
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


| HS Code: | 2935009090 |
|---|
130693-82-2..HCL
Dorzolamide (trade name Trusopt) is a carbonic anhydrase inhibitor. It is an anti-glaucoma agent, and acts by decreasing the production of aqueous humour.[1] It is optically applied in the form of a 2% eye drops.[2]
History
This drug, developed by Merck, was the first drug in human therapy (market introduction 1995) which resulted from structure-baseddrug design. It was developed to circumvent the systemic side effects of acetazolamide which has to be taken orally.[2]
Uses
Dorzolamide hydrochloride is used to lower increased intraocular pressure in open-angle glaucoma and ocular hypertension.
Pharmacodynamics
It lowers IOP by about 20%.[2]
Side effects
Ocular stinging, burning, itching and bitter taste.[2] it causes shallowing of the anterior chamber and leads to transient Myopia.
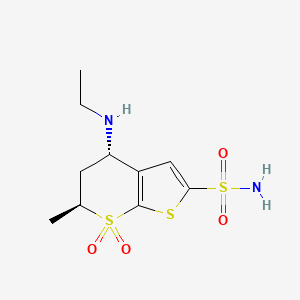

Dorzolamide Hydrochloride and its derivatives is known. U.S. Pat. No. 5,688,968 describes preparation of Dorzolamide HCl starting from chiral 5,6-dihydro-4-(S)-hydroxy-6-(S)-methyl-4H-thiopyran-7,7-dioxide, as depicted in scheme 1:

The process described in BP 0 296 879 (equivalent of U.S. Pat. No. 4,797,413) is of particular relevance. EP 0 296 879 describes the synthesis of Dorzolamide Hydrochloride starting from thiophene-2-thiol as depicted in scheme 2 and 3



The process described in EP 0,296,879 (scheme 2) has the following disadvantages: (a) The starting material Thiophene-2-thiol is unstable and undergoes oxidation to form disulfide, leading to lower yield of viii; (b) the yield of sulfonamide (xii) from sulphonic acid (x) is very poor (35%) and requires use of 18-crown-6 ether, which is expensive; (c) oxidation of alcohol (xiii) to sulfone is carried out using oxone which is expensive and hazardous; and separation of cis/trans isomer is done by column chromatography which is industrially inconvenient.
| Systematic (IUPAC) name | |
|---|---|
| (4S,6S)-2-ethylamino-4-methyl-5,5-dioxo- 5λ6,7-dithiabicyclo[4.3.0]nona-8,10-diene-8-sulfonamide |
|
| Clinical data | |
| Trade names | Trusopt |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602022 |
|
|
| Legal status | |
| Routes | Topical (eye drops) |
| Pharmacokinetic data | |
| Protein binding | ~33% |
| Half-life | 4 months |
| Identifiers | |
| CAS number | 130693-82-2 |
| ATC code | S01EC03 |
| PubChem | CID 5284549 |
| DrugBank | DB00869 |
| ChemSpider | 4447604 |
| UNII | 9JDX055TW1 |
| KEGG | D07871 |
| ChEBI | CHEBI:4702 |
| ChEMBL | CHEMBL218490 |
| Chemical data | |
| Formula | C10H16N2O4S3 |
| Molecular mass | 324.443 g/mol |
TRUSOPT® (dorzolamide hydrochloride ophthalmic solution) is a carbonic anhydrase inhibitor formulated for topical ophthalmic use.
Dorzolamide hydrochloride is described chemically as: (4S-trans)-4-(ethylamino)-5,6-dihydro-6methyl-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide monohydrochloride. Dorzolamide hydrochloride is optically active. The specific rotation is
Its empirical formula is C10H16N2O4S3•HCl and its structural formula is:
 |
Dorzolamide hydrochloride has a molecular weight of 360.9 and a melting point of about 264°C. It is a white to off-white, crystalline powder, which is soluble in water and slightly soluble in methanol and ethanol.
TRUSOPT Sterile Ophthalmic Solution is supplied as a sterile, isotonic, buffered, slightly viscous, aqueous solution of dorzolamide hydrochloride. The pH of the solution is approximately 5.6, and the osmolarity is 260-330 mOsM. Each mL of TRUSOPT 2% contains 20 mg dorzolamide (22.3 mg of dorzolamide hydrochloride). Inactive ingredients are hydroxyethyl cellulose, mannitol, sodium citrate dihydrate, sodium hydroxide (to adjust pH) and water for injection. Benzalkonium chloride 0.0075% is added as a preservative.
The dorzolamide hydrochloride product is prepared from the aminated intermediate of Formula IV by the following scheme.


[00056] Preparation of dorzolamide hydrochloride product from the animated intermediate of Formula IV
[00057] Fuming sulfuric acid (20%, 5 1) is cooled to -7°±2°C and the aminated intermediate of Formula IV (2.5 Kg) is added to it in portions during stirring. The temperature of the reaction mixture is increased to 20°+5°C during addition of the aminated intermediate of Formula IV. The reaction mixture is stirred for 22 hours at 20°±5°C. Thionyl chloride (20 1) is added to the stirred reaction mixture at 20±5°C. The reaction mixture is heated to 60°-65°C and stirred for 24 hours at this temperature. The mixture is cooled back to 40°±2°C and the excess amount of thionyl chloride is evaporated at this temperature under vacuum. (The volume of the residue: ~9 1.) The residue is cooled to -5°+2°C.
[00058] Ethyl acetate (75 1) is cooled to -10°±5°C and the residue is added to it at this temperature. The temperature of the diluted solution: 10°-25°C. Aqueous ammonia (25%, 75 1) is cooled to -10°±5°C and the residue is added to it at this temperature during effective stirring, while maintaining the temperature below 300C. The final pH: ~11. The slurry is cooled to 0°+2°C and stirred for 14 hours at this temperature. The formed ammonium sulfate is filtered and the cake is washed with ethyl acetate (2x 20 1 and 10 1). Ethyl acetate is evaporated from the filtrate at 38°±2°C under vacuum. The residue is heated to 38°±2°C, washed with toluene (3×37.5 1) at this temperature. Water (25 1) is added to the aqueous phase, cooled to 20°-25°C and extracted with ethyl acetate (3x 75 1, 37.5 1, and 37.5 1). The collected ethyl acetate phase is concentrated to ~ 100 1 at 38°±2°C under vacuum. The residue is cooled to 20°-25°C and hydrogen chloride in ethanol (5%, 10.8 1) is added to it during stirring. The formed slurry is stirred for 1 hour at 20°-25°C then cooled to 0°-4°C and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×20 1) and dried at 55°-60°C under vacuum for 4-8 hours to give Dorzolamide hydrochloride salt (~2 Kg).
[00059] Crude Dorzolamide hydrochloride salt (9 Kg) is solved in water (225 1) at 20°-25°C and the pH is set to 8.0-8.5 by addition of 25% of aqueous ammonia (2 1). The formed slurry is extracted with ethyl acetate (5×72 1). The collected ethyl acetate phase is concentrated to 180 1 by vacuum distillation. The residue is cooled to 20°-25°C, ethyl acetate (45 1) and hydrogen chloride in ethanol (5%, 22.5 1) are added to it during stirring (pH:~1.0). The formed slurry is stirred for 1 hour at 20°-25°C then cooled to 0°-4°C and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×30 1), and dried at 55°-60°C under vacuum for 4-8 hours to give purified Dorzolamide hydrochloride salt (~8.2Kg).
[00060] Purified Dorzolamide hydrochloride salt (8 Kg) dissolved in water
(24 1) at 95°-105°C and treated with active carbon (80 g). After filtration, the water solution is cooled gradually to 0°-4°C and stirred for 3-5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with cooled water (2×5 1) and dried at 55°-60°C under vacuum for 4-8 hours to give crystallized DRZ HCl salt (~6.6 Kg).
http://www.google.com/patents/US20060142595
The invention provides a process for preparing 5,6-dihydro-4-(S)-(ethylamino)-6-(S)methyl-4H-thieno[2,3b]thiopyran-2-sulphonamide-7,7-dioxide hydrochloride of formula (I), comprising of nine steps, as depicted in scheme 4 below:


Example 9Preparation of 5,6 dihydro-4H-4-(S)-ethylamino-6-(S)-methylthieno[2,3-b]thiopyran-2-sulfonamide-7,7 dioxide Hydrochloride (I)
A mixture of compound from example 8 (15 gm0.0462 mole) and di-p-toluyl-D-tartaric acid monohydrate (4.55 gm, 0.01125 mole) in n-propanol (1600 ml) was heated to boiling and hot solution filtered through a filter-aid pad with a layer of charcoal. The filtrate was concentrated by boiling to a volume of about (400 ml) and then allowed to crystallize. After standing overnight the crystals were filtered off and material recrystallized twice more from n-propanol (400 ml) to yield a 2:1 salt of free base to acid. Combined mother liquors from this recrystallization were saved for stage B. The salt was then treated with a saturated solution of sodium bicarbonate and mid extracted with ethyl acetate. The organic extract were dried, filtered and concentrated to dryness to yield (3.2 gm) of freebase. The hydrochloride salt was prepared from 5,6 N HCl ethanol and crystallized from methanol-isopropanol to yield (2.83 gm) of (+) isomer, SOR 8.23 (C 0.9 methanol) M.P. 283-285° C. The combine mother liquor was treated with saturated solution of sodium bicarbonate and mixture extracted with ethyl acetate. The organic exacts were dried, filtered and concentrated to dryness. The residue was treated with di-p-toluyl-L-tartaric acid monohydrate (4.55 gm, 0.01125 mole) in n-propanol (1600 ml) and the isomer separated by the process described previously to give title compound (I) (3.75 gm, 22.48%) SOR=−8.34 (C 1, Methaol) M.P. 283 to 285° C.,
………………………………………..
http://www.google.com/patents/WO2008135770A2?cl=en
Dorzolamide is chemically termed as (4S,6S)-4-(ethylamino)-5,6-dihydro-6-methyl-4H- thieno[2,3-/?]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride. Dorzolamide hydrochloride is represented by following structural Formula I:
HN ‘CH,

Formula I
Dorzolamide hydrochloride is known to be a carbonic anhydrase inhibitor useful in the treatment of ocular hypertension.
A process for the preparation of dorzolamide and its derivatives was first described in EP 0296879. The process of particular relevance is depicted in scheme 1. Scheme 1

(viϋ) (ix) Trans and Cis (x)

Trans (xi) Trans(+) (xii) ( I )
The process disclosed in scheme 1 has following disadvantages.
(a) The reduction of the ketone of sulfonamide (vi) using absolute ethanol is carried out at reflux and then stirred at room temperature for several hours to complete the reaction. This longer duration of reaction produces many impurities.
(b) Oxidation of alcohol (vii) to sulfone (viii) is carried out using oxone. The oxone has many disadvantages such as it is irritating to the eyes, skin, nose and throat. It should be used with adequate ventilation and exposure to its dust should be minimized. Traces of heavy metal salts catalyze the decomposition of oxone. It is practically insoluble in all organic solvents hence a phase transfer catalyst is required.
(c) Activation of the 4-hydroxy group of the sulfoaminated hydroxysulfone (viii) and nucleophilic substitution by desired ethylamine, results in all diastereomeric products (x) i.e. trans and cis isomers, which must be separated by column chromatography and resolved, further using resolving agent. As a result, product loss is greater when the desired product is the more active enantiomer.
An alternate route for the preparation of dorzolamide hydrochloride by the Ritter reaction is disclosed in EP0296879 and consists of the treatment of a aliphatic hydroxyl with a nitrile and a strong acid to form an amide. The process disclosed is as depicted in Scheme 2.
Scheme 2

(viii) (ix-a ) Trans and Cis (x)

Trans(+) (xii)
Trans (+/-) (xi) ( I )
The reaction involves conversion of hydroxysulfones (viii) to the corresponding acetoamidosulfones (ix-a) with retention of configuration followed by reduction of the amido group, chromatographic separation and resolution to obtain the desired trans isomer (I).
The prior art teaches the use of an excess quantity of sulfuric acid to carry out the Ritter reaction and hence a large quantity of ice is required for quenching the reaction mass. When the reaction mass in concentrated sulfuric acid comes into contact with ice, a large amount of localized heat is generated causing decomposition of material. Since a huge amount of water is required for quenching the reaction mass, the amount of ethyl acetate required for extraction is also substantially large. The work-up using water is not advisable nor applicable industrially.
United States Patent 5688968 describes an alternative route of preparation of dorzolamide hydrochloride starting from chiral 5,6-dihydro-4-(S)-hydroxy-6-(S)-methyl-4H-thiopyran-7,7- dioxide, as depicted in Scheme 3:
Scheme 3

(xiv) (XV)
(xiii)

(xvi) (xvii ) Trans:Cis:: 95: 5 (xviii)
HN CH,


(xix) ( I )
The process described in Scheme 3 has the following disadvantages: (a) Use of expensive chiral hydroxysulfone starting material. The process for the preparation of the chiral hydroxysulfone starting material is disclosed in U.S. Patents Nos. 5,157,129, 5,474,919 and 5,760,249. In these processes, the chiral hydroxysulfone is obtained by the asymmetric enzymatic reduction of the corresponding ketosulfone, or by cyclization of the chiral thienyl thiobutyric acid, obtained, in turn, from a chiral hydroxyester or lactone, and the subsequent stereospecific reduction of the resulting ketone, (b) The process according to this patent uses maleic acid to separate the undesired cis- isomer from dorzolamide. However this maleate salt formation to remove the cis isomer is only suitable when the ratio of trans/cis is greater than 95:5. That means, the maleate salt formation of dorzolamide does not the remove cis isomer exclusively when the cis isomer content is more than 5%. It sometimes requires repeated purification to achieve the desired chiral purity.
Another alternate route for the preparation of dorzolamide hydrochloride is disclosed in United States patent no.7109353 which involves the use of sodium perborate as an oxidant, as depicted in Scheme 4.
Scheme 4
chlorinating agent, cyclinization



Vl IV

VIl VlIl IX
The process disclosed in Scheme 4 has following disadvantages (a) Conversion of (i) to (ii) requires the mixture to be refluxed for 18-20 hrs which is time consuming and may cause impurity in the product.
(b) As the process uses the Ritter reaction to convert (vi) to (vii), a large amount of water is required to quench the hot mass of reaction which is not practical in an industrial set-up. (c) Sodium perborate is used as an oxidizing agent to convert (v) to (vi), which has got bleaching properties, and the handling of it may be injurious when done so for a prolonged period.
Yet another process for the preparation of dorzolamide is disclosed in United States publication no. 20060155132 which involves protecting the chiral 5,6-dihydro-4-(R)- hydroxy-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran-7,7-dioxide as depicted in Scheme 5.
Scheme 5
protected amination benzyl sulphonyl chloride



The process disclosed in Scheme 5 has the following disadvantages, (a) The conversion process of compound (II) to (III) requires a very low temperature which ranges from -30° to 00C. (b) The amination process requires 16- 20 hrs, which is time consuming and may cause impurity in the product. All these disadvantages of the prior art are overcome by the process in accordance with the present invention.
Scheme 8

Example 4
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide
A suspension of 5,6-dihydro-4H-4-acetylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide (83.25 gms, 0.24 moles) in THF (832 ml) was cooled to 00C and sodium borohydride (49.11 gms, 1.29 moles) was added in lots maintaining temperature below 5°C. Reaction mass was stirred for 15 minutes at 5°C and boron trifluoride diethyl- etherate (249.75 ml, 287.2 gms, 2.02 moles) was added below 5°C. The reaction mass was stirred for 5 hours at 0°C to 5°C. Temperature of the reaction mass was raised to 25°C to 300C and stirred for 18 hours. The reaction mass was quenched in 1M sulphuric acid solution (1082 ml) below 5°C, temperature raised to 25°C to 30°C and stirred for 1 hour. The solvent was distilled under reduced pressure at 800C. The reaction mass was cooled to 100C and p H adjusted to 7 – 8 using 50% sodium hydroxide solution. Material was extracted in 1665 ml ethyl acetate once and 832 ml twice. The combined organic layers were washed with saturated sodium chloride solution, dried over sodium sulphate, charcoalised, filtered on hyflo, distilled to get title compound (77.42 gms). HPLC: 80:20::Trans:Cis
Example 7
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide hydrochloride
(a) Dorzolamide di-p-toluyl-L-tartrate salt as prepared in example 6 (44.26 gms, 0.085 moles) was taken in ethyl acetate (557.0 ml), basified with saturated sodium bicarbonate solution. Reaction mass was stirred for 15 minutes at 25°C to 3O0C and aqueous layer was extracted with ethyl acetate (278 ml X 2). The organic layers were combined, washed with brine solution, dried over sodium sulphate, and charcoalized. To the clear solution, IPA + HCL (16.35 ml, 0.089 moles) was added, stirred for 30 minutes and ethyl acetate was removed by distillation at atmospheric pressure at 85°C to about 280 ml volume, cooled to 25-3O0C, stirred for 12 hours at same temperature and filtered to get 26.0 gms of dorzolamide hydrochloride. Trans (-) dorzolamide hydrochloride > 99.5% Trans (+) dorzolamide hydrochloride < 0.5% Cis Isomer <0.1%
(b) Dorzolamide hydrochloride was obtained in a similar manner in quantitative yield from the salt of example 6(b).
(c) Dorzolamide hydrochloride was obtained in a similar manner in quantitative yield from the salt of example 6(c).
Example 8
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide -7,7-dioxide hydrochloride without isolation of base
Dorzolamide di-p-toluyl-L-tartrate (50 gms, 0.096 moles) prepared as per example 6, was charged in a round bottom flask along with isopropanol (1000 ml). The reaction mass was heated to 800C and charged with IPA-HCI (20 ml) dropwise to pH 3 to 4. The reaction mass was heated to reflux for 5-10 minutes. The clear solution obtained was concentrated to 100 ml. The reaction mass was charged with 300 ml ethyl acetate, cooled to 25°C, stirred for 12 to 14 hours at same temperature. The resulting dorzolamide hydrochloride was isolated by filtration and washed with ethyl acetate (50 ml), dried under vacuum at 60- 65 0C for 5-6 hours. Yield- 30 gms.
Trans (-) dorzolamide hydrochloride > 99.5% Trans (+) dorzolamide hydrochloride < 0.5% Cis Isomer <0.1%
…………………………………………………………..
http://www.google.com/patents/EP0453288A1

………………………………………
http://www.google.com/patents/US20060155132
Dorzolamide hydrochloride, known chemically as 5,6-dihydro-4-(S)-ethylamino-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran-2-sulfonamide-7,7-dioxyde hydrochloride, is a topically effective carbonic anhydrase inhibitor useful in the treatment of ocular hypertension.
Dorzolamide hydrochloride has the structure of Formula I:

U.S. Pat. Nos. 4,677,155 and 4,797,413 disclose Dorzolamide. In the prior art synthesis of dorzolamide, a chiral hydroxysulfone is used as a starting material. The chiral hydroxysulfone starting material can be obtained using the processes disclosed in U.S. Pat. Nos. 5,157,129, 5,474,919, and 5,760,249. In the disclosed processes, the chiral hydroxysulfone is obtained by the asymmetric enzymatic reduction of the corresponding ketosulfone, or by cyclization of the chiral thienyl thiobutyric acid, obtained, in turn, from a chiral hydroxyester or lactone, and the subsequent stereospecific reduction of the resulting ketone.
Processes for the preparation of dorzolamide hydrochloride are described in U.S. Pat. Nos. 4,797,413, 5,157,129, and 5,688,968 and in U.S. patent application Publication Ser. No. 2003/0220509. The disclosed processes involve conversion of a hydroxysulfone to the corresponding acetamidosulfone by a Ritter reaction with retention of configuration, followed by introduction of a sulfonamido group, and the subsequent reduction of the amido group to an amine, providing the desired product.
The process disclosed in U.S. Pat. No. 4,797,413 includes activation of the 4-hydoxy group of the sulfonaminated hydroxysulfone with tosyl chloride and the introduction of the desired alkylamino group by nucleophilic substitution, resulting in all diastereomeric products, which must be separated and resolved. As a result, at least 75 percent of the product is lost when the desired product is the more active enantiomer.

EXAMPLE 2
Preparation of 5,6-dihydro-4-(S)-ethylamino-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran 7,7-dioxide hydrochloride salt (Formula IV)
Tetrahydrofuran (50 l) and triethyl amine (4.8 l) are added to 4-(R)-hydroxy-5,6-dihydro-6-(S)-methyl-4H-thieno[2,3b]thiopyran-7,7-dioxide (5 Kg) and stirred under a nitrogen atmosphere at room temperature. The solution is cooled to −10° C. Benzylsulfonyl chloride (5.4 Kg) solved in THF (15 l) is added to the DRZ-19 THF solution in portions while maintaining the temperature below 0° C. The feeding funnel is washed with THF (2 l). The reaction mixture is stirred at 0° C. for 2-4 hours. The formed TEA HCl is filtered and the cake is washed with THF (2×10 l) Ethylamine in THF (30%, 63.7 l) is added to the filtrate and the reaction mixture is stirred at 20°-25° C. for 16 hours. Ethylamine gas prepared by heating of 70% EtNH2water solution (50 l) is absorbed in cooled THF (30 l). Water (20 l) is added to the reaction mixture and THF is evaporated from the filtrate at 40°±5° C. under vacuum. The residue is cooled to 20°-25° C., ethyl acetate (60 l) is added to it and stirred vigorously. After phase separation, the organic phase is washed with water (20 l). The ethyl acetate phase is heated to 40°±2° C. and hydrochloric acid (4M, ˜8-10 l) is added to it during stirring to set pH 2.0-2.5. The formed slurry is cooled to −8°±2° C. and stirred for 3 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (30 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give the desired salt (˜5 Kg).
Preparation of dorzolamide hydrochloride product from the aminated intermediate of Formula IV
Fuming sulfuric acid (20%, 5 l) is cooled to −7°±2° C. and the aminated intermediate of Formula IV (2.5 Kg) is added to it in portions during stirring. The temperature of the reaction mixture is increased to 20°±5° C. during addition of the aminated intermediate of Formula IV. The reaction mixture is stirred for 22 hours at 20°±5° C. Thionyl chloride (20 l) is added to the stirred reaction mixture at 20°±5° C. The reaction mixture is heated to 60°-65° C. and stirred for 24 hours at this temperature. The mixture is cooled back to 40°±2° C. and the excess amount of thionyl chloride is evaporated at this temperature under vacuum. (The volume of the residue: ˜9 l.) The residue is cooled to −5°±2° C.
Ethyl acetate (75 l) is cooled to −10°±5° C. and the residue is added to it at this temperature. The temperature of the diluted solution: 10°-25° C. Aqueous ammonia (25%, 75 l) is cooled to −10°±5° C. and the residue is added to it at this temperature during effective stirring, while maintaining the temperature below 30° C. The final pH: ˜11. The slurry is cooled to 0°±2° C. and stirred for 14 hours at this temperature. The formed ammonium sulfate is filtered and the cake is washed with ethyl acetate (2×20 l and 10 l ). Ethyl acetate is evaporated from the filtrate at 38°±2° C. under vacuum. The residue is heated to 38°±2° C., washed with toluene (3×37.5 l) at this temperature. Water (25 l) is added to the aqueous phase, cooled to 20°-25° C. and extracted with ethyl acetate (3×75 l, 37.5 l, and 37.5 l). The collected ethyl acetate phase is concentrated to ˜100 l at 38°±2° C. under vacuum. The residue is cooled to 20°-25° C. and hydrogen chloride in ethanol (5%, 10.8 l) is added to it during stirring. The formed slurry is stirred for 1 hour at 20°-25° C. then cooled to 0°-4° C. and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×20 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give Dorzolamide hydrochloride salt (˜2 Kg).
Crude Dorzolamide hydrochloride salt (9 Kg) is solved in water (225 l) at 20°-25° C. and the pH is set to 8.0-8.5 by addition of 25% of aqueous ammonia (2 l). The formed slurry is extracted with ethyl acetate (5×72 l). The collected ethyl acetate phase is concentrated to 180 l by vacuum distillation. The residue is cooled to 20°-25° C., ethyl acetate (45 l) and hydrogen chloride in ethanol (5%, 22.5 l) are added to it during stirring (pH:˜1.0). The formed slurry is stirred for 1 hour at 20°-25° C. then cooled to 0°-4° C. and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×30 l), and dried at 55°-60° C. under vacuum for 4-8 hours to give purified Dorzolamide hydrochloride salt (˜8.2 Kg).
Purified Dorzolamide hydrochloride salt (8 Kg) dissolved in water (24 l) at 95°-105° C. and treated with active carbon (80 g). After filtration, the water solution is cooled gradually to 0°-4° C. and stirred for 3-5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with cooled water (2×5 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give crystallized DRZ HCl salt (˜6.6 Kg).
…………………………………………………………

Reaction of (I) with acetic anhydride-sulfuric acid in methylene chloride provided the sulfonic acid in 98% yield. Conversion to the sulfonyl chloride with phosphorous pentachloride in methylene chloride followed by treatment with aqueous ammonia gave the sulfonamide (II). Reduction of the carbonyl function with sodium borohydride and oxidation of the thiopyran sulfur with Oxone(R) yielded (IV). The 4-hydroxy substituent was converted to the acetylamino functionality under Ritter conditions. Reduction of (V) with borane-dimethylsulfide complex yielded (VI) as a mixture of diasteriomers. Chromatography on silica gel gave the trans-racemate, which was resolved into its individual enantiomers through the di-p-toluoyl-L-tartaric acid salt. The absolute configuration of the S,S-enantiomer, MK-507, was established by single crystal X-ray analysis.
……………………………………………………….
http://bonanzasite.com/synthesis-of-dorzolamide-hydrochloride

………………………..

//////////A new synthesis of MK-0507 has been described: The condensation of 3(R)-(tosyloxy)butyric acid methyl ester (I) with lithium 2-thienylmercaptide (II) in formamide-THF gives 3(S)-(2-thienylthio)butyric acid methyl ester (III), which is hydrolyzed with aqueous HCl to the corresponding free acid (IV). The intramolecular Friedel-Crafts’cyclization of (IV) with trifluoroacetic anhydride yields 6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-4-one (V), which is reduced with LiAlH4 in toluene to afford 4(R)-hydroxy-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran (VI). Epimerization of (VI) with sulfuric acid gives the alcohol (VII) in a cis:trans ratio of 24:76%. Oxidation of (VII) with H2O2 and sodium tungstate yields the 7,7-dioxide (VIII; cis-trans mixture), which is acetylated with acetic anhydride to the acetate (IX). The reaction of (IX) with acetonitrile and sulfuric acid affords N-[6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-4-yl]acetamide 7,7-dioxide (X; cis-trans mixture), which is sulfonated with chlorosulfonic acid and then treated with SOCl2 to give 4-acetamide-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonyl chloride 7,7-dioxide (XI; cis-trans mixture). The reaction of (XI) with concentrated aqueous NH4OH in THF yields the corresponding sulfonamide (XII), which by reduction with BH3-dimethylsulfide in THF affords 4-(ethylamino)-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide (XIII; cis-trans mixture). Finally, this mixture is treated with maleic acid in acetone and the resulting maleates are submitted to fractionated crystallization, giving the maleate of the (4S,6S)-isomer, which is treated first with NaHCO3 and then with HCl to give MK-0507; [alpha](25)589 -17.1 C (c 1, H2O).
 CAS NO. 130693-82-2, DORZOLAMIDE HCL H-NMR spectral analysis |
 CAS NO. 130693-82-2, DORZOLAMIDE HCL C-NMR spectral analysis |
References
- Dorzolamide at Drugs.com. Revised: 12/2011
- KD Tripari MD. Essentials of Medical Pharmacology (5 ed.). Jaypee Brothers Medical Publishers(P) Ltd. p. 88. ISBN 81-8061-187-6.
Further reading
- Kubinyi H (1999). “Chance favors the prepared mind–from serendipity to rational drug design”. J Recept Signal Transduct Res19 (1–4): 15–39. doi:10.3109/10799899909036635. PMID 10071748.
- Plummer C, MacKay E, Gelatt K (2006). “Comparison of the effects of topical administration of a fixed combination of dorzolamide-timolol to monotherapy with timolol or dorzolamide on IOP, pupil size, and heart rate in glaucomatous dogs”. Vet Ophthalmol 9 (4): 245–9. doi:10.1111/j.1463-5224.2006.00469.x. PMID 16771760.
- Grover S, Apushkin M, Fishman G (2006). “Topical dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa”. Am J Ophthalmol 141 (5): 850–8. doi:10.1016/j.ajo.2005.12.030. PMID 16546110.
- Almeida G, Faria e Souza S (2006). “Effect of topical dorzolamide on rabbit central corneal thickness”. Braz J Med Biol Res 39(2): 277–81. doi:10.1590/S0100-879X2006000200015. PMID 16470316.
Reference:
CIPLA LIMITED; CURTIS, Philip, Anthony Patent: WO2008/135770 A2, 2008 ; Location in patent: Page/Page column 21-22 ;
RAGACTIVES, S.L. Patent: US2003/220509 A1, 2003 ; Location in patent: Page/Page column 12 ;
WO2011/101704 A1, ;
Literature References:
Carbonic anhydrase inhibitor. Prepn: J. J. Baldwin et al., EP 296879; eidem, US 4797413 (1988, 1989 both to Merck & Co.). Mechanism of action study: R.-F. Wang et al., Arch. Ophthalmol. 109, 1297 (1991).
HPLC determn in plasma and urine: B. K. Matuszewski, M. L. Constanzer, Chirality 4, 515 (1992).
Clinical evaluations in glaucoma and ocular hypertension: E. A. Lippa et al., Ophthalmology 98, 308 (1991); E. A. Lippa et al., Arch. Ophthalmol. 110, 495 (1992).
| Reference | ||
|---|---|---|
| 1 | * | KAMEI K. ET AL.: ‘Chemical structure, physico-chemical properties and stability of dorzolamide hydrochloride‘ IYAKUHIN KENKYU vol. 25, no. 6, 1994, pages 438 – 452, XP008040715 |
| 2 | * | QUINT M.-P. ET AL.: ‘Dorsolamide hydrochloride‘ ANALYTICAL PROFILES OF DRUG SUBSTANCES AND EXCIPIENTS vol. 26, 1999, pages 283 – 316, XP008040718 |
| EP2128161A1 * | May 30, 2008 | Dec 2, 2009 | Ragactives, S.L. | Process for obtaining 4-hydroxy-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-7,7-dioxide and its enantiomers, and applications thereof |
| WO2008135770A2 * | May 7, 2008 | Nov 13, 2008 | Cipla Ltd | Process for preparing dorzolam ide |
| WO2009144263A2 * | May 28, 2009 | Dec 3, 2009 | Ragactives, S.L.U. | PROCESS FOR OBTAINING 4-HYDROXY-6-METHYL-5, 6-DIHYDRO-4H-THIENO [2,3-b] THIOPYRAN-7, 7-DIOXIDE AND ITS ENANTIOMERS, AND APPLICATIONS THEREOF |
| WO2014005943A1 * | Jun 28, 2013 | Jan 9, 2014 | Zach System S.P.A. | Process for preparing enantiomerically enriched oxamides |
| US8263787 | May 7, 2008 | Sep 11, 2012 | Cipla Limited | Process for preparing dorzolamide |
| WO1994021645A1 * | Mar 16, 1994 | Sep 29, 1994 | Thomas J Blacklock | ENANTIOSELECTIVE SYNTHESIS OF 5,6-DIHYDRO-(S)-4-(ETHYLAMINO)-(S)-6-METHYL-4H-THIENO[2,3-b]THIOPYRAN-2-SULFONAMIDE 7,7-DIOXIDE AND RELATED COMPOUNDS |
| EP0296879A1 * | Jun 23, 1988 | Dec 28, 1988 | Merck & Co., Inc. | Substituted aromatic sulfonamides as antiglaucoma agents |
| US5474919 * | Sep 13, 1994 | Dec 12, 1995 | Merck & Co., Inc. | Bioconversion process for the synthesis of transhydroxy sulfone by Rhodotorula rubra or Rhodotorula piliminae |
| US5760249 * | Aug 28, 1996 | Jun 2, 1998 | Merck & Co., Inc. | Synthesis of hydroxysulfone and related compounds |
| US20060142595 * | Dec 28, 2004 | Jun 29, 2006 | Council Of Scientific & Industrial Research | Starting by reacting a 2-halothiophene with a Grignard reagent in a solvent in situ with sulfur, triethylamine hydrochloride, crotonic acid and a base; product is chlorinated, cyclized, chlorosulfonated and aminated, reduced, oxidized, amidated, hydrogenated, neutralized, recrystallized and resolved |
| US5157129 | Apr 18, 1990 | Oct 20, 1992 | Merck & Co., Inc. | Enantiospecific synthesis of s-(+)-5,6-dihydro-4-(r-amino)-4h-thieno(2,3-b)thiopyran-2-sulfonamide-7,7-dioxide |
| US5474919 | Sep 13, 1994 | Dec 12, 1995 | Merck & Co., Inc. | Bioconversion process for the synthesis of transhydroxy sulfone by Rhodotorula rubra or Rhodotorula piliminae |
| US5688968 | Jan 6, 1995 | Nov 18, 1997 | Merck & Co., Inc. | Enantioselective synthesis of 5,6-dihydro-(S)-4-(ethylamino)-(S)-6-methyl-4H-thieno 2,3-B!thiopyran-2-sulfonamide 7,7-dioxide |
| US5760249 | Aug 28, 1996 | Jun 2, 1998 | Merck & Co., Inc. | Synthesis of hydroxysulfone and related compounds |
| US7109353 | Dec 28, 2004 | Sep 19, 2006 | Council Of Scientific And Industrial Research | Process for preparing 5,6-dihydro-4-(S)-(ethylamino)-6-(S) methyl-4H-thieno[2,3b]thiopyran-2-sulphonamide-7,7-dioxide HCl |
| US20060155132 | Jan 6, 2006 | Jul 13, 2006 | Kovacs Laszlo Z | Method of making dorzolamide hydrochloride |
| EP0296879A1 | Jun 23, 1988 | Dec 28, 1988 | Merck & Co., Inc. | Substituted aromatic sulfonamides as antiglaucoma agents |
| WO1994021645A1 | Mar 16, 1994 | Sep 29, 1994 | Thomas J Blacklock | ENANTIOSELECTIVE SYNTHESIS OF 5,6-DIHYDRO-(S)-4-(ETHYLAMINO)-(S)-6-METHYL-4H-THIENO[2,3-b]THIOPYRAN-2-SULFONAMIDE 7,7-DIOXIDE AND RELATED COMPOUNDS |
| WO2008135770A2 | May 7, 2008 | Nov 13, 2008 | Cipla Ltd | Process for preparing dorzolam ide |


