
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
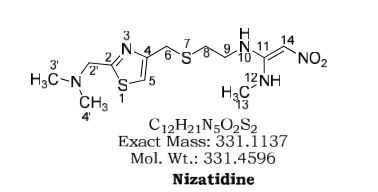
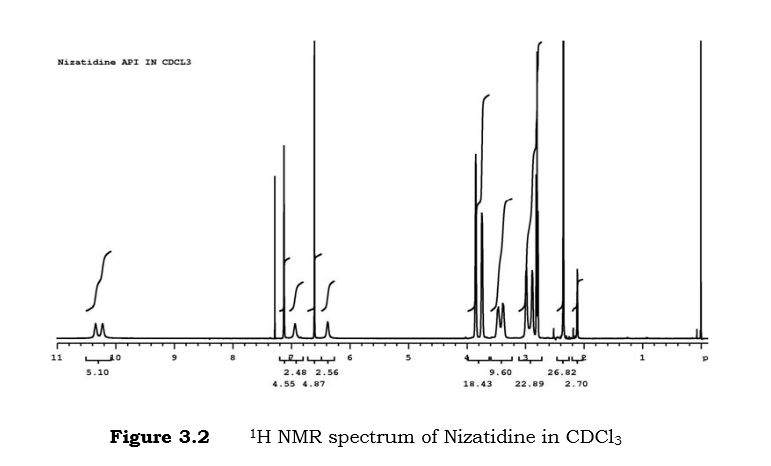

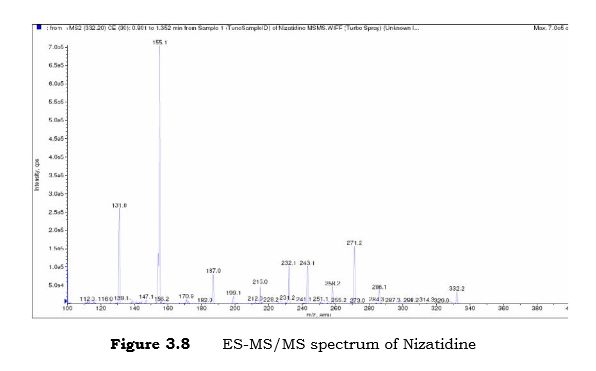

Nizatidine is a histamine H2 receptor antagonist that inhibits stomach acid production, and is commonly used in the treatment of peptic ulcer disease and gastroesophageal reflux disease. It was developed by Eli Lilly and is marketed under the brand names Tazac and Axid.
Clinical use
Nizatidine is used to treat duodenal ulcers, gastric ulcers, and gastroesophageal reflux disease (GERD/GORD), and to prevent stress ulcers.[1]
Adverse effects
Side effects are uncommon, usually minor, and include diarrhea, constipation, fatigue, drowsiness, headache, and muscle aches.[1]
History and development
Nizatidine was developed by Eli Lilly, and was first marketed in 1987. It is considered to be equipotent with ranitidine and differs by the substitution of a thiazole ring in place of the furan ring in ranitidine. In September 2000, Eli Lilly announced they would sell the sales and marketing rights for Axid to Reliant Pharmaceuticals.[2] Subsequently, Reliant developed the oral solution of Axid, marketing this in 2004, after gaining approval from the U.S. Food and Drug Administration (FDA).[3] However, a year later, they sold rights of the Axid Oral Solution (including the issued patent[4] protecting the product) to Braintree Laboratories.[5]
Nizatidine proved to be the last new histamine H2 receptor antagonist introduced prior to the advent of proton pump inhibitors.’
Nizatidine, the systematic chemical name of which is N-[2-[[[2-[ imemylammo)memyl]-4-tl iazolyl]memyl]mio]e yl]–N’- methyl-2-nitro-l,l-ethenecliamine, which has the formula (I).This compound is a histamine H2-receptor antagonist which is useful as anti- ulcer agents capable of inmbiting gastric acid secretion in mammals.

United States Patent No. 4,375,547; 4587344, 4777260; 4,904,792 and 5334725 discloses Nizatidine and other related products. The synthesis of nizatidine disclosed in US patent No. 4,904,792 involves a multi-step process. The first step of the process comprises reacting dimethylaminotmoacetamide hydrochloride with ethyl bromopyruvate to obtain 2-(dinιethylaminon ethyl)-4-thiazolecarboxylate. Reduction of this 4- tbiazolecarboxylate derivative with lithium triethylborohydride gives 2-
(<-Umethylaminoπιethyl)-4-tI-ύazolenιethanol, which is then converted into 4- (2-ammoetϊhyl)ti omethyl-2-d by reacting with
2-aminoethanethiol hydrochloride (cysteamine hydrochloride). This 2- ό-imetihylan-ιinoπιethylthiazol derivative is then converted into Nizatidine by reacting .with N-met-hyl-l-methyltHo-2-mt-coet-hyleneamine in the presence of an acid United States Patent No. 4,382,090 describes a method to prepare 4-
(2-aminoethyl)tMome1_hyl-2-din ethylaminon etihyltl iazol by fusing 4- cmoronιe yl-2-d- nethylaminonιet-hylthiazole with cysteamine hydrochloride at above 100 °C.
United States Patent No. 4,468,517 described a method to prepare 4- cldoronιethyl-2-<-ιimethylaminon et-hylt-lιiazole. The method described in this patent involved reaction of dimet-hylaminotmoacetamide hydrochloride with 1,3-dichloroacetone in haloalkane (1,2-dichloroethane) as a solvent to obta 4-cHoromethyl^-hydroxy-2-dimet^ This 2-thiazoline derivative is then dehydrated with a dehydrating agent like PC13, PBr3, SOCl2, POCl3 etc., to get 4-chloromethyl-2- din etihyl-in monietihylthiazole.
European Patent Application EP 0,515,121 and EP 0,960,880 describe the process for the preparation of 2-(dim.et-hylarninomethyl)-4- thiazolemethanol. The process consists of reacting (-Umethylaminothioacetamide hydrochloride with 1,3-dichloroacetone in toluene to get 4-chlorometiιyl-4-hyαioxy-2-d-methylaminomethyl-2- thiazoline, which is then reacted with alkali metal base in an inert solvent such as toluene to get 2-(dimethylam-m.omethyl)-4-thiazolemethanol.
The methods described in United States Patent No. 4,468,517 for the synthesis of 4-chloromethyl-4-hy( oxy-2-dimethyl-ui-momethyl-2- thiazoline, requires complete evaporation of the solvent 1,2-dichloroethane to get the crude product; it is then washed with ethyl acetate to obtain a pure product. Evaporation of the solvent to complete dryness is an inconvenient and inappropriate operation in large-scale manufacturing. Such evapprations in large-scale operations would produce the solids as lumps; further washing such lumps with solvents would be ineffective due to improper -mixing of -the solid -with solvent. The method described in EP 0,515,121 and EP 0,960,880 for the synthesis of 4-cHorometlιyl-4-hyc oxy*-2-α_im requires isolation of the product from the reaction mixture by precipitation of the product from the mother liquor by the addition of petroleum ether. The crude product obtained by the precipitation is then subjected to an additional purification step by crystallisation from toluene.
A number of procedures are described for the preparation of dimethylammotmoacetamide. Examples are Japanese Patent No. JP 62,273,948, JP 62,273,949, JP 02,264,755 and Org. Prep. Proced. Int., 1992, 24, P.66-7. All the procedures described in the literature- or the preparation of dirnethylaminotitioacetamide from dimethylam oacetomtrile involve the use of hydrogen sulfide under pressure in the presence of promoters or catalysts. The disadvantage with the use of hydrogen sulfide is the difficulty it poses in handling commercial quantities, as it is a very toxic gas. The object of the present invention is to provide an improved manufactxiring process for 4-chloromethyl-4-hydr xy-2- di–netihylam omethyl-2-tibiazoline..
 .
.
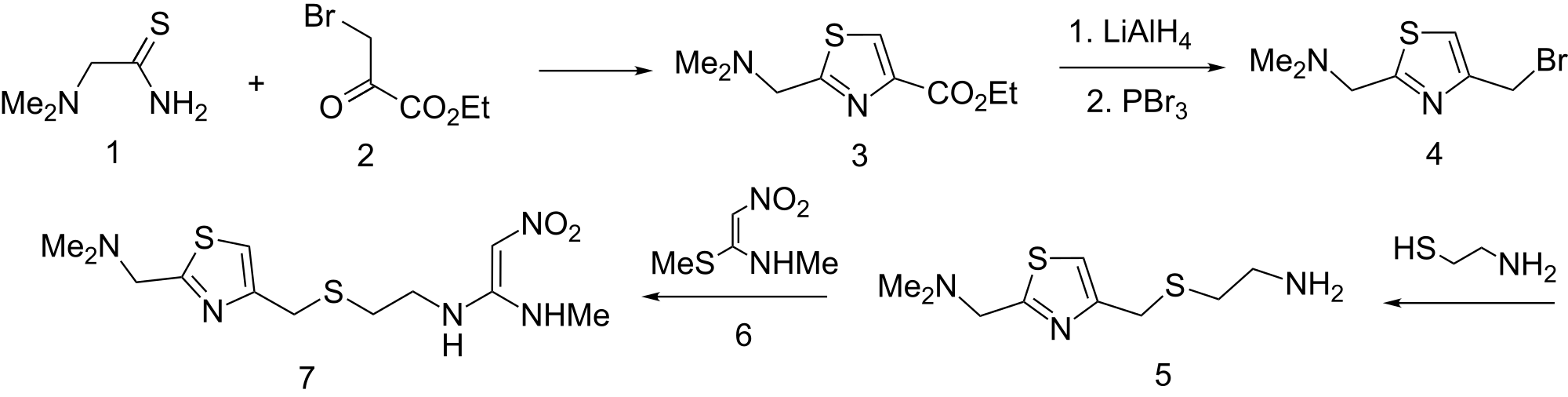
SYN2
The cyclization of dimethylaminothioacetamide (I) with ethyl bromopyruvate (II) in refluxing ethanol gives ethyl 2-(dimethylaminomethyl)-4-thiazolecarboxylate (III), which is reduced with lithium triethyl borohydride in THF yielding 2-(dimethylaminomethyl)-4-thiazolemethanol (IV). The condensation of (IV) with 2-aminoethanethiol (V) by means of 48% HBr affords 2-(dimethylaminomethyl)-4-(2-aminoethylthiomethyl)thiazole (VI), which is finally condensed with 1-(methylthio)-2-nitro-N-methylethyleneamine (VII) in water.

PATENT
http://www.google.com/patents/WO2004069817A1?cl=en
Example No: 1 Preparation of dirnethylaniinothioacetaniide hydrochloride Into water (3000 ml), phosphorus pentasulfi.de (1302 g; 2.93 mol) and dimethylam oacetonitrile (1000 g; 11.88 mol) are added one after another at 10°C. The mixture is then slowly warmed to 70°C and maintained for 3 hrs to complete the reaction. The reaction mixture is then cooled to 20°C and sodiu hydroxide (53% w/w, 2200 g, 29.15 mol) is added into it below 20°C. The reaction mixture is then warmed to 50°C and extracted with toluene (2 x 2000 l). Isopropanolic hydrochloric acid (12% w/w; 3700 ml) is added into the extract at 25 to 30°C to adjust the pH to 2 and the mass stirred for 1 h to precipitate the product. The slurry is filtered, washed with isopropyl alcohol (1000 ml) and dried to get (1360 g) dimethyl ammotMoacetamide hydrochloride. Yield = 74.0%, HPLC purity = 97.6% Example No: 2
Preparation of 4-chloromethyl-4-hydr oxy-2-dimethylaminomethyl-2- thiazoline
Dimethylam othioacetamide hydrochloride (1000 g; 6.472 mol) is suspended in diisopropyletiier (4000 ml). Added into this suspension is sodium bicarbonate (1200 g; 14.28 mol) and sodium sulphate (1000 g). The slurry is heated to 55-60° C and stirred for 1 hr. Into this suspension is added 1,3 dichloroacetone (1000 g; 7.87 mol) dissolved in diisopropylether (1000 ml). The reaction is continued at 50-55° C for 2 h. The progress of the reaction is monitored by a qualitative HPLC analysis. Upon completion of the reaction, the reaction mixture is* filtered hot at 50-55° C to remove insoluble inorganic salts. The mother liquor is cooled slowly to 0-5° C to crystallize out the product. The product is then filtered and washed with precooled diisopropylether (250 ml). The product is dried at 50° C under reduced pressure to obtain 1120 g. Yield = 83%; HPLC purity = 98.2%. The following example illustrates the process to convert this pure 4- cHoromethyl-4-hyσ-roxy-2-ά-imet^^ Nizatidine. Example No 3: Preparation of N- [2- [ [ [2- [(Dimethylaι-nino)methyl] -4- thiazolyl] methyl] thio] ethyl] -N’-methyl-2-nitro-l,l-ethenediamine. A. Preparation of 4-chloromethyl-2-ααmethylam onιethylthiazole Hydrochloride.
Thionyl chloride (430 ml; 5.9 mol) is added into chloroform (1000 ml) and cooled to 20° C. Into this solution is added 4-chloromethyl-4- hyά^oxy-2-dinιethylam ome yl-2-thiazoline (1000 g; 4.79 mol), dissolved in chloroform (4000 ml). The reaction mixture is further gradually heated to 60-65° C and maintained at this temperature till qualitative HPLC analysis shows the completion of the reaction. The reaction mixture is then cooled slowly to 30° C to get the product crystallized out. The product is filtered, washed and dried under reduced pressure to obtain 900 g of pure product. Yield = 83.3 %. B. Preparation of 4-(2-am oethyl)thiomethyl-2- ά-imethylam omethylthiazole.
2-A-minoethanetl iol hydrochloride (cysteamine hydrochloride, 520 g; 4.5 mol) is suspended in water (500 ml). This suspension is cooled to 5° C and sodium hydroxide solution (45 % w/w, 870 ml; 14.7 mol) is added into it at 5-10° C. Into this suspension, hydroxylamine sulphate (100 g; 0.6 mol) is added and stirred. A solution of 4-chloromethyl-2- di-n ethyl- inomethylthiazole hydrochloride (1000 g; 4.43 mol) dissolved in water (1250 ml) is prepared separately. This solution is added into the said suspension below 10° C and the reaction continued at 10° C for another 1 h. The completion of the reaction is determined by qualitative HPLC. The reaction mixture is then diluted with water (2000 ml), heated to 40-45° C and extracted with toluene (2 x 2000 ml). The toluene extract is treated with activated carbon at 40-45° C for 30 min. Activated carbon is removed by filtration through hyflo bed and evaporated toluene from the filtrate under reduced pressure at 60° C to obtain 910 g of the product. Yield = 88 %. C. Preparation of N-(2-(((2-(Dimethylamino)methyl)-4- tltiazolyl)m.ethyl)tltio)elhyl)-N’-methyl-2-nitro-l ,1 -etheneά-iamine (Nizatidine).
N-methyl-l-methyltHo-2-mtroethyleneamine (NMSM, 610 g; 4.12 mol) is mixed with water (1500 ml), and the mixture is cool to 20-25° C. 4- (2-Am-hoethyl)d omethyl-2-<^ (1000 g; 4.32 mol) dissolved in water (1500 ml) is added into this suspension at 20-25° C. The reaction mixture is warmed to 30-35° C and continued the reaction for 8 h. The progress of the reaction is monitored by qualitative HPLC analysis. The reaction mixture is extracted with toluene (2 x 1000 ml), and the aqueous layer is treated with activated carbon (50 g) at 55-60° C for 30 min. Activated carbon is removed by filtration through hyflo bed and the aqueous filtrate is extracted with chloroform (4 x 1000 ml)rThe cHorόform extract is concentrated under reduced pressure at less than 50° C; ethyl acetate (3000 ml) is added into the concentrate and reconcentrated. Acetone (300 ml), ethyl acetate (300 ml) is added into the concentrate and cooled to 0-5° C to crystallize the product. The product is filtered, washed with precooled ethyl acetate (250 ml), and dried to obtain pure Nizatidine 1160 g. Yield = 81.0%; HPLC purity -= 99.3%.
References
- [1] Archived May 26, 2008 at the Wayback Machine
- [2] Archived December 26, 2013 at the Wayback Machine
- “United States Patent: 6930119”. Patft.uspto.gov. Retrieved 2015-10-11.
- [3] Archived August 14, 2007 at the Wayback Machine
External links
| US4468517 * | May 12, 1983 | Aug 28, 1984 | Eli Lilly And Company | Synthesis of thiazoles |
| US5457206 * | Jul 1, 1994 | Oct 10, 1995 | Eli Lilly And Company | Process for preparing intermediates to nizatidine and related compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015002150A1 | Jun 30, 2014 | Jan 8, 2015 | Shin Nippon Biomedical Laboratories, Ltd. | Novel compound, organic cation transporter 3 detection agent, and organic cation transporter 3 activity inhibitor |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(E)-1-N‘-[2-[[2-[(dimethylamino)methyl]-1,3-thiazol-4-yl]methylsulfanyl]ethyl]-1-N-methyl-2-nitroethene-1,1-diamine
|
|
| Clinical data | |
| Trade names | Axid |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a694030 |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | >70% |
| Protein binding | 35% |
| Metabolism | Hepatic |
| Biological half-life | 1–2 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | 76963-41-2 |
| ATC code | A02BA04 |
| PubChem | CID 3033637 |
| IUPHAR/BPS | 7248 |
| DrugBank | DB00585 |
| ChemSpider | 2298266 |
| UNII | P41PML4GHR |
| KEGG | D00440 |
| ChEBI | CHEBI:7601 |
| ChEMBL | CHEMBL653 |
| Chemical data | |
| Formula | C12H21N5O2S2 |
| Molar mass | 331.46 g/mol |


{kind=link}