
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
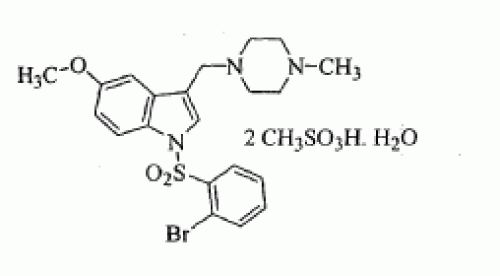
SUVN-502
CAS OF MONOHYDRATE MESYLATE 1791396-45-6
CAS MESYLATE 1791396-46-7
1-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-1-piperazinyl)methyl]-1H-indole dimesylate monohydrate
l-{(2-BROMOPHE YL) SULFONYLJ-5-METHOXY-3- [(4-METHYL-l-PIPERAZINYL) METHYLJ-1H-INDOLE DIMESYLATE MONOHYDRATE
l-[(2- bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l-piperazinyl)methyl]-lH-indoIe dimesylate monohydrate
MF OF DIMESYLATE – C21 H24 Br N3 O3 S . 2 C H4 O3 S
Serotonin 6 receptor antagonists

……………..BASE form of SUVN-502
1 -[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l -piperazinyl)methyl]-lH-indole
CAS OF BASE 701205-60-9, 478.40, C21 H24 Br N3 O3 S
1H-Indole, 1-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-1-piperazinyl)methyl]-, methanesulfonate (1:2)
5-HT 6 receptor antagonist
SUVN-502 (in phase II)
https://www.clinicaltrials.gov/ct2/show/NCT02580305
Suven Life Sciences Ltd
IN 2013CH05537

Used as 5-HT 6 receptor antagonist for treating Alzheimer’s disease, attention deficit hyperactivity disorder, Parkinson’s disease and schizophrenia.
SUVN-502
SUVN-502 is a pure 5-HT6 receptor antagonist with >1200-fold selectivity over 5-HT2A receptor with a superior profile that differentiates from competitor 5-HT6 antagonists. SUVN-502 has an excellent human pharmacokinetics for once a day treatment.
The Phase 2A trial is designed to evaluate the safety, tolerability, pharmacokinetics and efficacy of SUVN-502 for the treatment of moderate Alzheimer’s Disease (AD).This trial is expected to enrol 537 patients and the primary objective of the study is to evaluate the efficacy of a serotonin receptor subtype 6 (5-HT6) antagonist, SUVN-502, at daily doses of 50 mg or 100 mg compared to placebo, as adjunct treatment in subjects with moderate Alzheimer’s disease (Mini-Mental State Examination [MMSE] score of 12 to 20) currently treated with the acetylcholinesterase inhibitor, Donepezil Hydrochloride (HCl) and the N-methyl-D-aspartic acid (NMDA) antagonist, MemantineHCl. Efficacy will be assessed by the 11-item Alzheimer’s Disease Assessment Scale for Cognitive Behaviour (ADAScog-11) after 26 weeks of treatment. The trial is likely to complete by end of second quarter 2017, subject to the achievement of estimated 12 months’ enrolment goal in USA.
Secondary objectives of this POC study are to further evaluate the efficacy of these treatments usingClinical Dementia Rating (CDR) Scale, Sum of Boxes (CDR-SB), MMSE, Alzheimer’s Disease Co-operative Study Activity of Daily Living (ADCS-ADL), Neuropsychiatric Inventory (NPI) 12 item and Cornell Scale for Depression and Dementia (C-SDD).
This study is being coordinated by Dr. Jeffrey Cummings, MD, Director, Cleveland Clinic Lou RuvoCenter for Brain Health, Las Vegas, NV, USA.
Prior to the initiation of Phase 2A study, SUVN-502 has successfully undergone two phase 1 studies in Switzerland and USA on 122 healthy young and elderly male populations with no major adverse events and no serious adverse events.
5-HT6 receptor is one of the potential therapeutic target for the development of cognitive enhancers for the treatment of Alzheimer’s disease (AD) and schizophrenia. 5-HT6 receptor is localized exclusively in central nervous system, in areas important for learning and memory. In recent years several studies (Brain Research, 1997, 746, 207-219; Journal of
Neuroscience, 1998, 18(15), 5901-5907; International Review of Neurobiology Volume 96, 201 1 , 27-47 & Annual Reviews in Pharmacology and Toxicology, 2000, 40, 319-334a) have reported that 5-HT6 receptor antagonists show beneficial effect on cognition in animal models.
PATENT
WO2015083179
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015083179
l-[(2- bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l-piperazinyl)methyl]-lH-indoIe dimesylate monohydrate of formula (I) of the present invention is illustrated by the Sc eme-1 as given below:

Mannich Adduct

Scheme-1
Example 1: Preparation of l-[(2-bromophenyI)suIfonyl]-5-methoxy-3-[(4-methyl-l-piperazinyI)methyl]-lH-indole dimesylate monohydrate
Step (i) & (u): Preparation of 5-methoxy-3-[(4-methyl-l-piperazinyI)methyl]-lH-indole
Step (i):
1-Methylpiperazine (15 Kg, 0.15 Kg Mole) was charged into a reactor. The mass was cooled to 5 °C – 10 °C. Demineralised water (12 Kg) was added to the above mass slowly, maintaining the mass temperature 10 °C – 20 °C, over a period of 30 minutes. Then added acetic acid (6.16 Kg, 0.103 Kg Mole) to the above mass in 30 minutes, maintaining the mass temperature at 10 °C – 20 °C. The mass was further stirred for another 15 – 20 minutes at 10 °C – 20 °C and aqueous formaldehyde solution (15.67 Kg, 30 % w/v, 0.1567 Kg Mole) was added in 60 minutes maintaining the mass temperature at 15 °C – 20 °C. The resultant thick, red colored reaction mass was stirred for another 2 hours at 20 °C – 30 °C to obtain the mannich adduct.
Step (ii):
Simultaneously in a separate reactor 125 Kg of methanol was charged at 25 °C – 35 °C. 5-methoxyindole (20 Kg, 0.1359 Kg Mole) was added and the mass was stirred to obtain a clear solution. The mass was cooled to 8 °C – 10 °C in 1.5 hours by circulating brine in the reactor jacket. The Mannich adduct, prepared as above, was charged into the reactor containing cooled methanolic solution of 5-methoxyindole from an addition tank over a period of 50 – 60 minutes, while maintaining the temperature of the reaction mass at 8 °C – 16 °C. After completion of addition, the mass temperature was allowed to rise to 20 °C – 35 °C. Then the reaction mass was further stirred for 3 hours at 20 °C – 35 °C. After completion of the reaction (thin layer chromtography), the reaction mass was discharged into clean and dry containers.
Another reactor was charged with 400 L of demineralised water followed by the addition of 20 Kg of lye solution at 20 °C – 35 °C. The content was cooled to 10 °C – 15 °C under stirring. The above reaction mass in the containers was added to the reactor, maintaining the mass temperature at 10 °C – 15 °C in 30 – 40 minutes. The final pH of the solution was adjusted to 9 – 12, if necessary by adding some more lye solution. Then the product was extracted with ethyl acetate (1 x 260 L & 4 x 160 L) maintaining the mass temperature at 10 °C – 15 °C during the entire operations. The pH of aqueous layer was adjusted to 9 – 12 before each extraction.
The combined organic layer was washed with (2 x 170 Kg) of brine solution (the brine solution was prepared by adding 95 Kg of vacuum salt to 245 Kg of demineralised water) at 20 °C – 35 °C. The total organic extracts, obtained after the brine washing, were dried over 35 Kg of anhydrous sodium sulfate under stirring for 30 minutes at 20 °C – 35 °C.
The organic layer was filtered and charged into another clean reactor. The solvent was removed totally under 500 – 600 mm of Hg vacuum, at 20 °C – 45 °C.
The residual mass, thus obtained, was cooled to room temperature and charged 60 L toluene and stirred the contents at 20 °C – 45 °C for 15 minutes. The solvent was distilled off under reduced pressure (500 – 700 mm of Hg vacuum) at 45 °C – 65 °C. The operation was repeated again by the addition of 60 L toluene and stirring the contents at 20 °C – 45 °C for 15 min. The solvent was distilled off under reduced pressure (500 – 700 mm of Hg vacuum) at 45 °C – 65 °C again to ensure total removal of ethylacetate to avoid losses during recrystallization step. The residual technical product, 5-methoxy-3-[(4-methyl-l- piperazinyl)methyl]-lH-indole, thus obtained, was recrystallized twice, as per the details given below, to obtain the product of desired purity.
Step (Hi): Crystallization of 5-methoxy-3-[(4-methyI-l-piperazinyl)methyl]-lH-indoIe
Charged 61 Kg of toluene into the above reactor which contains the technical product, 5-methoxy-3-[(4-methyl-l-piperazinyl)methyl]-lH-indole. The contents were heated to 85 °C – 95 °C and maintained for an hour at 85 °C – 95 °C. The clear solution, thus obtained, was allowed to cool to 30 °C – 40 °C by circulating room temperature water in the reactor jacket. The mass was further cooled to 10 °C – 15 °C and maintained for 3 hours at the same temperature. The crystalline solid mass was filtered through nutsche and the solid on the nutsche was washed with 18 L of chilled (10 °C – 15 °C) toluene and sucked well. The material was further washed with 20 L of n-hexane and sucked dry to obtain 22.7 Kg of crystalline material.
Step (iv): Recrystallization of 5-methoxy-3-[(4-methyI-l-piperazinyI)methyl]-lH-indole
Charged 40 Kg of toluene into a reactor followed by the addition of the 5-methoxy- 3-[(4-methyl-l-piperazinyl)methyl]-l H-indole (22.7 Kg) obtained in the first crystallization step under stirring. The contents were heated to 95 °C – 105 °C and maintained for 2 hours to obtain a clear solution. The mass was allowed to cool to 35 °C -40 °C by circulating room temperature water in the jacket. It was further cooled to 10 °C -15 °C and maintained for 3 hours at 10 °C – 15 °C. The crystalline solid mass was filtered through nutsche and the solid on the nutsche was washed with 8 L of chilled (10 °C – 15 °C) toluene and sucked well. The material was further washed with 15 L of n-hexane and sucked dry. The material was further dried in tray driers at 20 °C – 25 °C to obtain the title product, as off white crystalline powder.
Weight of the crystallized material: 19.95 Kg;
Yield (based on 5-methoxyindole charged): 56.6 %;
HPLC purity: 99.74 %;
Total impurities: 0.26 %;
Assay: 100.6 %;
Moisture content: 0.24 %;
Melting range (°C): 139 – 140.6;
IR spectra (cm“1): 3125, 2951, 1875, 1622, 1585, 1492, 1351, 1288, 1215, 1059, 930, 654; Ή – NMR (CDCI3, δ ppm): 2.30 (3H, s), 2.5 (8H, bs), 3.71 (2H, s), 3.86 (3H, s), 6.83 -6.86 (1H, dd, J = 8.81, 2.7 Hz), 7.01 (1H, d, J = 2.06 Hz), 7.18 – 7.20 (2H, m), 8.91 (1H, s); 13C – NMR (CDCI3, δ ppm): 45.89, 52.79, 53.39, 55.1 1, 55.83, 101.3, 1 1 1.39, 11 1.75, 1 11.81, 124.88, 128.45, 131.48, 153.77;
Mass [M+H]+: 260.3.
Step (v): Preparation of l-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l-piperazinyl)methyI]-lH-indoIe
Tetrahydrofuran (85.78 Kg) was charged into a reactor at 20 °C – 35 °C. Then charged the crystallized 5-methoxy-3-[(4-methyl-l-piperazinyl)methyl]-lH-indole (21.5 Kg, 0.0829 Kg Mole) into the reactor at 20 – 35 °C and stirred the mass well. The mass was cooled to 10 °C – 20 °C with chilled water in the jacket. Charged powdered potassium hydroxide (16.1 1 Kg) to the above suspension at 10 °C – 20 °C in 10 minutes under stirring. Slight exotherm was observed. Mass temperature rose from 15.1 °C to 16.3 °C. The mass was further stirred for 60 minutes at 10 °C – 20 °C. A solution of 2-bromobenzenesulfonyl chloride (27.71 Kg, 0.1084 Kg Mole) in 41.72 Kg tetrahydrofuran was added through addition tank at a constant rate in 60 minutes at 10 °C – 30 °C. The reaction was exothermic and the mass temperature went up from 16 °C to 30 °C. Then removed the chilled water from the jacket and stirred the mass for 3 hours at 25 °C – 35 °C. As the reaction was progressing the mass thickened due to formation of potassium chloride. The progress of the reaction was monitored by thin layer chromatography (Eiuent system: Chloroform and Methanol in 8:2 ratio and the product is relatively non-polar). Since thin layer chromatography shows the presence of starting material (5-methoxy-3-[(4-methyl-l-piperazinyl)methyl]-lH-indole), another lot of 2-bromo benzenesulfonyl chloride (4.5 Kg, 0.0176 Kg Mole) dissolved in 13.71 Kg tetrahydrofuran was added to the reaction mass at 30 °C in 25 minutes. No exotherm observed. The reaction mass was further stirred for 60 minutes at 30 °C – 35 °C. Since the starting material was absent as per thin layer chromatography, it was taken for further workup.
In the mean while charged 360 L demineralised water into another reactor and cooled the contents to 10 °C – 15 °C. The above reaction mass was quenched into chilled water in 60 minutes (mass temperature was 12.1 °C). The pH of the reaction mass was adjusted to ~ 9.5 with an aqueous solution of potassium hydroxide. The product was extracted with (4 x 155 L) ethyl acetate maintaining the mass temperature at 10 °C – 15 °C. The pH of aqueous layer was adjusted to ~ 9.5 before each extraction. The combined organic layer was taken for extraction of the product into aqueous acetic acid. …. j
Acetic acid (8.69 Kg, 0.1448 Kg mole) was dissolved in 137 L of demineralised water and cooled the mass to 10 °C – 15 °C. Charged the above organic extracts into it and stirred for 30 minutes at 10 °C – 15 °C. The mass was allowed to settle for 20 minutes and separated the bottom aqueous acetic acid extract containing the product into a fresh clean reactor.
Further, the extraction and separation process with fresh aqueous acetic acid solution was repeated thrice using 3 x 145 Kg of aqueous acetic acid solution (prepared by dissolving 25.74 Kg, 0.429 Kg Mole of acetic acid in 412 L of demineralised water) following the similar procedure mentioned above, maintaining mass temperature at 10 °C -15 °C. The combined aqueous acetic acid extracts (containing the product) were taken into the reactor. It was washed with 44 L of ethyl acetate by stirring the mass at 10 °C – 15 °C for 15 minutes, followed by 15 minutes settling. The aqueous product layer was separated. The pH of the aqueous solution was found to be 4.5. The mass was cooled to 10 °C – 15 °C and the pH of the solution was adjusted to ~ 9.5 with chilled caustic lye solution (31 Kg). The product was extracted with (4 x 155 L) of ethyl acetate, maintaining the mass temperature at 10 °C – 15 °C. The pH of aqueous layer was adjusted to ~ 9.5 before each extraction.
The organic layer was washed with (2 x 1 12 Kg) brine solution (prepared from 51.6 Kg vacuum salt and 175 L water) at 10 °C – 15 °C. The organic layer was dried over 32 Kg of anhydrous sodium sulfate at 20 °C – 35 °C and filtered into another clean reactor.
Solvent was removed under 500 – 600 mm Hg by circulating 50 °C – 55 °C water in the jacket of the reactor.
To the residual mass in the reactor after solvent removal, charged 36 L of methanol followed by 72 L of isopropanol. The reaction mass was heated to reflux temperature (65 °C – 75 °C). At mass temperature ~ 70 °C a clear solution was obtained. The mass was allowed to cool to 35 – 45 °C with room temperature water circulation in the reactor jacket. Further, it was cooled to 15 °C – 20 °C by circulating brine in the jacket and maintained under stirring for 2 hours at 15 °C – 20 °C. The solids were filtered through nutsche and sucked well under vacuum. The cake was washed with 36 L of isopropanol (15 °C – 20 °C) and sucked well. The wet solid material (37.76 Kg) was taken in tray drier and air dried at 25 °C – 35 °C for 60 minutes. Further, it was dried at 40 °C – 45 °C for 6 hours to obtain 32.64 Kg of the title product.
Overall Yield: 82.3 % (based on Mannich base charged);
HPLC purity: 99.36 %;
Single major impurity: 0.29 %;
Total impurities: 0.64 %;
Assay: 100.5 %;
Loss on drying at 105 °C: 0.21 %;
Melting range (°C): 128.1 – 129.2;
IR spectra (cm‘1): 2931, 2786, 1607, 1474, 1369, 1222, 1 178, 1032, 737, 597;
Ή – NMR (CDC13, δ ppm): 2.29 (3H, s), 2.32 – 2.50 (8H, bs), 3.62 (2H, s), 3.83 (3H, s),
6.83 – 6.86 (1H, dd, J = 8.98, 2.46 Hz), 7.19 – 7.20 (1H, d, J = 2.42 Hz), 7.36 – 7.40 (1 H, dt,
J.= 7.68, 1.56 Hz), 7.45 – 7.47 (1H, t, J = 7.50 Hz), 7.53 – 7.55 (1H, d, J = 9.00, Hz), 7.64 – 7.66 (2H, m), 8.03 – 8.05 (1H, dd, J = 7.89, 1.54 Hz);
13C – NMR (CDCI3, δ ppm): 45.94, 53.07, 53.33, 55.17, 55.60, 103.28, 1 13.20, 1 13.69,
117.83, 120.42, 127.05, 127.69, 129.57, 131.16, 131.57, 134.48, 135.90, 138.09, 156.12;
Mass [M+Hf: 478.1, 480.1.
Step (vi): Preparation of l-[(2-bromophenyl)sulfonyI]-5-methoxy-3-[(4-methyI-l-piperazinyl)methyI]-lH-indoIe dimesylate
Charged 182.5 Kg of absolute ethanol into a reactor at 20 °C – 35 °C. Then charged l-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l-piperazinyl)methyl]-lH-indole -(obtained in the above step, 32.02 Kg, 0.067 Kg Mole) under stirring in a single lot at 20 °C – 35 °C (mass temperature), added methanesulfonic acid (13.9 Kg, 0.1446 Kg Mole) slowly to the above reaction mass from a holding tank in 60 minutes, maintaining mass temperature at 20 °C – 35 °C. No clear solution was obtained at any stage. The mass became thick, but stirrable. The reaction mass was stirred for 24 hours maintaining mass temperature between 25 °C – 30 °C. The mass was filtered through nutsche under nitrogen atmosphere and it was sucked well. The cake, thus obtained, was washed thoroughly with 48 L of ethyl alcohol (slurry wash), sucked well and the cake was again washed with 18 L of ethyl alcohol (spray wash) followed by washing with n-hexane (27 L). It was sucked dry to obtain 70.23 Kg wet cake. The wet cake was taken in a tray drier and dried at 20 °C – 35 °C for 10 hours to obtain 49.43 Kg product (LOD: ~ 9.57 %).
Weight of product on dry basis: 44.65 Kg
Yield of salt: Quantitative (based on l -[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methy 1- 1 -piperaziny l)methy 1]- 1 H- indo le charged) ;
HPLC purity: 99.69 %;
Total impurities: 0.31 %;
Salt content: 27.39 %.
Step (vii): Preparation of l-[(2-bromop enyl)sulfonyI]-5-methoxyr3-[(4-methyI-l-piperazinyl)methyl]-lH-indole dimesylate monohydrate
Charged 415 Kg of aqueous ethanol (95 % ethanol & 5 % water) into a reactor, followed by the addition of l-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l-piperazinyl)methyl]-lH-indole dimesylate (44.65 Kg, 0.0666 Kg Mole, obtained from the above step) at 20 °C – 35 °C. In the meanwhile carbon slurry was prepared separately by adding 6.7 Kg of carbon powder into 18 Kg of aqueous ethanol (95 % ethanol & 5 % water). Then the carbon slurry was transferred to the reactor and the reaction mass was heated at 75 °C – 80 °C by circulating 80 °C – 90 °C hot water in the reactor jacket for 45 minutes. The mass was filtered hot into another clean reactor, washed the carbon bed with 54.25 Kg of aqueous ethanol (95% ethanol & 5% water) at 75 °C – 80 °C. The contents of the reactor were heated at reflux temperature (76 PC – 78 °C) for 30 minutes to obtain a clear solution. The mass was allowed to cool on its own to 45 °C in 10 hours by applying compressed air in the reactor jacket. It was further cooled to 10 °C – 15 °C with chilled water circulated in the jacket and maintained under stirring for 3 hours. Filtered the crystalline material through a centrifuge and the material on the centrifuge was washed with 18.6 Kg of aqueous ethanol (95 % ethanol & 5 % water) (10 °C – 15 °C) and spin dried. The whole material was air dried in a tray drier for 14 hours at 20 °C – 35 °C. The material was milled, sieved and collected in poly bag to obtain 37.7 Kg of the title product. The uniform material was sampled for analysis.
Weight of dry product: 37.7 Kg;
Yield of salt: 82.2 %;
HPLC purity: 99.7 %;
Single impurity: 0.3 %;
Assay: 99.9 %;
Moisture content: 2.61 %;
Salt content (Dimesylate) 27.56 %;
Melting range (°C): 218.0 – 220.0;
IR spectra (cm“1): 3148, 3012, 161 1, 1590, 1471, 1446, 1439, 1382, 1220, 1 194, 1 180, 1045, 775, 596;
Ή – NMR (D20, δ ppm): 2.65 (6H, s), 2.89 (3H, s), 3.52 (8H, bs), 3.70 (3H, s), 4.46 (2H, s), 6.75 – 6.78 (1H, dd, J = 9.07, 2.02 Hz), 7.10 – 7.1 1 (1H, d, J = 1.9 Hz), 7.32 – 7.38 (2H, m), 7.44 – 7.47 (1H, t, J = 7.6 Hz), 7.54 – 7.56 (1H, dd, J = 7.79 Hz), 8.04 (1H, s), 8.14 -8.16 (lH, d, J = 7.94 Hz);
, C – NMR (δ ppm): 38.42, 42.79, 48.19, 50.35, 55.80, 102.57, 108.20, 113.72, 114.07, 1 19.62, 128.25, 128.56, 130.17, 131.80, 132.15, 135.28, 135.95, 156.21 ;
Mass [M+H]+: 478, 480.
PATENT………on metabolite and not the drug
caution……….drug has a methyl
Suven Life Sciences Ltd is developing l-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl- l -piperazinyl)methyl]-lH-indole dimesylate monohydrate, which is a selective 5-HT6 receptor antagonists intended for the symptomatic treatment of AD and other disorders of memory and cognition like attention deficient hyperactivity, parkinson’s and schizophrenia. 1 -[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l -piperazinyl)methyl]-lH-indole, and its pharmaceutically acceptable salts were disclosed by Ramakrishna et al. in WO 2004/048330. l -[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l-piperazinyl)methyl]-lH-indole dimesylate;monohydrate has already completed Phase 1 clinical trials. Based on phase I clinical trials results, we confirmed l -[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(l -piperazinyl)methyl]-lH-indole of formula (I) as an active metabolite of l -[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl- 1 -piperazinyl)methyl]- 1 H-indoIe dimesylate monohydrate in human volunteers.
The development and understanding of the metabolism of l-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-l -piperazinyl)methyl]-lH-indole dimesylate monohydrate is desirable for progression of science and necessary step in the commercialization of this compound. Therefore, there is a need to understand regarding metabolism and metabolites of l-t(2-bromophenyl)sulfonyI]-5-methoxy-3-[(4-methyl-l -piperazinyl)methyl]-lH-indole dimesylate monohydrate.
In order to improve pharmaceutical properties and efficacy of active metabolite, we performed salt selection program for l -[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[( l -piperazinyl)methyl]-lH-indole. Based on the results obtained, dimesylate dihydrate salt of 1-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(l-piperazinyl)methyl]-lH-indole of formula (Π) is selected for further development along with the compound of formula (I).
 l -[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[( l -piperazinyl)methyl]-lH-indole. NOTE THE DRUG IS WITH A METHYL
l -[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[( l -piperazinyl)methyl]-lH-indole. NOTE THE DRUG IS WITH A METHYL

SCHEME 1

SCHEME2
Example 1: Preparation of l-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(l-piperazinyl)methyl]-lH-indo

Step (i) & (ii): Preparation of 3-[(l-t-Butyloxycarbonyl piperazin-4-yl)methyI]-5-methoxy-lH-indole

Step (i):
Demineralized water (DM water) (660 mL) and N-Boc piperazine ( 150.0 grams, 0.8034 moles) were charged into a 2 Litres three necked round bottomed flask provided with a mechanical stirrer and a thermometer pocket. The mass was stirred for 10 minutes at 25 °C, to obtain a clear solution. Then acetic acid (32.5 mL, 0.5416 moles) was added to the above mass while maintaining the mass temperature at ~ 25 °C in 10 minutes. After completion of addition, the clear solution was stirred at 25 °C for 30 minutes.
To the above stirred mass at 25 °C, aqueous formaldehyde solution (81 mL, 30 % w/v, 0.81 moles) was added slowly through an addition funnel over a period of 30 minutes maintaining the mass temperature below 25 °C. During the addition, white slurry mass was formed. The resultant white slurry mass was stirred for another 1 hour at 25 – 30 °C. Methanol (MeOH) (300 mL) was added to the above mass to obtain a clear solution. The solution was further stirred for 30 minutes at 25 °C to obtain Mannich adduct.
Step (ii):
5-Methoxyindole (106.4 grams, 0.7238 moles) and methanol (550 mL) were charged into a 4 necked round bottom flask. The mass was stirred for 10 minutes at 25 °C to obtain a clear solution and then cooled the mass to 18 – 20 °C. The mannich adduct (prepared in above step) was added to the flask through an addition funnel maintaining mass temperature below 20 °C, over a period of 1 hour. The mass was further stirred for a period of 1 hour at 25 – 30 °C, while monitoring the progress of the reaction by thin layer chromatography (TLC).
After completion of the reaction (1 hour), DM water (2.2 Litres) and ethyl acetate (1
Litre) were added to the reaction mass and pH adjusted to 10.5 (on pH paper) with lye solution (80 mL) maintaining the mass temperature at 20 – 24 °C. The organic (product) layer was separated and the aqueous layer was further extracted with ethyl acetate (2 x 500 mL). The combined organic layer was washed with saturated brine solution (300 mL) and dried over anhydrous sodium sulfate. The organic layer was filtered free of sodium sulfate and concentrated under reduced pressure. n-Hexane (300 mL) was added to the residual mass and further concentrated under vacuum for removal of traces of ethyl acetate to obtain 272.2 grams of technical product.
Purity: 96.16 %;
Ή – NMR (CDC13, δ ppm): 1.45 (9H, s), 2.44 (4H, bm), 3.41 – 3.43 (4H, bm), 3.69 (2H, s), 3.87 (3H, s), 6.85 – 6.88 (1H, dd, J = 8.75, 2.23 Hz), 7.10 ( 1 H, d, J = 0.96 Hz), 7.19 (1 H, d, J = 2.24 Hz), 7.24 – 7.26 (1H, d), 8.04 (1H, bs);
Mass [M+H]+: 346.2.
Step (iii): Purification of 3-[(l-t-Butyloxycarbonyl piperazin-4-yl)methyI]-5-methoxy-lH-indole
n-Hexane (1.25 Litres) was taken in 2 Litres four necked round bottom flask equipped with thermometer pocket and mechanical stirrer and charged the above obtained technical compound (270.9 grams). The mass was stirred for 1 hour at 25 °C. The product was filtered through Buckner funnel under vacuum. The compound was washed with n-hexane (2 x 125 mL), sucked well and air dried at 25 °C for 20 hours to obtain 240.0 grams of above title compound. Yield: 96 %;
Purity: 97.09 %;
Ή – NMR (CDCI3, δ ppm): 1.45 (9H, s), 2.45 (4H, s), 3.43 (4H, s), 3.69 (2H, s), 3.86 (3H, s), 6.85 – 6.88 (1H, dd, J = 8.7, 2.2 Hz), 7.08 – 7.09 (1H, d, J = 1 .57 Hz), 7.19 ( 1 H, d, J = 2.2 Hz), 7.23 – 7.25 (l H, d, J = 8.77 Hz), 8.25 (lH, bs); –
Mass [M+H]+: 346.2.
Step (iv): Preparation of l-[(2-BromophenyI)sulfonyl]-5-methoxy-3-[(l-t-butyloxycarbonyl piperazin-4-yl)methyI]-lH-indole

Tetrahydrofuran (THF) (4.6 Litres) was charged into a reactor at 25 °C, followed by the addition of powdered potassium hydroxide (860.6 grams, 85 %, 13.06 moles) at 25 °C under stirring. THF (3 Litres) was charged into a 5 Litres, three necked round bottom flask, provided with a mechanical stirrer and thermometer pocket. 3-[(l -t-Butyloxycarbonyl piperazin-4-yl) methyl]-5-methoxy-lH-indole (obtained in above step) (1287.7 grams, 3.7324 moles) was charged into the flask at 25 °C and stirred the mass well for complete dissolution. Then the clear 3-[(l-t-Butyloxycarbonyl piperazin-4-yl) methyl]-5-methoxy-l H-indole solution, prepared as above, was slowly transferred to the reactor containing potassium hydroxide under stirring, maintaining the mass temperature below 25 °C. After completion of the addition, the reaction mass was stirred at 25 °C for 2 hours. A solution of 2-bromophenylsulfonyl chloride (1293.04 grams, 5.062 moles) dissolved in THF (2.0 Litres) was added to the reaction mass through an addition funnel at a constant rate in 30 minutes, maintaining the mass temperature at 20 – 32 °C. The reaction was exothermic in nature. The mass was further stirred for 1 hour at 25 – 30 °C.
As the reaction was progressing the mass thickened due to formation of potassium chloride. The progress of the reaction was monitored by TLC (Eluent system: Ethyl acetate) and the product is relatively non-polar. The starting material was absent as per TLC. A second lot of 2-bromophenylsuIfonyl chloride (52.5 grams, dissolved in 100 mL of THF) was added to the reaction mass at 28 °C and further stirred the mass at 28 °C for another hour to ensure completion of the reaction, The reaction mass was unloaded into neat carboys.
Ice-water (40 Litres) was charged into a clean reactor and the reaction mass unloaded in the carboys was quenched into the reactor under stirring and the pH of the resulting solution was found to be 1 1.5 (pH paper). The product was extracted with (15 Litres + 7.5 Litres + 7.5 Litres) ethyl acetate. The combined organic layer was washed with saturated brine solution (2 x 5 L) and dried over anhydrous sodium sulfate. Total volume of the organic layer was 30 Litres. A small portion of the organic layer was concentrated in laboratory and the solid obtained was analyzed to check the quality of the technical product.
Purity: 91.46 %;
Ή – NMR (CDC13, 5 ppm): 1.45 (9H, s), 2.42 – 2.43 (4H, bs), 3.42 (4H, bs), 3.62 (2H, s), 3.81 (3H, s), 6.83 – 6.86 (1H, m), 7.18 – 7.19 (1H, m), 7.38 – 7.45 (2H, m), 7.52 – 7.55 (1H, m), 7.64
– 7.66 (2H, m), 8.06 – 8.08 (1H, d, J = 7.76 Hz);
Mass [M+Hf : 564.3, 566.4.
The organic layer.was taken for further workup and the technical product was purified without isolation.
Step (v): Purification of l-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(l-t-butyloxycarbonyl piperazin-4-yI)methyI]-lH-indole
The above organic layer was filtered (30 Litres) and charged into a reactor. Solvent was distilled off under vacuum at 40 – 45 °C to obtain solids. Isopropanol (14 Litres) and methanol (7 Litres) were charged into the reactor containing the solid product. The reaction mass was heated to reflux temperature (70.5 °C) under stirring and further stirred the mass at reflux for two hours to ensure formation of clear solution.
Reaction mass was then slowly cooled to room temperature (30 minutes) with room temperature water circulation in the jacket. It was further cooled to 18 °C and stirred for 1 hour. The product was centrifuged and the cake on the centrifuge was washed with isopropanol / methanol mixture (1.6 Litres + 0.8 Litres). It was sucked well and air dried at 40 – 45 °C for 4 hours in tray driers.
Weight of compound: 1554.8 grams, Cream colored crystalline powder, Yield: 77.7 %
Purity: 99.42 %;
Ή – NMR (CDCI3, δ ppm): 1.45 (9H, s), 2.42 (4H, bs), 3.42 (4H, bs), 3.63 (2H, s), 3.82 (3H, s), 6.83 – 6.86 (lH, dd, J = 8.34, 2.09 Hz), 7.19 (1 H, d, J = 2.0 Hz), 7.36 – 7.40 (1 H, t, J = 7.14 Hz), 7.43 – 7.47 (1H, t, J = 7.56 Hz), 7.52 – 7.55 (1 H, d, J = 8.95 Hz), 7.64 – 7.66 (2H, m), 8.06
– 8.08 ( 1H, d, J = 7.87 Hz); Mass: [M+H]+: 564.3, 566.3.
Step (vi): Preparation of l-((2-bromophenyl)snlfonyI]-5-methoxy-3-[(l-piperazinyl)methyl]-lH-indole dihydrochloride

S
l-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(4-t-butyloxycarbonyl-l -piperazinyl)methyl]-lH-indole (20.2 grams, 0.03578 M, obtained in the above step) was suspended in 250 mL of absoliite ethanol at 25 °C and then added 20 mL of 30 % (w/w) aqueous hydrochloric acid drop wise under stirring over a period of 30 minutes, whereby a clear solution was obtained. The reaction was exothermic and temperature went upto 38 °C. The mass was further heated at reflux for 4 hours. During this period solids separated. The mass was stirred for another 2 hours at reflux. The progress of the reaction was monitored by thin layer chromtography. After completion of the reaction, the mass was cooled to 25 °C and filtered the solids under suction. The solid on the filter was washed with 30 mL of absolute ethanol and the mass was dried under rotavacuum at 40 – 45 °C for 1 hour to obtain l-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[( 1 -piperazinyl)methyl]- 1 H-indole dihydrochloride (19.28 grams).
Purity: 99.8 %,
Mass: [M+H]+: 464.2, 466.2.
Step (vii): Preparation of l-[(2-bromophenyl)sulfonyl]-5-methoxy-3-[(l-piperazinyl)methyl]-lH-indole
The above obtained compound (19.09 grams) was suspended in demineralised water (300 mL) and cooled to 15 – 20 °C. The mass was basified to pH 10.5 to 1 1.0 by adding 40 % (w/w) lye solution, maintaining mass temperature below 20 °C under nitrogen atmosphere. The product was extracted with (2 x 150 mL) ethylacetate. The combined organic layer was washed with (100 mL) saturated brine solution, dried over anhydrous sodium sulfate and
solvent removed under rotavacuum at 40 – 45 °C to obtain the title compound (15.91 grams).
Yield: 96. 4 %
Purity: 99.89 %,
DSC (5 °C / minutes): 99.6 °C;
TGA (5 °C / minutes): 0.76 %;
Ή – NMR (CDCI3, δ ppm): 1.85 (1H, s), 2.44 (4H, bs), 2.86 – 2.88 (4H, t), 3.59 (2H, s), 3.76 (3H, s), 6.82 – 6.84 (lH, dd, J = 9.0, 2.45 Hz), 7.20 – 7.21 (1H, d, J = 2.28 Hz), 7.33 – 7.37 (1H, dt, J = 7.48 Hz), 7.41 – 7.44 (1 H, t), 7.52 – 7.54 (1H, d, J = 7.65 Hz), 7.62 – 7.64 (2H, m), 8.01 – 8.03 (1H, dd, J = 7.98, 1.15 Hz);
Mass: [M+H]+: 464.2, 466.2.
Example 2: Preparation of l-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(l-piperazinyl)methyl]-lH-in

Step (i) & (ii): Preparation of 3-[(l-t-Butyloxycarbonyl piperazin-4-yl)methyl]-5-methoxy-lH-indoIe

Step (i):
Demineralized water (DM water) (660 mL) and N-Boc piperazine ( 150.0 grams, 0.8034 moles) were charged into a 2 Litres three necked round bottomed flask provided with a mechanical stirrer and a thermometer pocket. The mass was stirred for 10 minutes at 25 °C, to obtain a clear solution. Then acetic acid (32.5 mL, 0.5416 moles) was added to the above mass while maintaining the mass temperature at ~ 25 °C in 10 minutes. After completion of addition, the clear solution was stirred at 25 °C for 30 minutes.
To the above stirred mass at 25 °C, aqueous formaldehyde solution (81 mL, 30 % w/v, 0.81 moles) was added slowly through an addition funnel over a period of 30 minutes maintaining the mass temperature below 25 °C. During the addition, white slurry mass was formed. The resultant white slurry mass was stirred for another 1 hour at 25 – 30 °C. Methanol (MeOH) (300 mL) was added to the above mass to obtain a clear solution. The solution was further stirred for 30 minutes at 25 °C to obtain Mannich adduct.
Step (ii):
5-Methoxy indole (106.4 grams, 0.7238 moles) and methanol (550 mL) were charged into a 4 necked round bottom flask. The mass was stirred for 10 minutes at 25 °C to obtain a clear solution and then cooled the mass to 18 – 20 °C. The mannich adduct (prepared in above step) was added to the flask through an addition funnel maintaining mass temperature below 20 °C, over a period of 1 hour. The mass was further stirred for a period of 1 hour at 25 – 30 °C, while monitoring the progress of the reaction by thin layer chromatography (TLC).
After completion of the reaction (1 hour), DM water (2.2 Litres) and ethyl acetate (1 Litre) were added to the reaction mass and pH adjusted to 10.5 (on pH paper) with lye solution (80 mL) maintaining the mass temperature at 20 – 24 °C. The organic (product) layer was separated and the aqueous layer was further extracted with ethyl acetate (2 x 500 mL). The combined organic layer was washed with saturated brine solution (300 mL) and dried over anhydrous sodium sulfate. The organic layer was filtered free of sodium sulfate and concentrated under reduced pressure. n-Hexane (300 mL) was added to the residual mass and further concentrated under vacuum for removal of traces of ethyl acetate to obtain 272.2 grams of technical product.
Purity: 96.16 %;
Ή – NMR (CDC13, δ ppm): 1.45 (9H, s), 2.44 (4H, bm), 3.41 – 3.43 (4H, bm), 3.69 (2H, s), 3.87 (3H, s), 6.85 – 6.88 (1H, dd, J = 8.75, 2.23 Hz), 7.10 (1Ή, d, J = 0.96 Hz), 7.19 (1H, d, J = 2.24 Hz), 7.24 – 7.26 (1 H, d), 8.04 (1H, bs);
Mass [M+H]+: 346.2.
Step (iii): Purification of 3-[(l-t-ButyloxycarbonyI piperazin-4-yl)methyl]-5-methoxy-lH-indole
n-Hexane (1.25 Litres) was taken in 2 Litres four necked round bottom flask equipped with thermometer pocket and mechanical stirrer and charged the above obtained technical compound (270.9 grams). The mass was stirred for 1 hour at 25 °C. The product was filtered through Buckner funnel under vacuum. The compound was washed with n-hexane (2 x 125 mL), sucked well and air dried at 25 °C for 20 hours to obtain 240.0 grams of above title compound. Yield: 96 %;
Purity: 97.09 %;
Ή – N R (CDC13, δ ppm): 1.45 (9H, s), 2.45 (4H, s), 3.43 (4H, s), 3.69 (2H, s), 3.86 (3H, s), 6.85 – 6.88 (lH,jdd, J = 8.7, 2.2 Hz), 7.08 – 7.09 (1 H, d, J = 1.57 Hz), 7.19 ( 1H, d, J = 2.2 Hz),
7.23 – 7.25 (1H, d, J = 8.77 Hz), 8.25 (1H, bs);
Mass [M+H]+: 346.2.
Step (iv): Preparation of l-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(l-t-butyloxycarbonyl pipera

Tetrahydrofuran (THF) (4.6 Litres) was charged into a reactor at 25 °C, followed by the addition of powdered potassium hydroxide (860.6 grams, 85 %, 13.06 moles) at 25 °C under stirring. THF (3 Litres) was charged into a 5 Litres, three necked round bottom flask, provided with a mechanical stirrer and thermometer pocket. 3-[( 1 -t-Butyloxycarbonyl piperazin-4-yl)methyl]-5-methoxy-lH-indole (obtained in above step) (1287.7 grams, 3.7324 moles) was charged into the flask at 25 °C and stirred the mass well for complete dissolution. Then the clear 3-[(l-t-Butyloxycarbonyl piperazin-4-yl)methyl]-5-methoxy-l H-indole solution, prepared as above, was slowly transferred to the reactor containing potassium hydroxide“ under stirring, maintaining the mass temperature below 25 °C. After completion of
the addition, the reaction mass was stirred at 25 °C for 2 hours. A solution of 2- bromophenylsulfonyl chloride (1293.04 grams, 5.062 moles) dissolved in THF (2.0 Litres) was added to the reaction mass through an addition funnel at a constant rate in 30 minutes, maintaining the mass temperature at 20 – 32 °C. The reaction was exothermic in nature. The mass was further stirred for 1 hour at 25 – 30 °C.
As the reaction was progressing the mass thickened due to formation of potassium chloride. The progress of the reaction was monitored by TLC (Eluent system: Ethyl acetate) and the product is relatively non-polar, The starting material was absent as per TLC. A second lot of 2-bromophenylsulfony] chloride (52.5 grams, dissolved in 100 mL of THF) was added to the reaction mass at 28 °C and further stirred the mass at 28 °C for another hour to ensure completion of the reaction. The reaction mass was unloaded into neat carboys.
Ice-water (40 Litres) was charged into a clean reactor and the reaction mass unloaded in the carboys was quenched into the reactor under stirring and the pH of the resulting solution was 11.5 (pH paper). The product was extracted with (15 Litres + 7.5 Litres + 7.5 Litres) ethyl acetate. The combined organic layer was washed with saturated brine solution (2 x 5 L) and dried over anhydrous sodium sulfate. Total volume of the organic layer was 30 Litres. A small portion of the organic layer was concentrated in laboratory and the solid obtained was analyzed to check the quality of the technical product.
Purity: 91.46 %;
Ή – NMR (CDC , δ ppm): 1.45 (9H, s), 2.42 – 2.43 (4H, bs), 3.42 (4H, bs), 3.62 (2H, s), 3.81 (3H, s), 6.83 – 6.86 (1 H, m), 7.18 – 7.19 (1H, m), 7.38 – 7.45 (2H, m), 7.52 – 7.55 (1 H, m), 7.64 – 7.66 (2H, m), 8.06 – 8.08 (1 H, d, J = 7.76 Hz);
, Mass [M+H : 564.3, 566.4.
The organic layer was taken for further workup and the technical product was purified without isolation.
Step (v): Purification of l-[(2-BromophenyI)suIfonyl]-5-methoxy-3-[(l-t- butyloxycarbonyl piperazin-4-yl)methyl]-lH-indole
The above organic layer was filtered (30 Litres) and charged into a reactor. Solvent was distilled off under vacuum at 40 – 45 °C to obtain solids. Isopropanol (14 Litres) and
methanol (7 Litres) were charged into the reactor containing the solid product. The reaction mass was heated to reflux temperature (70.5 °C) under stirring and further stirred the mass at reflux for two hours to ensure formation of clear solution.
Reaction mass was then slowly cooled to room temperature (30 minutes) with room temperature water circulation in the jacket. It was further cooled to 18 °C and stirred for 1 hour. The product was centrifuged and the cake on the centrifuge was washed with isopropanol / methanol mixture (1 .6 Litres + 0.8 Litres). It was sucked well and air dried at 40
– 45 °C for 4 hours in tray driers.
Weight of compound: 1554.8 grams, Gream colored crystalline powder, Yield: 77.7 %
Purity: 99.42 %;
Ή – NMR (CDQlj, δ ppm): 1.45 (9H, s), 2.42 (4H, bs), 3.42 (4H, bs), 3.63 (2H, s), 3.82 (3H, s), 6.83 – 6.86 (1H, dd, J =.8.34* 2.09 Hz), 7.19 (1H, d, J = 2.0 Hz), 7.36 – 7.40 (1H, t, J = 7.14 Hz), 7.43 – 7.47 (1H, t, J = 7÷56 Hz), 7.52 – 7.55 (lH, d, J = 8.95 Hz), 7.64 – 7.66 (2H, m), 8.06
– 8.08 (1 H, d, J = 7.87 Hz); Mass: [M+H]+: 564.3, 566.3.
Step (vi): Preparation of l-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(l-piperazinyl)methyl)-l

9
l-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(l -t-butyIoxycarbonyl piperazin-4-yl)methyl]-lH-indole (obtained in the above step, 1540 grams, 2.73 mole) was dissolved in acetone (30.8 Litres) and charged into a glass lined reactor. The temperature of the reaction mass was raised to reflux temperature (56 °C). Methanesulfonic acid (920 grams, 9.57 moles) diluted with acetone (6 Litres) was added to the above mass at reflux temperature, slowly over a period of 30 minutes, through an addition funnel. During addition vigorous reflux was observed. The reaction mass was a clear solution before and after the addition of methanesulfonic acid solution. After stirring for ~ 90 minutes at reflux, thick mass of solids separated out. The progress of the reaction was monitored by TLC. The reaction was completed in 4 hours. Then the mass was cooled to 25 °C and further stirred for two hours at 25 °C. The product was filtered through nutsche filter under vacuum. The product on the nutsche filter was washed with acetone (8 Litres). The material was unloaded into trays and air dried at 30-35 °C for 4 hours in a tray drier. Weight of the product: 1.61 Kg (off white with pinkish tinge).
Yield: 90 %;
Salt content (dimesylate): 32.1 % w/w;
Purity: 99.97 %;
Ή – NMR (D20, 5 ppm): 2.64 (6H, s), 3.48 (4H, bs), 3.53 (4H, bs), 3.70 (3H, s), 4.50 (2H, s), 6.75 – 6.78 (1H, dd, J = 8.97, 1.92 Hz), 7.11 (1H, d, J = 1.78 Hz), 7.32 – 7.34 ( 1H, t, J = 9.28 Hz), 7.34 – 7.38 (lH, t, J = 7.63 Hz), 7.44 – 7.48 ( 1H, d, 3 = 7.76 Hz), 7.54 – 7.56 (2H, d, J = 7.85 Hz), 8.06 (1H, s), 8.15 – 8.17 (2H, d, J = 7.87 Hz);
Mass: [M+H]+: 464.2, 466.2.
Step (vii): Preparation of l-{(2-Bromophenyl)suIfonyl]-5-methoxy-3-[(l-piperazinyl)methyl]-l

Acetone (24.15 L) was taken in a Glass Lined Reactor at 25-30 °C, followed by l-[(2-Bromo phenyl)sulfonyl]-5-methoxy-3-[(l-piperazinyl)methyl]-lH-indole dimesylate (obtained in the above step) (1.61 Kg) and the resulting mass was stirred To obtain slurry. DM water (4.0 L) was added to the reactor and then the mass temperature was raised to reflux temperature (56.0-57.5 °C). A clear solution was obtained at reflux. It was maintained for 15 minutes. The mass was cooled to 45-50 °C and added activated carbon (161 grams) to the mass and stirred the mass for 45 minutes at reflux temperature: It was filtered hot into another reactor, which was maintained at 50 °C. The clear filtrate was allowed to cool on its own, under nitrogen
blanket. Solids separated when the mass temperature was ~ 44 °C. The mass was allowed to cool to room temperature (30-35 °C) and then it was further cooled at 10-12 °C for 2 hours. The product was centrifuged, washed with acetone (5 L) and sucked well. The wet product (weight: 1.5 Kg) was spread into trays and dried in a tray drier at 40-45 °C for 7.5 hours, till organic volatile impurities are below the allowable limits. Weight of the dry product obtained: 1.3 Kg. Yield: – 76.5 %
Purity: 99.98 %;
Melting range (°C): 203.8 – 205.3;
Salt content (Dimesylate): 28.26 %;
Moisture Content: 5.2 %;
TGA: 4.9 %; ,
Ή – NMR (D20, δ ppm): 2.65 (6H, s), 3.48 (8H, bm), 3.71 (3H, s), 4.48 (2H, s), 6.77 – 6.80 (1H, dd, J = 9.18, 2.24 Hz), 7.12 – 7.13 (1 H, d, J = 2.12 Hz), 7.35 – 7.37 (1H, d, J = 9.06 Hz), 7.37 – 7.41 (1 H, t, J = 7.98 Hz), 7.46 – 7.50 (1 H, t, J = 7.66 Hz), 7.57 – 7.58 (1 H, d, J = 7.86 Hz), 8.06 ( 1H, s), 8.17 – 8.20 (1H, dd, J = 7.95, 0.87 Hz),
Mass [M+H]+: 464.2, 466.1 ;
PATENT
WO 2004/048330
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004048330
REFERENCES
http://www.avarx.com/search/showOpportunityDetails?asset_id=2424
Phase II
Alzheimer’s disease; Schizophrenia
Phase I
Attention-deficit hyperactivity disorder; Cognition disorders; Parkinson’s disease
05 Jan 2016
Suven Life Sciences has patent protection for chemical entities targeting serotonin receptors for the treatment of neurodegenerative disorders in Canada, Africa and South Korea
11 Dec 2015
Suven Life Sciences receives patent allowance for chemical entities targeting serotonin receptors in Eurasia, Europe, Israel and Macau
01 Oct 2015
Phase-II clinical trials in Schizophrenia in USA (PO)
////////
Brc1ccccc1S(=O)(=O)n4cc(CN2CCN(C)CC2)c3cc(ccc34)OC


Thanks for the detailed explanation.
LikeLike