
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
SNS 032, BMS-387032
N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide
Cas 345627-80-7, MP 165-167° C
M.Wt:380.53, Formula:C17H24N4O2S2
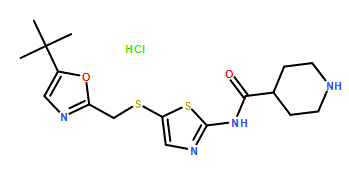
SNS 032, BMS-387032 HYDROCHLORIDE
| Formula | C17H24N4O2S2 . HCl |
|---|---|
| MW | 380.5 . 36.5 |
| CAS | 345627-90-9 |
A potent and selective Cdk inhibitor
Potent inhibitor of cyclin-dependent kinases (cdks) 9, 2 and 7 (IC50 values are 4, 38 and 62 nM respectively). Displays no activity against 190 additional kinases (IC50 >1000 nM). Arrests the cell cycle at G2/M; inhibits transcription, proliferation and colony formation, and induces apoptosis in RPMI-8226 multiple myeloma cells. Prevents tumor cell-induced VEGF secretion and in vitro angiogenesis. SNS-032 (BMS-387032) has firstly been described as a selective inhibitor of CDK2 with IC50 of 48 nM in cell-free assays and is 10- and 20-fold selective over CDK1/CDK4. It is also found to be sensitive to CDK7/9 with IC50 of 62 nM/4 nM, with little effect on CDK6. Phase 1.
Quality Control & MSDS
COA NMR HPLC Datasheet SDS/MSDS
- COA (Certificate Of Analysis)
- HPLC
- NMR (Nuclear Magnetic Resonance)
- MSDS (Material Safety Data Sheet)
SNS-032 (BMS-387032) is a potent and selective inhibitor of cyclin-dependent kinases (CDKs) 2, 7, and 9 [1], with IC50 values of 38 nM, 62 nM and 4 nM, respectively [2].
CDKs mean a family of serine/threonine kinases regulating cell cycle process. Some CDKs are related to transcription control and are often perturbed in cancer cells [3].
Decrease in the phosphorylation at Ser5 and Ser2 in the C-terminal domain (CTD) of RNA Pol II can indicate the inhibition to CDK9 and CDK7 [1]. Chronic lymphocytic leukemia (CLL) cells treated with SNS-032 for 6 or 24 hours showed a decrease in the phosphorylation of Ser2 and Ser5 of the CTD of RNA Pol II, this appeared to be both time- and concentration- dependent, and remarkably consistent among samples. For the phosphorylation of Ser2, the inhibition of SNS-032 was greater than that for the phosphorylation of Ser5, this was consistent with the fact that IC50 for the inhibition of CDK9 was lower compared with that for the inhibition of CDK7 (4 nM vs 62 nM). After 6 hours of SNS-032 exposure, protein levels of CDK7 and CDK9 were stable, but declined at 24 hours [4].
In patients with chronic lymphocytic leukemia (CLL), infusion of SNS-032 in a total dose of 75 mg/m2 resulted in a decrease in the phosphorylation at Ser5 and Ser2 in the C-terminal domain of RNA Pol II. This indicated the inhibition to Cdk9 and Cdk7 by SNS-032. This inhibition was first seen 2 hours after the beginning of the infusion with SNS-032, was pronounced after 6 hours and returned to baseline after 24 hours [1].

The cell cycle-regulated cyclin-dependent kinases (CDKs), CDK1, 2, and 4 have been extensively studied as potential therapeutic targets in cancer. Recent research has additionally underscored the potential role of several constitutively active CDKs including CDK7 and 9 as cancer targets. Phosphorylation of the c-terminal domain (CTD) of RNA Polymerase II by CDK7 and 9 are critical steps in transcriptional regulation. Inhibition of these kinases is predicted to have the greatest effect on the expression of proteins with short t½ and short-lived mRNA, including proteins involved in apoptotic regulation. CDK7 also activates cell-cycle CDKs 1, 2, 4 and 6. SNS-032 (formerly BMS-387032) has previously been described as a selective inhibitor of CDK2 with potent antitumor activity in animal models. Here we show that in addition to inhibition of CDK2, SNS-032 also inhibits CDK7/cyclinH and CDK9/cyclinT at low nanomolar concentrations in biochemical assays. The compound is highly selective for CDK inhibition; in a panel of 208 kinases, only four non-CDK proteins were inhibited by >50% at 1 μM SNS-032. The cellular pharmacology of SNS- 032 mirrors the biochemical data. Cells treated with SNS-032 show a rapid cell cycle arrest and onset of cell death that corresponds with inhibition of multiple substrates of CDK2, 7, and 9. For instance, inhibition of Rb phosphorylation, accumulation of cyclin E protein and cell-cycle arrest at GI and G2 are observed in multiple cell lines in a time and dose-dependent manner, consistent with inhibition of CDK2 and CDK7. Furthermore, SNS-032 inhibits CDK9-mediated phosphorylation of Ser2 in the CTD with an IC50 = 200 nM. Corresponding with inhibition of RNA polymerase II, the short half-life, anti-apoptotic protein Mcl-1 is rapidly depleted from cells, coincident with the phosphorylation of p53. Expression of Mcl-1 is a candidate predictor of aggressive disease and resistance to chemotherapy in CLL and is essential for survival of B-cell lymphoma and multiple myelomas, supporting the use of SNS-032 as a treatment for these diseases. SNS-032, a selective inhibitor of multiple CDKs involved in apoptosis and cell cycle regulation, has potential for antitumor activity in both solid and hematological cancers. SNS-032 is currently in phase 1 clinical studies.
SNS-032, was designed as a selective CDK2 inhibitor. Here, we show that in addition to CDK2, CDK 7 and 9 inhibitory activities also contribute to the biological activity of the molecule. The CDK2/cyclin E complex regulates entry of cells into S phase by phosphorylating Rb, a negative regulator of the transcription factor E2F. CDK2 phosphorylates a number of additional substrates, including cyclin E, signaling its degradation. Inhibiting CDK2 should therefore arrest cells in G1 and stabilize cyclin E. The cellcycle CDKs (CDK1, 2 4 and 6) are activated by phosphorylation by CDK7/cyclin H (also called CAK). Inhibition of CDK7 would therefore also result in cell-cycle arrest at multiple points in the cell cycle due to failure to activate the cell cycle CDKs. CDK 7 and 9 activate transcription by phosphorylating the CTD of RNA pol II. Inhibition of CTD phosphorylation has been shown to inhibit transcription and reduce expression of short lived proteins, including those involved in apoptosis regulation. Stalling of RNA polymerase has also been shown to activate p53, leading to apoptosis. Thus, the CDK7 and 9 inhibitory activities of SNS-032 are expected to cause cytotoxicity via induction of apoptosis.
SNS-032 is a selective CDK inhibitor, preferentially targeting CDK2, CDK7 and CDK9 in vitro. • In cell models, SNS-032 shows dual activity, targeting both cell cycle progression and apoptosis pathway proteins. • SNS-032 Inhibited CDK9 and 7-mediated phosphorylation of ser 2 and ser 5 of the CTD of RNA pol II and in turn downregulates the antiapoptotic protein Mcl-1. • SNS-032 induced a cell cycle arrest, and increased cyclin E levels are consistent with inhibition of cell cycle CDKs • Mcl-1 is a key survival factor in many B-cell malignancies. SNS-032 is being pursed as treatment for these diseases.
| Biological Activity | ||||||
|---|---|---|---|---|---|---|
| Description | SNS-032 is a novel, potent and selective CDK inhibitor of CDK2, CDK7 and CDK9 with IC50 of 38 nM, 62 nM and 4 nM, respectively. | |||||
| Targets | CDK2 | CDK7 | CDK9 | |||
| IC50 | 38 nM | 62 nM | 4 nM [1] | |||
| In Vitro | SNS-032 has low sensitivity to CDK1 and CDK4 with IC50 of 480 nM and 925 nM, respectively. SNS-032 effectively kills chronic lymphocytic leukemia cells in vitro regardless of prognostic indicators and treatment history. Compared with flavopiridol and roscovitine, SNS-032 is more potent, both in inhibition of RNA synthesis and at induction of apoptosis. SNS-032 activity is readily reversible; removal of SNS-032 reactivates RNA polymerase II, which led to resynthesis of Mcl-1 and cell survival. [1] SNS-032 inhibits three dimensional capillary network formations of endothelial cells. SNS-032 completely prevents U87MG cell–mediated capillary formation of HUVECs. In addition, SNS-032 significantly prevents the production of VEGF in both cell lines, SNS-032 prevents in vitro angiogenesis, and this action is attributable to blocking of VEGF. Preclinical studies have shown that SNS-032 induces cell cycle arrest and apoptosis across multiple cell lines. [2] SNS-032 blocks the cell cycle via inhibition of CDKs 2 and 7, and transcription via inhibition of CDKs 7 and 9. SNS-032 activity is unaffected by human serum. [3]SNS-032 induces a dose-dependent increase in annexin V staining and caspase-3 activation. At the molecular level, SNS-032 induces a marked dephosphorylation of serine 2 and 5 of RNA polymerase (RNA Pol) II and inhibits the expression of CDK2 and CDK9 and dephosphorylated CDK7. [4] | |||||
| In Vivo | SNS-032 prevents tumor cell-induced VEGF secretion in a tumor coculture model. [2] SNS-032, a new CDK inhibitor, is more selective and less cytotoxic and has been shown to prolong stable disease in solid tumors. [4] | |||||
| Clinical Trials | SNS-032 currently in phase I clinical trial for chronic lymphocytic leukemia (CLL) and multiple myeloma (MM). | |||||
| Description | SNS-032 is a selective inhibitor of CDK2 with IC50 of 48 nM. | |||||
| Targets | CDK2 | CDK7 | CDK9 | |||
| IC50 | 48 nM | 62 nM | 4 nM | |||
CLIP
http://www.mdpi.com/1420-3049/19/9/14366/htm#B39-molecules-19-14366
SNS032, previously called BMS-387032, has been developed by Sunesis. This compound, which contains a thiazole unit, selectively inhibits CDK2 (IC50: 38 nM), CDK7 (IC50: 62 nM) and CDK9 (IC50: 4 nM) [39]. Preclinical studies demonstrated that SNS032 was able to inhibit cell cycle activity along with transcription [20].
SNS032 is in phase I clinical trials for the treatment of chronic lymphoid leukemia along with multiple myeloma, and the mode of administration is intravenous [39]. The purpose is to evaluate the dose-escalation of SNS-032 along with its safety, pharmacokinetics, pharmacodynamic activity and clinical efficacy. Biomarker analyses demonstrated mechanism-based pharmacodynamic activity with inhibition of CDK7 and CDK9, although limited clinical activity in heavily pretreated patients was observed [39].
Tong, W.G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective CDK2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. ASCO Annual Meeting. J. Clin. Oncol. 2010, 28, 3015–3022.

![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://journals.prous.com/journals/dof/20083311/html/df330932/images/sch01.gif)
SNS-032 (formerly BMS-387032) is a small-molecule cyclin-dependent kinase (CDK) inhibitor currently in phase I clinical trials for the treatment of B-cell malignancies and advanced solid tumors. Preclinical studies have shown that SNS-032 is a specific and potent inhibitor of CDK2, 7 and 9 which induces cell cycle arrest and apoptosis in tumor cell lines. It was shown to inhibit in vitro angiogenesis and prostaglandin E2 (PGE2) production, both strongly associated with tumorigenesis. Phase I clinical trials support the safety and tolerability of SNS-032 as evaluated in dose-escalation studies. The compound is currently administered by i.v. infusion but has shown promising potential for oral delivery.
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://journals.prous.com/journals/dof/20083311/html/df330932/images/sch02.gif)
 NMR
NMR
CLIP
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://i0.wp.com/www.rcsb.org/pdb/images/56H_600.gif)
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://i0.wp.com/www.mdpi.com/ijms/ijms-14-21805/article_deploy/html/images/ijms-14-21805f2-1024.png)
The structures of representative protein kinases inhibitors based on the aminopyrazole scaffold.http://www.mdpi.com/1422-0067/14/11/21805/htm
CLIP
N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,

N-Acyl-2-aminothiazoles with nonaromatic acyl side chains containing a basic amine were found to be potent, selective inhibitors of CDK2/cycE which exhibit antitumor activity in mice. In particular, compound 21 {N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide, BMS-387032}, has been identified as an ATP-competitive and CDK2-selective inhibitor which has been selected to enter Phase 1 human clinical trials as an antitumor agent. In a cell-free enzyme assay, 21 showed a CDK2/cycE IC50 = 48 nM and was 10- and 20-fold selective over CDK1/cycB and CDK4/cycD, respectively. It was also highly selective over a panel of 12 unrelated kinases. Antiproliferative activity was established in an A2780 cellular cytotoxicity assay in which 21 showed an IC50 = 95 nM. Metabolism and pharmacokinetic studies showed that 21 exhibited a plasma half-life of 5−7 h in three species and moderately low protein binding in both mouse (69%) and human (63%) serum. Dosed orally to mouse, rat, and dog, 21showed 100%, 31%, and 28% bioavailability, respectively. As an antitumor agent in mice, 21administered at its maximum-tolerated dose exhibited a clearly superior efficacy profile when compared to flavopiridol in both an ip/ip P388 murine tumor model and in a sc/ip A2780 human ovarian carcinoma xenograft model.
CLIP

http://pubs.rsc.org/en/content/articlehtml/2016/md/c6md90040b
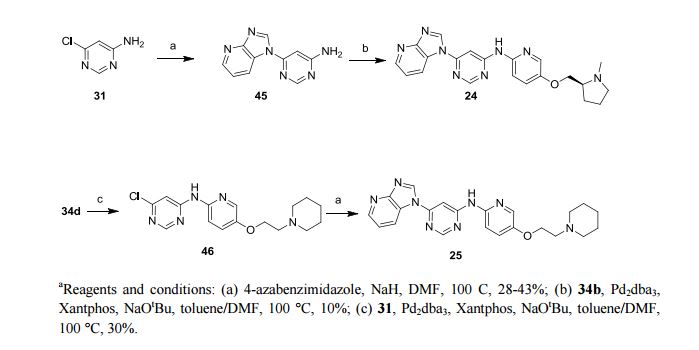
Heat shock factor 1 (HSF1) is a transcription factor that plays key roles in cancer, including providing a mechanism for cell survival under proteotoxic stress. Therefore, inhibition of the HSF1-stress pathway represents an exciting new opportunity in cancer treatment. We employed an unbiased phenotypic screen to discover inhibitors of the HSF1-stress pathway. Using this approach we identified an initial hit (1) based on a 4,6-pyrimidine scaffold (2.00 μM). Optimisation of cellular SAR led to an inhibitor with improved potency (25, 15 nM) in the HSF1 phenotypic assay. The 4,6-pyrimidine 25 was also shown to have high potency against the CDK9 enzyme (3 nM).

Discovery of 4,6-disubstituted pyrimidines as potent inhibitors of the heat shock factor 1 (HSF1) stress pathway and CDK9
E-mail: Paul.Workman@icr.ac.uk, Keith.Jones@icr.ac.uk
DOI: 10.1039/C6MD00159A
COMPD 25
1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.21 (s, 1H), 8.74 (d, J = 0.9 Hz, 1H), 8.62 (dd, J = 8.2, 1.5 Hz, 1H), 8.56 (dd, J = 4.7, 1.5 Hz, 1H), 8.16-8.13 (m, 2H), 7.64 (br d, J = 8.6 Hz, 1H), 7.52-7.47 (m, 2H), 4.14 (t, J = 5.9 Hz, 2H), 2.66 (t, J = 5.9 Hz, 2H), 2.47-2.42 (m, 4H), 1.53-1.47 (m, 4H), 1.42 – 1.33 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 160.74, 158.32, 156.72, 154.88, 150.74, 146.47, 145.38, 143.74, 134.21, 125.02, 124.16, 122.29, 119.60, 114.32, 94.06, 66.49, 57.35, 54.35, 25.54, 23.88. HRMS (ESI+ ): calcd for C22H25N8O (M + H)+ , 417.2146; found 417.2163.
NOTE, THERE IS ERROR IN STRUCTURE ABOVE OF SNS 032
References
References:
[1]. Tong W.G., Chen R., Plunkett W., et al. Phase I and Pharmacologic Study of SNS-032, a Potent and Selective Cdk2, 7, and 9 Inhibitor, in Patients With Advanced Chronic Lymphocytic Leukemia and Multiple Myeloma. Journal of Clinical Oncology, 2010, 28(18):3015- 3022.
[2]. Chipumuro E., Marco E., Christensen C.L., et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell, 2014, 159:1-14.
[3]. Meng H., Jin Y.M., Liu H., et al. SNS-032 inhibits mTORC1/mTORC2 activity in acute myeloid leukemia cells and has synergistic activity with perifosine against Akt. Journal of Hematology & Oncology, 2013, 6:18.
[4]. Chen R., Wierda W.G., Chubb S., et al. Mechanism of action of SNS032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood, 2009, 113(19):4637-4645.Chen et al (2010) Responses in mantle cell lymphoma cells to SNS-032 depend on the biological context of each cell line. Cancer Res. 70 6587. PMID: 20663900.
Conroy et al (2009) SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemother.Pharmacol. 64 723. PMID: 19169685.
Ali et al (2007) SNS-032 prevents tumor cell-induced angiogenesis by inhibiting vascular endothelial growth factor. Neoplasia 9 370. PMID: 17534442.
Misra et al (2004) N-(Cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J.Med.Chem. 47 1719. PMID: 15027863.
Abstract
SNS-032, a CDK inhibitor, exhibited modest to high anti-neuroblastoma activity against a panel of 109 neuroblastoma cell lines in the range of the therapeutic plasma levels reported for SNS-032 through a mechanism involving CDK7 and CDK9 inhibition-mediated down-regulation of XIAP, Mcl-1, BIRC2, cIAP-1 and surviving.
Abstract
The anti-AML mechanism of SNS-032, a cyclin-dependent kinase inhibitor, has been identified though characterizing in vitro effects of SNS-032 alone or in combination with perifosine.
Abstract
Although it induces apoptosis in cancer cells, SNS-032 has no significant effects on normal HSC and HPC in terms of self-renewal inhibition, differentiation suppression and apoptosis induction.
Abstract
The CDK7/9 inhibitor SNS-032-induced down-regulation of FIP1L1-PDGFRα and Bcr-Abl has the potential to be used to decrease the acquired resistant to imatinib.
Abstract
SNS-032, a CDK inhibitor, alone or in combination with Ara-C exhibited potent anti-AML activity, where down-regulation of antiapoptotic genes, cluding BCL2, XIAP amd MCL1, was associated with the synergistic anti-AML effect of the combination treatment.
CC(C)(C)C1=CN=C(O1)CSC2=CN=C(S2)NC(=O)C3CCNCC3

