H), 6.80 (broad s, 1H), 6.91-6-94 (m, 1H), 7.77-7.82 (m, 1H), 7.88-8.01 (m, 2H), 8.05-8.09 (m, 1H), 8.31 (d, 1H), 13.10 (broad s, 1H).
Example 4. Crystallization of N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((S)- 1 -hy droxy ethyl)- lH-pyrazole-3-carboxamide (la)
N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((S)- 1 -hydroxyethyl)-lH-pyrazole-3-carboxamide (la) (5.00 g; 12.54 mmol) was mixed with 47.5 ml of ACN and 2.5 ml of water. The mixture was heated until compound (la) was fully dissolved. The solution was allowed to cool slowly to RT to form a precipitate. The mixture was then further cooled to 0 °C and kept in this temperature for 30 min. The mixture was filtered and the precipitate was dried under vacuum to obtain 4.50 g of crystalline title compound which was used in the X-ray diffraction study.
Example 6. Synthesis of N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)-propan-2-yl)-5-((R)- 1 -hy droxy ethyl)- lH-pyrazole-3-carboxamide (lb)
a) Ethyl-5 -((R)- 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxylate


Potassium dihydrogen phosphate buffer (Solution I) was prepared by dissolving potassium dihydrogen phosphate (11.350 g, 54.89 mmol) to water (333 ml) and adjusting pH of the solution to 7.0 by addition of 5 M solution of NaOH. MgS04 x 7 H20 (1.650 g), NAD monosodium salt (0.500 g), D(+)-glucose (10.880 g) and optimised enzyme CDX-901 lyophilised powder (0.200 g) were added to Solution I. To this solution (Solution II) were added KRED-NADH- 110 (0.467 g), ethyl-5-acetyl-1 H-pyrazole-3 -carboxylate (10.00 g; 54.89 mmol) and 2-methyltetrahydro-furan (16 ml). The mixture was agitated at 30° C for 11 h and allowed to cool to RT overnight. The pH of the mixture was kept at 7 by addition of 5 M solution of NaOH. The mixture was evaporated to a smaller volume. The evaporation residue was agitated for 10 min with diatomaceous earth (40 g) and activated charcoal (0.54 g), and filtered. Material on the filter was washed with water (40 ml) and the washings were combined with the filtrate. Layers were separated and aqueous phase was extracted with EtOAc (450 ml and 2×270 ml). Combined organic phases were dried over Na2S04, filtered and evaporated to dryness to give 9.85 g of the title compound (100 % ee).
b) Ethyl-5 -((R)- 1 -((tert-butyldiphenylsilyl)oxy)ethyl)- 1 H-pyrazole-3 -carboxylate


Imidazole (5.32 g; 78.08 mmol) was added to a DCM (67 ml) solution of the compound of Example 6(a) (9.85 g; 53.48). The mixture was stirred until all reagent was dissolved and tert-butyldiphenyl chlorosilane (13.21 ml; 50.80 mmol) was added to the mixture. The mixture was stirred for 1.5 h, 70 ml of water was added and stirring was continued for 15 min. Layers were separated and organic phase was washed with 2×70 ml of water and dried over Na2S04, filtered and concentrated to give 22.07 g of crude title compound.
c) 5 -((R)- 1 -((tert-Butyldiphenylsilyl)oxy)ethyl)- 1 H-pyrazole-3 -carboxylic acid


Compound of Example 6(b) (11.3 g; 26.74 mmol; theoretical yield from the previous step) was dissolved in 34 ml of THF and 50 ml of 2 M NaOH (aq.) was added. The mixture was heated under reflux temperature for 70 min. The mixture was extracted with 2×55 ml of EtOAc and combined organic phases were washed with brine, dried over Na2S04, filtered and concentrated. Evaporation residue was triturated in 250 ml of n-heptane, filtered and dried to give 17.58 g of crude title compound.
d) 5-((R)- 1 -((tert-Butyldiphenylsilyl)oxy)ethyl)-N-((S)- 1 -(3-(3-chloro-4-cyano-phenyl)- 1 H-pyrazol- 1 -yl)propan-2-yl)- 1 H-pyrazole-3 -carboxamide


A mixture of the compound of Example 6(c) (11.14 g; 26.75 mmol; theoretical yield from the previous step), 91 ml of DCM, HBTU (1.52 g; 4.01 mmol), EDCIxHCl
(5.90 g; 30.76 mmol), (S)-4-(l-(2-aminopropyl)-lH-pyrazol-3-yl)-2-chlorobenzo-nitrile (6.97 g; 26.75 mmol) and DIPEA (3.98 g; 30.76 mmol) was stirred at RT for 3 h and at 30° C for 22 h. The mixture was washed with 2×90 ml of 0.5 M HC1 and 4×90 ml of water, dried over Na2S04, filtered and concentrated. Crude product was purified by flash column chromatography (n-heptane-EtOAc) to give 16.97 g of title compound.
e) N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((R)- 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxamide (lb)
A mixture of the compound of Example 6(d) (6.09 g; 9.56 mmol), 61 ml of THF and TBAF was stirred at 40 °C for 6.5 h. The mixture was concentrated and 61 ml of EtOAc was added to the evaporation residue. Solution was washed with 2×50 ml of 0.5 M HC1 and 4×50 ml of water, dried over Na2S04, filtered and concentrated. Crude product was purified by flash column chromatography (n-heptane-EtOAc) to give 1.71 g of the title compound. 1H-NMR (400MHz; d6-DMSO; 300K): Major tautomer (~85%): 5 1.10 (d, 3H), 1.38 (d, 3H), 4.14-4.57 (m, 2H), 5.42 (d, 1H),
6.39(s, 1H), 6.86-6.98 (m, 1H), 7.74-7.84 (m, 1H), 7.86-8.02 (m, 2H), 8.08 (s, 1H), 8.21 (d, 1H), 13.04 (broad s, 1H). Minor tautomer (-15%) δ 0.95-1.24 (m, 3H), 1.25-1.50 (m, 3H), 4.14-4.57 (m, 2H), 4.60-4.90 (m, 1H), 5.08 (d, 1H), 6.78 (broad s, 1H), 6.86-6.98 (m, 1H), 7.77-7.84 (m, 1H), 7.86-8.02 (m, 2H), 8.02-8.12 (m, 1H), 8.32 (d, 1H), 13.1 1 (broad s, 1H).
Example 7. Crystallization of N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((R)- 1 -hy droxy ethyl)- 1 H-pyrazole-3 -carboxamide (lb)
N-((S)- 1 -(3-(3-chloro-4-cyanophenyl)- lH-pyrazol- 1 -yl)propan-2-yl)-5-((R)- 1 -hydroxyethyl)-l H-pyrazole-3 -carboxamide (lb) (3.7 g; 9.28 mmol) was mixed with 70 ml of ACN and 3.5 ml of water. The mixture was heated to reflux temperature until compound (lb) was fully dissolved. The solution was allowed to cool slowly. The mixture was filtered at 50 °C to obtain 6.3 mg of the precipitate. Mother liquid was cooled to 41 °C and filtered again to obtain 20.7 mg of the precipitate. Obtained mother liquid was then cooled to 36 °C and filtered to obtain 173 mg of the precipitate. The final mother liquid was cooled to RT, stirred overnight, cooled to 0 °C, filtered, washed with cold ACN: water (1 : 1) and dried to obtain 2.71 g of the precipitate. The precipitates were checked for optical purity and the last precipitate of crystalline title compound (optical purity 100 %) was used in the X-ray diffraction study.
Example 9. Synthesis of Ethyl-5 -((S) 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxylate


HO
Zinc trifluoromethanesulfonate (0.259 g; 0.713 mmol) and (S)-(-)-3-butyn-2-ol (0.25 g; 3.57 mmol) were added to 0.75 ml (5.35 mmol) of Et3N under nitrogen
atmosphere. Ethyldiazoacetate (0.45 ml; 4.28 mmol) was added slowly and the
mixture was heated at 100 °C for 2 h. The mixture was cooled to RT and 5 ml of water was added. The mixture was washed with 15 ml of DCM, 5 ml of water was added and phases were separated. Water phase was washed twice with DCM, all organic layers were combined, dried with phase separator filtration and evaporated to dryness to give 0.523 g of crude material. The product was purified by normal phase column chromatography (0-5 % MeOH:DCM) to give 0.165 mg of the title compound. 1H-NMR (400MHz; d6-DMSO; temp +300 K): Tautomer 1 (major 77%): δ 1.28 (t, 3H), 1.39 (d, 3H), 4.20-4.28 (m, 2H), (d, 1H), 4.75-4.85 (m, 1H) 5.43 (broad d, 1H), 6.54 (broad s, 1H), 13.28 (broad s, 1H). Tautomer 2 (minor 23%): δ 1.28 (t, 3H), 1.39 (d, 3H), 4.20-4.28 (m, 2H), 4.66-4.85 (m, 1H), 5.04-5.15 (broad s, 1H), 6.71 (broad s, 1H), 13.60 (broad s, 1H).
Exam le 10. Ethyl-5 -((R)- 1 -hydroxy ethyl)- 1 H-pyrazole-3 -carboxylate

Zinc trifluoromethanesulfonate (1.037 g; 2.85 mmol) and (R)-(+)-3-butyn-2-ol (1.00 g; 14.27 mmol) were added to 2.98 ml (21.40 mmol) of Et3N under nitrogen atmosphere. Ethyldiazoacetate (1.80 ml; 21.40 mmol) was added slowly and then refluxed for 3 h. The mixture was cooled to RT and 45 ml of water was added. The mixture was extracted with 3×50 ml of DCM, organic layers were combined, dried with phase separator filtration and evaporated to dryness to give 2.503 g of crude material which was purified by normal phase column chromatography (0-10 % MeOH:DCM) to give 0.67 lmg of the title compound. 1H-NMR (400MHz; d6-DMSO; temp +300 K): Tautomer 1 (major 78%): δ 1.28 (t, 3H), 1.39 (d, 3H), 4.18-4.35 (m, 2H), (d, 1H), 4.75-4.85 (m, 1H) 5.42 (broad d, 1H), 6.54 (s, 1H), 13.29 (broad s, 1H). Tautomer 2 (minor 22%): δ 1.28 (t, 3H), 1.39 (d, 3H), 4.18-4.35 (m, 2H), 4.66-4.85 (m, 1H), 5.09 (broad s, 1H), 6.71 (broad s, 1H), 13.61 (broad s, 1H).
References
- “Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies.”. Sci Rep. 5: 12007. 2015. doi:10.1038/srep12007. PMC 4490394
 . PMID 26137992.
. PMID 26137992.
- Fizazi K, Albiges L, Loriot Y, Massard C (2015). “ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer”. Expert Rev Anticancer Ther. 15(9): 1007–17. doi:10.1586/14737140.2015.1081566. PMID 26313416.
- Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, Nykänen PS, Törmäkangas OP, Palvimo JJ, Kallio PJ (2015). “Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies”. Sci Rep. 5: 12007.doi:10.1038/srep12007. PMC 4490394. PMID 26137992.
- “ODM-201 is safe and active in metastatic castration-resistant prostate cancer”. Cancer Discov. 4 (9): OF10. 2014. doi:10.1158/2159-8290.CD-RW2014-150. PMID 25185192.
- Pinto Á (2014). “Beyond abiraterone: new hormonal therapies for metastatic castration-resistant prostate cancer”. Cancer Biol. Ther. 15 (2): 149–55. doi:10.4161/cbt.26724.PMC 3928129. PMID 24100689.
- Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, Garcia JA, Protheroe A, Tammela TL, Elliott T, Mattila L, Aspegren J, Vuorela A, Langmuir P, Mustonen M (2014). “Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial”. Lancet Oncol. 15 (9): 975–85. doi:10.1016/S1470-2045(14)70240-2. PMID 24974051.
- Agarwal N, Di Lorenzo G, Sonpavde G, Bellmunt J (2014). “New agents for prostate cancer”. Ann. Oncol. 25 (9): 1700–9. doi:10.1093/annonc/mdu038. PMID 24658665.
External links
Fenner A. Prostate cancer: ODM-201 tablets complete phase I. Nat Rev Urol. 2015 Dec;12(12):654. doi: 10.1038/nrurol.2015.268. Epub 2015 Nov 3. PubMed PMID: 26526759.
2: Massard C, Penttinen HM, Vjaters E, Bono P, Lietuvietis V, Tammela TL, Vuorela A, Nykänen P, Pohjanjousi P, Snapir A, Fizazi K. Pharmacokinetics, Antitumor Activity, and Safety of ODM-201 in Patients with Chemotherapy-naive Metastatic Castration-resistant Prostate Cancer: An Open-label Phase 1 Study. Eur Urol. 2015 Oct 10. pii: S0302-2838(15)00964-1. doi: 10.1016/j.eururo.2015.09.046. [Epub ahead of print] PubMed PMID: 26463318.
3: Fizazi K, Albiges L, Loriot Y, Massard C. ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev Anticancer Ther. 2015;15(9):1007-17. doi: 10.1586/14737140.2015.1081566. PubMed PMID: 26313416; PubMed Central PMCID: PMC4673554.
4: Bambury RM, Rathkopf DE. Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide. Urol Oncol. 2015 Jul 7. pii: S1078-1439(15)00269-0. doi: 10.1016/j.urolonc.2015.05.025. [Epub ahead of print] Review. PubMed PMID: 26162486.
5: Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, Nykänen PS, Törmäkangas OP, Palvimo JJ, Kallio PJ. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. 2015 Jul 3;5:12007. doi: 10.1038/srep12007. PubMed PMID: 26137992; PubMed Central PMCID: PMC4490394.
6: Thibault C, Massard C. [New therapies in metastatic castration resistant prostate cancer]. Bull Cancer. 2015 Jun;102(6):501-8. doi: 10.1016/j.bulcan.2015.04.016. Epub 2015 May 26. Review. French. PubMed PMID: 26022286.
7: Bjartell A. Re: activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Eur Urol. 2015 Feb;67(2):348-9. doi: 10.1016/j.eururo.2014.11.019. PubMed PMID: 25760250.
8: De Maeseneer DJ, Van Praet C, Lumen N, Rottey S. Battling resistance mechanisms in antihormonal prostate cancer treatment: Novel agents and combinations. Urol Oncol. 2015 Jul;33(7):310-21. doi: 10.1016/j.urolonc.2015.01.008. Epub 2015 Feb 21. Review. PubMed PMID: 25708954.
9: Boegemann M, Schrader AJ, Krabbe LM, Herrmann E. Present, Emerging and Possible Future Biomarkers in Castration Resistant Prostate Cancer (CRPC). Curr Cancer Drug Targets. 2015;15(3):243-55. PubMed PMID: 25654638.
10: ODM-201 is safe and active in metastatic castration-resistant prostate cancer. Cancer Discov. 2014 Sep;4(9):OF10. doi: 10.1158/2159-8290.CD-RW2014-150. Epub 2014 Jul 9. PubMed PMID: 25185192.
11: Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, Garcia JA, Protheroe A, Tammela TL, Elliott T, Mattila L, Aspegren J, Vuorela A, Langmuir P, Mustonen M; ARADES study group. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol. 2014 Aug;15(9):975-85. doi: 10.1016/S1470-2045(14)70240-2. Epub 2014 Jun 25. PubMed PMID: 24974051.
12: Agarwal N, Di Lorenzo G, Sonpavde G, Bellmunt J. New agents for prostate cancer. Ann Oncol. 2014 Sep;25(9):1700-9. doi: 10.1093/annonc/mdu038. Epub 2014 Mar 20. Review. PubMed PMID: 24658665.
13: Pinto Á. Beyond abiraterone: new hormonal therapies for metastatic castration-resistant prostate cancer. Cancer Biol Ther. 2014 Feb;15(2):149-55. doi: 10.4161/cbt.26724. Epub 2013 Nov 1. Review. PubMed PMID: 24100689; PubMed Central PMCID: PMC3928129.
14: Yin L, Hu Q, Hartmann RW. Recent progress in pharmaceutical therapies for castration-resistant prostate cancer. Int J Mol Sci. 2013 Jul 4;14(7):13958-78. doi: 10.3390/ijms140713958. Review. PubMed PMID: 23880851; PubMed Central PMCID: PMC3742227.
15: Leibowitz-Amit R, Joshua AM. Targeting the androgen receptor in the management of castration-resistant prostate cancer: rationale, progress, and future directions. Curr Oncol. 2012 Dec;19(Suppl 3):S22-31. doi: 10.3747/co.19.1281. PubMed PMID: 23355790; PubMed Central PMCID: PMC3553559.
//////////// Bayer HealthCare, Orion, Antineoplastics, Androgen receptor antagonists, Phase III, Prostate cancer, BAY 1841788, ODM-201, даролутамид , دارولوتاميد , 达罗他胺 , دارولوتاميد , ダロルタミド
O=C(N[C@@H](C)Cn1ccc(n1)c2ccc(C#N)c(Cl)c2)c3cc(nn3)C(O)C


 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....