
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
BMS 986001
Censavudine, Festinavir
Has anti-HIV activity. IN PHASE 2
CAS: 634907-30-5, UNII: 6IE83O6NGA, OBP 601, 4′-Ethynyl D4T, 4′-Ed4T, TDK-4-114
Molecular Formula, C12-H12-N2-O4, Molecular Weight, 248.2368
2′,3′-Didehydro-3′-deoxy-4′-ethynylthymidine,
1-((2R,5R)-5-Ethynyl-5-(hydroxymethyl)-2H-furan-2-yl)-5-methyl-pyrimidine-2,4-dione,
2′,3′-Didehydro-3′-deoxy-4′-ethynylthymidine
INNOVATOR= YALE UNIVERSITY
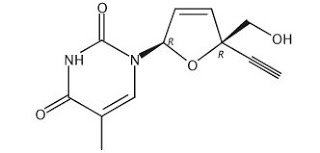
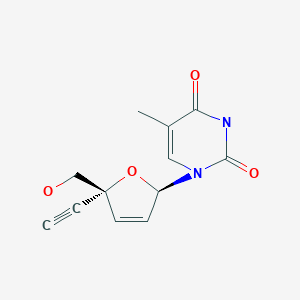
![]()
Festinavir is a nucleoside reverse transcriptase inhibitor
(NRTI) which is being developed for the treatment of HIV infection. The drug has shown considerable efficacy in early development, and with perhaps less toxicity than some other NRTIs, such as the drug stavudine (marketed under the trade name ZERIT®).
Festinavir has the chemical form and the structural formula:

Festinavir was developed by Yale University in conjunction with two Japanese research scientists, and is protected by U.S. Patent No. 7,589,078, the contents of which are incorporated herein by reference. The ‘078 patent sets forth the synthesis of the primary compound, and other structural analogs. In addition, Oncolys BioPharma, Inc. of Japan has now published US 2010/0280235 for the production of 4′ ethynyl D4T. As starting raw material, the Oncolys method utilizes a substituted furan compound, furfuryl alcohol. In another publication by Nissan Chemical Industries of Japan, and set forth in WO 201 1/099443, there is disclosed a method for producing a beta-dihydrofuran deriving compound or a beta-tetrahydrofuran deriving compound. In this process, a diol compound is used as the starting material. Nissan has also published WO 2011/09442
directed to a process for the preparation of a β-glycoside compound. Two further publications, each to Hamari Chemicals of Japan, WO 2009/1 19785 and
WO 2009/125841, set forth methods for producing and purifying ethynyl thymide compounds. Pharmaset, Inc. of the U.S. has also published US 2009/0318380,
WO 2009/005674 and WO 2007/038507 for the production of 4’ -nucleoside analogs for treating HIV infection. Reference is also made to the BMS application entitled
“Sulfilimine and Sulphoxide Methods for Producing Festinavir” filed as a PCT application, PCT/US2013/042150 on May 22, 2013 (now WO2013/177243).
PAPER
Haraguchi, Kazuhiro; Bioorganic & Medicinal Chemistry Letters 2003, V 13(21), PG 3775-3777
http://dx.doi.org/10.1016/j.bmcl.2003.07.009
http://www.sciencedirect.com/science/article/pii/S0960894X0300831X
Compounds having methyl, vinyl, and ethynyl groups at the 4′-position of stavudine (d4T: 2′,3′-didehydro-3′-deoxythymidine) were synthesized. The compounds were assayed for their ability to inhibit the replication of HIV in cell culture. The 4′-ethynyl analogue (15) was found to be more potent and less toxic than the parent compound stavudine.
Graphic


- Physical data for 15 are as follows: solid (mp 207–209 °C);
- UV (MeOH) λmax 264 nm (ε 10800), λmin 235 nm (ε 4800);
- 1H NMR (CDCl3) δ 1.83 (3H, s, Me), 2.63 (1H, s, C≡CH), 3.47 (1H, br, OH), 3.88 (1H, d,Jgem=12.5 Hz, H-5′a), 3.96 (1H, d, Jgem=12.5 Hz, H-5′b), 5.91 (1H, dd, J1′,2′=1.1 Hz and J2′,3′=5.9 Hz, H-2′), 6.30 (1H, dd, J1′,3′=2.0 Hz and J2′,3′=5.9 Hz, H-3′), 7.16–7.17 (1H, m, H-1′), 7.44 (1H, d, J6,Me=1.1 Hz, H-6), 9.06 (1H, br, NH);
- FAB-MS m/z 249 (M++H). Anal. calcd for C12H12N2O4·1/6H2O: C, 57.37; H, 4.95; N, 11.15. Found: C, 57.36; H, 4.69; N, 10.98.
- PAPER
- Scalable Synthesis of the Potent HIV Inhibitor BMS-986001 by Non-Enzymatic Dynamic Kinetic Asymmetric Transformation (DYKAT)
Angewandte Chemie, International Edition (2015), 54, (24), 7185-7188. - http://onlinelibrary.wiley.com/doi/10.1002/anie.201502290/abstract
- http://onlinelibrary.wiley.com/store/10.1002/anie.201502290/asset/supinfo/anie_201502290_sm_miscellaneous_information.pdf?v=1&s=9c516d28bb61a8b090de88c2a75f5f50f060aaa9
-
Scalable Synthesis of the Potent HIV Inhibitor BMS-986001 by Non-Enzymatic Dynamic Kinetic Asymmetric Transformation (DYKAT)†
Described herein is the synthesis of BMS-986001 by employing two novel organocatalytic transformations: 1) a highly selective pyranose to furanose ring tautomerization to access an advanced intermediate, and 2) an unprecedented small-molecule-mediated dynamic kinetic resolution to access a variety of enantiopure pyranones, one of which served as a versatile building block for the multigram, stereoselective, and chromatography-free synthesis of BMS-986001. The synthesis required five chemical transformations and resulted in a 44 % overall yield.

white crystalline solid. 1: Rf = 0.8 (silica, MeOH:CH2Cl2,1:4);
M.P. = 196-207°C;
1 H NMR (d6-DMSO, 500 MHz): δ = 11.34 (s, 1 H), 6.88 (s, 1 H), 6.35 (d, J = 6.0 Hz, 6.05 (d, J = 6.0 Hz, 1 H), 5.45 (t, J = 5.5 Hz, 1 H), 3.69 (dd, J = 12.0, 1.5 Hz, 1 H), 3.64 (s, 1 H), 3.59 (dd, J = 12.0, 1.5 Hz, 1 H) 1.70 (s, 3 H) ppm;
13C NMR (d6-DMSO, 125 MHz): δ = 163.85, 150.82, 136.81, 135.54, 127.13, 109.04, 88.94, 86.60, 81.45, 77.39, 65.76, 12.23 ppm;
HRMS calcd for C12H12N2O4H+ [M + H+] 249.09 found 249.08.
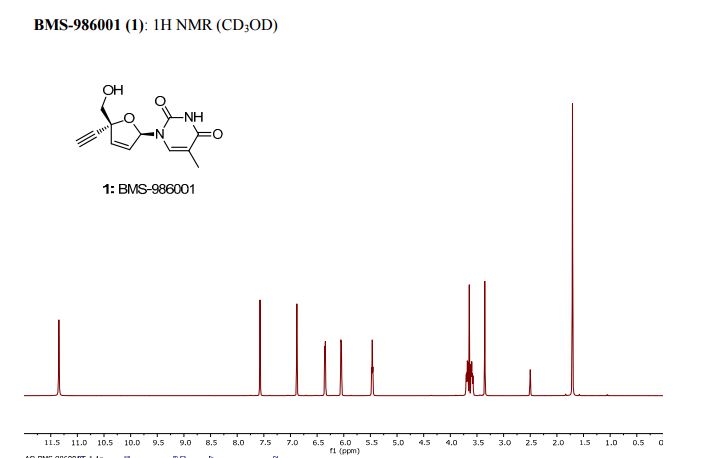

PATENT
WO 2014172264
https://www.google.ch/patents/WO2014172264A1?cl=en
invention:

Step#l: Acetal Formation

Compound 1
85% yield
The starting material is 5-methylurdine, which is commercially available. The first step of the process is an acetal formation. 5-methyluridine is utilized and is treated with H2SO4 and acetaldehyde. Other acids available to the scientist, such as perchloric acid, will also work for this transformation. The solvent utilized for this step is acetonitrile (ACN), and other solvents may also be utilized as well. Once the starting material is consumed, a slurry is obtained and the product can be simply filtered off and dried to provide Compound 1 as a solid.

Acetal formation
Preparation of l-((3aR,4R,6R,6aR)-6-(hydroxymethyl)-2-methyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl)-5-methylpyrimidine-2,4(lH,3H)-dione
The following were added to a flask: 5-methyluridine (10 g, 38.70 mmol), acetonitrile (20 mL) and 70% perchloric acid (4.01 mL, 47.63 mmol). A solution of acetaldehyde (3.26 mL, 58.10 mmol) in acetonitrile (20 mL) was added dropwise over 1 h. The resulting solution was allowed to stir at 20 °C for 18 h. The resulting slurry was filtered and dried (50 °C, 25 mmHg) to afford Acetal (9.30 g, 84% yield) as white solid
XH NMR (400MHz, DMSO-d6) δ = 11.39 (s, 1H), 7.72 – 7.63 (m, 1H), 5.82 (d, J=3.0 Hz, 1H), 5.21 – 5.07 (m, 2H), 4.84 (dd, J=6.6, 2.5 Hz, 1H), 4.68 (dd, J=6.6, 3.0 Hz, 1H), 4.12 – 4.05 (m, 1H), 3.65 – 3.51 (m, 2H), 3.36 (s, 2H), 1.77 (s, 3H), 1.37 (d, J=5.1 Hz, 3H) 13C NMR (101MHz, DMSO-d6) δ = 163.77, 150.32, 137.64, 109.39, 104.50, 90.79, 86.16, 83.83, 81.37, 61.25, 19.76, 12.06
Step #2: Acetate protection

Compound 2
85% yield
The next step of the sequence is installation of a 4-biphenylacetate. Without being bound by any particular theory, this protecting step may be chosen for two reasons:
1) To provide a solid intermediate that can be easily isolated, and
2) Act as a directing group in the next step (set forth later on).
This reaction consists of reacting Compound 1 with 4-biphenyl acid chloride and pyridine in acetonitrile. In this reaction, pyridine is preferred as it allows the reaction to occur only at the -OH moiety of the molecule. It should also be noted that other polar solvents could be used, but acetonitrile allowed the desired product Compound 2 to be isolated as s solid.

Ac lation
Preparation of ((3aR,4R,6R,6aR)-2-methyl-6-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)tetrahydrofuro[3,4-d] [l,3]dioxol-4-yl)methyl [1,1′-biphenyl]-4-carboxylate.
Acetal (9.30 g, 32 mmol) was dissolved into acetonitrile (100 mL). Pyridine (1.3 eq) was added followed by the addition of 4-biphenylcarbonyl chloride (1.05 eq). The solution was heated to 50 °C and held for 2 h. The slurry was cooled to 20 °C and held for 2 h. The slurry was filtered and washed with acetonitrile (100 mL). The solids were dried (50 °C, 25 mmHg) to Compound 2 (85% yield).
XH NMR (400MHz, CHLOROFORM-d) δ = 8.10 (d, J=8.1 Hz, 2H), 7.62 (d, J=7.6 Hz, 2H), 7.67 (d, J=8.1 Hz, 2H), 7.55 – 7.36 (m, 3H), 7.09 (s, 1H), 5.71 (s, 1H), 5.26 (q, J=4.7 Hz, 1H), 5.03 (dd, J=6.6, 2.0 Hz, 1H), 4.91 (dd, J=6.7, 3.2 Hz, 1H), 4.73 – 4.63 (m, 1H), 4.61 – 4.50 (m, 2H), 2.02 (s, 3H), 1.85 – 1.76 (m, 3H), 1.52 (d, J=4.8 Hz, 3H)
1JC MR (101MHz, CHLOROFORM-d) δ = 164.02, 161.94, 148.20, 144.18, 137.85, 135.89, 128.20, 127.05, 126.36, 126.30, 125.35, 125.26, 1 14.49, 109.20, 103.88, 92.51, 83.36, 83.29, 79.87, 75.45, 75.13, 74.81, 62.54, 17.92, 10.32, -0.01

With the acetal and 4-biphenylacetate groups in place, the next reaction is a regioselective acetal opening utilizing TMSOTf (Trimethylsilyl trifluoromethane sulfonate, or other available Lewis acids)/Et3N to afford the corresponding silyl ether, which is cleaved in situ, to afford the 2-vinyloxy compound as Compound 3. Compound 3 may be prepared in a step-wise fashion (shown below), but in order to reduce the number of steps, it is possible to take Compound 3 and selectively form the desired 2-vinyl oxy regioisomer Compound 3. Those skilled in the art may recognize that the 4-biphenylacetate can be important to obtain high selectivity for this transformation.


![]()
Although a variety of Lewis acids may be utilized, TMSOTf is generally found to be more effective. Et3 is also a preferred reactant, as other amine bases are generally less effective. The ratio of TMSOTf to Ets is preferably within the range of about 1 : 1.3; if the reaction medium became acidic, Compound 3 would revert back to Compound 2. In terms of solvents, DCM (Dichloromethane) may be particularly effective, but toluene, CF3-PI1, sulfolane, and DCE (Dichloroethene) are also effective. The reaction can be worked up using aqueous acid, preferably K2HP04, or methanolic NH4F to quench the reaction, as well as remove the TMS-ether in situ.

TMSOTf-opening
Preparation of ((2R,3R,4R,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-4-(vinyloxy)tetrahydrofuran-2-yl)methyl [1,1′-biphenyl]-4-carboxylate
Compound 2 (20 g, 43.06 mmol) was dissolved into DCM (160 mL). Triethylamine (78 mL, 560 mmol) was added followed by the addition of TMSOTf (80.30 mL, 431 mmol). This solution was heated to 45 °C and held there until complete by HPLC analysis (6 h). Once complete, this solution was added to ammonium acetate (66.40 g, 861 mmol) in water (200 mL). After stirring for 20 min, the layers were separated. The organics were concentrated and the resulting residue was dissolved into EtOAc (200 mL). The organics were washed with the following solution (potassium phosphate monobasic (118 g, 861 mmol) in water (400 mL). The organics were then dried ( a2S04), filtered and concentrated. The resulting residue was purified by column chromatography [Silica gel; 20% to 90% EtOAc in Hexanes] to afford Compound 3 (15.8 g, 79% yield) as a solid.
XH NMR (400MHz, CHLOROFORM-d) 6 = 9.18 (br. s., IH), 8.18 – 8.06 (m, 2H), 7.73 -7.56 (m, 4H), 7.55 – 7.38 (m, 3H), 7.24 (d, J=1.3 Hz, IH), 6.59 (dd, J=14.0, 6.4 Hz, IH), 5.81 (d, J=2.0 Hz, IH), 4.84 (dd, J=12.6, 2.5 Hz, IH), 4.63 (dd, J=12.5, 4.2 Hz, IH), 4.59 – 4.44 (m, 3H), 4.40 – 4.26 (m, 2H), 1.70 (d, J=1.0 Hz, 3H)
13C MR (101MHz, CHLOROFORM-d) δ = 166.13, 163.65, 150.00, 149.67, 146.39, 139.66, 135.67, 130.16, 129.01, 128.40, 128.06, 127.32, 127.28, 111.43, 91.93, 89.44, 81.60, 80.19, 69.32, 63.06, 12.32
Step #4: Iodiiiation

Compound 4
Compound 3 75% yie|d
Next, Compound 3 is transformed into the iodide compound which is Compound 4. This can be accomplished by treating Compound 3 with (2.0 eq), PPI13 (2.0 eq.) and imidazole (4.0 eq). Other methods to install the iodide may also be utilized, such as mesylation/Nal, etc., but these may be less preferred. In addition, other halogen-bearing compounds such as Br2 and CI2 may be considered by the skilled scientist. Premixing imidazole, , and PPh3, followed by addition of Compound 3 in THF and heating at 60 °C allows smooth conversion to Compound 4. It is highly preferred to add all reagents prior to the addition of Compound 3; if not, the vinyloxy group will be cleaved. Other solvents, such as 2-MeTHF and PhMe may be utilized, but THF often provides the best yield.

Iodiiiation
Preparation of ((2R,3S,4S,5R)-3-iodo-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-4-(vinyloxy)tetrahydrofuran-2-yl)methyl [l,l’-biphenyl]-4-carboxylate
The following were added to a flask: imidazole (8.79 g, 129 mmol),
triphenylphosphine (16.94 g, 65 mmol), iodine 16.39 g, 65 mmol) and THF (525 mL). A solution of Compound 3 (15 g, 32 mmol) in THF (375 mL) was added. The solution was heated to 60 °C and was held at 60 °C for 4 h. Once complete by HPLC analysis (4 h), the solution was concentrated and the residue was purified by column chromatography [Silica gel; 10% to 60% EtOAc in Hexanes] to afford Compound 4 (17.0 g, 92% yield) as a solid.
XH NMR (400MHz, CHLOROFORM-d) δ = 9.25 (br. s., IH), 8.16 (d, J=8.3 Hz, 2H), 7.75 – 7.61 (m, 5H), 7.54 – 7.40 (m, 3H), 7.32 – 7.24 (m, 2H), 7.23 – 7.16 (m, 2H), 6.56 -6.45 (m, IH), 6.06 (d, J=1.5 Hz, IH), 4.89 (s, IH), 4.66 (dd, J=12.0, 6.9 Hz, IH), 4.56 (dd, J=12.0, 3.9 Hz, IH), 4.46 (d, J=4.0 Hz, IH), 4.39 – 4.26 (m, 2H), 4.13 (dt, J=7.1, 3.8 Hz, 1H), 2.06 – 1.97 (m, 3H)
1JC MR (101MHz, CHLOROFORM-d) δ = 165.96, 163.94, 150.27, 149.29, 146.28, 139.81, 137.88, 135.84, 130.37, 129.06, 129.01, 128.34, 128.25, 127.94, 127.31, 127.22, 125.32, 1 11.07, 91.37, 90.32, 89.18, 78.43, 69.15, 25.81, 21.49, 12.71
Step #5: Iodide Elimination

Compound 4
The next step of the sequence is to install the allyic moiety. Heating a solution of Compound 4 in toluene in the presence of DABCO (l,4-Diazabicyclo[2.2.2]octane) allows for elimination of the iodide. Other solvents, such as THF and DCE may be utilized, but toluene often provides the best conversion and yield. Other amine bases may be used in this transformation, but generally DABCO is preferred.

Elimination
Preparation of ((4R,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l (2H)-yl)-4-(vinyloxy)-4,5-dihydrofuran-2-yl)methyl [l,l’-biphenyl]-4-carboxylate
Compound 4 (17 g, 30 mmol) was dissolved into toluene (255 niL), and DABCO (10 g, 89 mmol) was added. The solution was heated to 90 °C and held there for 2 h. Once complete, the organics were washed with sat. aq. a2S203 (200 mL). The organics were then dried ( a2S04), filtered, and concentrated. The resulting residue was purified by column chromatography [Silica gel; 5% to 60% EtOAc in Hexanes] to yield
Compound 5 (10.9, 85% yield) as a foam.
XH NMR (400MHz, CHLOROFORM-d) δ = 8.93 (br. s., IH), 8.18 – 8.11 (m, 2H), 7.75 -7.61 (m, 5H), 7.55 – 7.39 (m, 4H), 6.95 (d, J=1.0 Hz, IH), 6.54 (d, J=2.0 Hz, IH), 6.46 (dd, J=14.3, 6.7 Hz, IH), 5.53 (d, J=2.5 Hz, IH), 5.09 (d, J=2.8 Hz, IH), 5.04 (d, J=6.6 Hz, 2H), 4.29 (dd, J=14.3, 2.4 Hz, IH), 4.23 (dd, J=6.7, 2.4 Hz, IH), 1.88 (d, J=1.0 Hz, 3H)
1JC MR (101MHz, CHLOROFORM-d) δ = 165.73, 159.58, 149.10, 146.49, 139.70, 134.51, 132.17, 132.07, 131.94, 131.92, 130.30, 129.01, 128.56, 128.44, 128.40, 127.73, 127.30, 127.28, 112.50, 99.16, 90.57, 90.23, 84.81, 58.68, 12.44
Step #6: Claisen Rearrangement

An important reaction in the sequence is the Claisen rearrangement. This reaction is utilized to install the quaternary stereocenter and the olefin geometry in the ring. Heating Compound 5 in benzonitrile at 190 °C for 2-3 hours allows for smooth conversion to Compound 6, and after chromatography, a 90% yield can be achieved.
Toluene (110 °C, 8 h) also works to provide the desired Compound 6 as a solid by simply cooling the reaction to 20 °C (no chromatography). Other solvents with boiling points over about 100°C may also be utilized.

Claisen Rearrangement
Preparation of ((2S,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2-(2-oxoethyl)-2,5-dihydrofuran-2-yl)methyl [l,l’-biphenyl]-4-carboxylate
Compound 5 (1 mmol) was dissolved into benzonitrile (10 mL). The solution was heated to 190 °C for 3 h. After cooling to 20 °C, the solution was purified by column chromatography [silica gel, 50:50 Hexanes:EtOAc] to afford Compound 6 (1 mmol).
Alternatively, Compound 5 (1 mmol) was dissolved into toluene (10 mL). The solution was heated to 110 °C and held for 12 h. Upon cooling to 20 °C, a slurry formed. The solids were filtered, washed (PhMe) and dried (50 °C, 25 mmHg) to afford
Compound 6 (1 mmol) as a white solid.
XH NMR (400MHz, CHLOROFORM-d) δ = 9.84 (t, J=1.8 Hz, 1H), 8.53 (br. s., 1H), 8.13 – 8.03 (m, J=8.3 Hz, 2H), 7.73 – 7.67 (m, 2H), 7.67 – 7.60 (m, 2H), 7.56 – 7.38 (m, 3H), 7.14 (d, J=1.3 Hz, 1H), 7.04 (t, J=1.5 Hz, 1H), 6.57 (dd, J=6.1, 2.0 Hz, 1H), 6.02 (dd, J=5.9, 1.1 Hz, 1H), 4.68 – 4.52 (m, 2H), 3.06 – 2.89 (m, 2H), 1.59 (d, J=1.0 Hz, 3H)
13C MR (101MHz, CHLOROFORM-d) δ = 198.33, 165.83, 163.35, 150.65, 146.56, 139.63, 136.24, 135.02, 130.21, 129.04, 128.44, 127.86, 127.49, 127.41, 127.28, 111.59, 90.03, 89.61, 67.33, 50.06, 12.06
ne Formation via elimination of Enol Nonaflate

The alkyne formation is performed by first treating Compound 6 with TMSCl (Trimethylsilyl chloride)/Et3N. NfF (Nonafluoro- 1 -butanesulfonyl fluoride) and P-base () are then added at -20 °C. After warming to 20 °C, the desired alkyne Compound 7 can be isolated in about 80 % yield. Initially, TMSCl is presumed to react at the NH moiety. NfF/P-base then reacts with the aldehyde to form the enol Nonaflate. Upon warming to 20 °C in the presence of P-base, the enol Nonaflate eliminates smoothly to the alkyne Compound 7. Without the TMSCl/Et3N, the yields are only -25%.

Alkyne formation
Preparation of ((2R,5R)-2-ethynyl-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2,5-dihydrofuran-2-yl)methyl [l,l’-biphenyl]-4-carboxylate
Compound 6 (1 g, 2.24 mmol) was dissolved into DMF (Dimethylformamide) (5 mL). (Other polar solvents could also have been used.) Triethylamine (406 uL, 2.91 mmol) was added and the solution was cooled to 0 °C. TMSCl (314 uL, 2.46 mmol) was added and the solution was allowed to stir at 0 °C for 30 min. The solution was then cooled to -20 °C, and NfF (484 uL, 2.69 mmol) was added and the solution was allowed to stir at -20 °C for 5 min. Phosphazane P l-base (1.54 mL, 4.93 mmol) was added
dropwise over 20 min. The solution was then allowed to warm to 20 °C and held for 20 h. The solution was then poured into water (50 mL) and extracted with DCM (100 mL). The organics were concentrated and the resulting residue was purified by column chromatography [Silica gel; 10% to 60% EtOAc in Hexanes] to afford Compound 7 (816 mg, 85% yield) as a solid.
XH NMR (400MHz, DMSO-d6) δ = 11.46 (s, 1H), 8.08 – 7.97 (m, J=8.6 Hz, 2H), 7.92 -7.80 (m, 2H), 7.73 (d, J=7.1 Hz, 2H), 7.59 – 7.39 (m, 3H), 7.06 (d, J=1.0 Hz, 1H), 6.89 (d, J=1.5 Hz, 1H), 6.61 (dd, J=5.6, 2.0 Hz, 1H), 6.23 (dd, J=5.6, 1.0 Hz, 1H), 4.66 (d, J=12.1 Hz, lH), 4.57 (d, J=11.6 Hz, 1H), 3.87 (s, 1H), 1.37 (s, 3H)
13C MR (101MHz, DMSO-d6) δ = 164.89, 163.57, 150.61, 145.13, 138.73, 135.30, 134.40, 129.94, 129.12, 128.49, 127.84, 127.78, 127.18, 126.98, 110.01, 89.37, 83.69, 80.01, 78.23, 66.89, 11.46

90% yield
The final step of the sequence is to remove the aromatic ester protecting group. This consists of hydrolysis by NaOH in aq. THF solution. The API is extracted into THF and then crystallized from THF/PhMe.

Deprotection
Preparation of l-((2R,5R)-5-ethynyl-5-(hydroxymethyl)-2,5-dihydrofuran-2-yl)-5-methylpyrimidine-2,4(lH,3H)-dione (Ed4T)
Compound 7 (10 g, 23.40 mmol) was dissolved into THF (100 mL). 3N NaOH (10 mL) was added. The solution was allowed to stir at 20 °C for 12 h. The layers were split and the organics were kept. The organics were concentrated to reach a KF <1 wt%. Toluene (100 mL) was added, and solids crashed out of solution. The solids were filtered and washed with Toluene (100 mL). The solids were then dried (50 °C, 25 mmHg) to afford Festinavir (5.21 g, 90% yield) as a white solid.
XH NMR (400MHz, DMSO-d6) δ = 1 1.36 (s, 1H), 7.58 (s, 1H), 6.89 (s, 1H), 6.36 (d, J=6.1 Hz, 1H), 6.05 (d, J=6.1 Hz, 1H), 5.48 (t, J=5.6 Hz, 1H), 3.78 – 3.49 (m, 3H), 3.46 3.31 (m, 1H), 1.71 (s, 3H)
1JC MR (101MHz, DMSO-d6) δ = 163.80, 150.76, 136.75, 135.47, 127.06, 108.98, 88.87, 86.52, 81.37, 77.33, 65.68, 12.17.
PAPER
Tetrahedron (2009), 65(36), 7630-7636.
Volume 65, Issue 36, 5 September 2009, Pages 7630–7636

Synthesis of (±)-4′-ethynyl-5′,5′-difluoro-2′,3′-dehydro-3′-deoxy- carbocyclic thymidine: a difluoromethylidene analogue of promising anti-HIV agent Ed4T
PAPER
Nucleophilic Substitution at the 4‘-Position of Nucleosides: New Access to a Promising Anti-HIV Agent 2‘,3‘-Didehydro-3‘-deoxy-4‘-ethynylthymidine
Journal of Organic Chemistry (2006), 71(12), 4433-4438.
http://pubs.acs.org/doi/abs/10.1021/jo060194m

For the synthesis of 2‘,3‘-didehydro-3‘-deoxy-4‘-ethynylthymidine (8: 4‘-Ed4T), a recently reported promising anti-HIV agent, a new approach was developed. Since treatment of 1-(2,5-dideoxy-β-l–glycero-pent-4-enofuranosyl)thymine with Pb(OBz)4 allowed the introduction of the 4‘-benzoyloxy leaving group, nucleophilic substitution at the 4‘-position became feasible for the first time. Thus, reaction between the 4‘-benzoyloxy derivative (14) and Me3SiC⋮CAl(Et)Cl as a nucleophile led to the isolation of the desired 4‘-“down”-ethynyl derivative (18) stereoselectively in 62% yield. As an application of this approach, other 4‘-substituted nucleosides, such as the 4‘-allyl (24a) and 4‘-cyano (26a) derivatives, were synthesized using organosilicon reagents. In these instances, pretreatment of 14 with MeAlCl2 was necessary.

PATENTS
US75890782009-09-15Anti-viral nucleoside analogs and methods for treating viral infections, especially HIV infections
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016060252 | 2016-03-03 | 5-METHYLURIDINE METHOD FOR PRODUCING FESTINAVIR |
| US2015140610 | 2015-05-21 | SULFILIMINE AND SULPHOXIDE METHODS FOR PRODUCING FESTINAVIR |
| US2015104511 | 2015-04-16 | Pharmaceutical Antiretroviral Combinations Comprising Lamivudine, Festinavir and Nevirapine |
| US8927237 | 2015-01-06 | Method for producing acyloxypyranone compound, method for producing alkyne compound, and method for producing dihydrofuran compound |
| US2012322995 | 2012-12-20 | beta-DIHYDROFURAN DERIVING COMPOUND, METHOD FOR PRODUCING beta-DIHYDROFURAN DERIVING COMPOUND OR beta-TETRAHYDROFURAN DERIVING COMPOUND, beta-GLYCOSIDE COMPOUND, METHOD FOR PRODUCING beta GLYCOSIDE COMPOUND, AND METHOD FOR PRODUCING 4′-ETHYNYL D4T AND ANALOGUE COMPOUNDS THEREOF |
| US2012252751 | 2012-10-04 | ANTI-VIRAL NUCLEOSIDE ANALOGS AND METHODS FOR TREATING VIRAL INFECTIONS, ESPECIALLY HIV INFECTIONS |
| US8193165 | 2012-06-05 | Anti-viral nucleoside analogs and methods for treating viral infections, especially HIV infections |
| US2011312880 | 2011-12-22 | POTENT CHIMERIC NRTI-NNRTI BIFUNCTIONAL INHIBITORS OF HIV-1 REVERSE TRANSCRIPTASE |
| US2011054164 | 2011-03-03 | PRODUCTION PROCESS OF ETHYNYLTHYMIDINE COMPOUNDS FROM 5-METHYLURIDINE AS A STARTING MATERIAL |
| US2010280235 | 2010-11-04 | METHOD FOR PRODUCING 4’ETHYNYL d4T |
/////////BMS 986001, 634907-30-5, UNII: 6IE83O6NGA, OBP 601, 4′-Ethynyl D4T, 4′-Ed4T, TDK-4-114, PHASE 2
Cc1cn(c(=O)[nH]c1=O)[C@H]2C=C[C@](O2)(CO)C#C

