
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
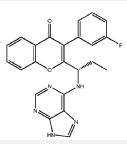 RP 6530
RP 6530(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one (Compound A1 is RP 6530).
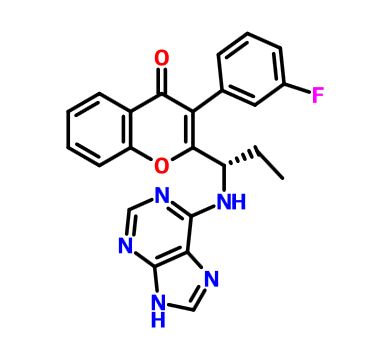
RP 6530
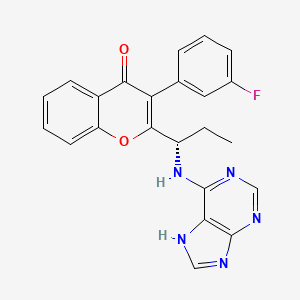
RP 6530, RP6530, RP-6530
Tenalisib
RP6530-1401, NCI-2015-01804, 124584, NCT02567656
(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
3-(3-fluorophenyl)-2-[(1S)-1-(7H-purin-6-ylamino)propyl]chromen-4-one
MW415.4, C23H18FN5O2
CAS 1639417-53-0, 1693773-94-2
RP6530 demonstrated high potency against PI3Kδ (IC50 =24.5 nM) and γ (IC50 = 33.2 nM) enzymes with selectivity over α (>300-fold) and β (>100-fold) isoforms. Cellular potency was confirmed in target-specific assays, namely anti-FcεR1-(EC50=37.8 nM) or fMLP (EC50 = 39.0 nM) induced CD63 expression in human whole blood basophils, LPS induced CD19+ cell proliferation in human whole blood (EC50=250 nM), and LPS induced CD45R+ cell proliferation in mouse whole blood (EC50=101 nM).
A PI3K inhibitor potentially for the treatment of hematologic malignancies.
An inhibitor of phosphoinositide-3 kinase (PI3K) δ/γ isoforms and anti-cellular proliferation agent for treatment of hematol. malignancies
Rhizen Pharmaceuticals is developing RP-6530, a PI3K delta and gamma dual inhibitor, for the potential oral treatment of cancer and inflammation In November 2013, a phase I trial in patients with hematologic malignancies was initiated in Italy ]\. In September 2015, a phase I/Ib study was initiated in the US, in patients with relapsed and refractory T-cell lymphoma. At that time, the study was expected to complete in December 2016
PATENTS……..WO 11/055215 , WO 12/151525.
Inventors
| Inventors | Meyyappan Muthuppalaniappan, Srikant Viswanadha, Govindarajulu Babu, Swaroop Kumar V.S. Vakkalanka, |
| Incozen Therapeutics Pvt. Ltd., Rhizen Pharmaceuticals Sa |
- Antineoplastics; Small molecules
- Mechanism of Action Phosphatidylinositol 3 kinase delta inhibitors; Phosphatidylinositol 3 kinase gamma inhibitors
- Phase I Haematological malignancies
- Preclinical Multiple myeloma
| Swaroop K. V. S. Vakkalanka, | |
| COMPANY | Rhizen Pharmaceuticals Sa |
https://clinicaltrials.gov/ct2/show/NCT02017613
PI3K delta/gamma inhibitor RP6530 An orally active, highly selective, small molecule inhibitor of the delta and gamma isoforms of phosphoinositide-3 kinase (PI3K) with potential immunomodulating and antineoplastic activities. Upon administration, PI3K delta/gamma inhibitor RP6530 inhibits the PI3K delta and gamma isoforms and prevents the activation of the PI3K/AKT-mediated signaling pathway. This may lead to a reduction in cellular proliferation in PI3K delta/gamma-expressing tumor cells. In addition, this agent modulates inflammatory responses through various mechanisms, including the inhibition of both the release of reactive oxygen species (ROS) from neutrophils and tumor necrosis factor (TNF)-alpha activity. Unlike other isoforms of PI3K, the delta and gamma isoforms are overexpressed primarily in hematologic malignancies and in inflammatory and autoimmune diseases. By selectively targeting these isoforms, PI3K signaling in normal, non-neoplastic cells is minimally impacted or not affected at all, which minimizes the side effect profile for this agent. Check for active clinical trials using this agent. (NCI Thesaurus)
| Company | Rhizen Pharmaceuticals S.A. |
| Description | Dual phosphoinositide 3-kinase (PI3K) delta and gamma inhibitor |
| Molecular Target | Phosphoinositide 3-kinase (PI3K) delta ; Phosphoinositide 3-kinase (PI3K) gamma |
| Mechanism of Action | Phosphoinositide 3-kinase (PI3K) delta inhibitor; Phosphoinositide 3-kinase (PI3K) gamma inhibitor |
| Therapeutic Modality | Small molecule |

Dual PI3Kδ/γ Inhibition By RP6530 Induces Apoptosis and Cytotoxicity In B-Lymphoma Cells
RP6530 is a potent and selective dual PI3Kδ/γ inhibitor that inhibited growth of B-cell lymphoma cell lines with a concomitant reduction in the downstream biomarker, pAKT. Additionally, the compound showed cytotoxicity in a panel of lymphoma primary cells. Findings provide a rationale for future clinical trials in B-cell malignancies.

PI3K Dual Inhibitor (RP-6530)
| Therapeutic Area | Respiratory , Oncology – Liquid Tumors , Rheumatology | Molecule Type | Small Molecule |
|---|---|---|---|
| Indication | Peripheral T-cell lymphoma (PTCL) , Non-Hodgkins Lymphoma , Asthma , Chronic Obstructive Pulmonary Disease (COPD) , Rheumatoid Arthritis | ||
| Development Phase | Phase I | Rt. of Administration | Oral |
Description
Rhizen is developing dual PI3K gamma/delta inhibitors for liquid tumors and inflammatory conditions.
Mechanism of Action
While alpha and beta isoforms are ubiquitous in their distribution, expression of delta and gamma is restricted to circulating hematogenous cells and endothelial cells. Unlike PI3K-alpha or beta, mice lacking expression of gamma or delta do not show any adverse phenotype indicating that targeting of these specific isoforms would not result in overt toxicity. Dual delta/gamma inhibition is strongly implicated as an intervention strategy in allergic and non-allergic inflammation of the airways and other autoimmune diseases. Scientific evidence for PI3K-delta and gamma involvement in various cellular processes underlying asthma and COPD stems from inhibitor studies and gene-targeting approaches. Also, resistance to conventional therapies such as corticosteroids in several COPD patients has been attributed to an up-regulation of the PI3K delta/gamma pathway. Disruption of PI3K-delta/gamma signalling therefore provides a novel strategy aimed at counteracting the immuno-inflammatory response. Due to the pivotal role played by PI3K-delta and gamma in mediating inflammatory cell functionality such as leukocyte migration and activation, and mast cell degranulation, blocking these isoforms may also be an effective strategy for the treatment of rheumatoid arthritis as well.
Given the established criticality of these isoforms in immune surveillance, inhibitors specifically targeting the delta and gamma isoforms would be expected to attenuate the progression of immune response encountered in airway inflammation and rheumatoid arthritis.

Clinical Trials
Rhizen has identified an orally active Lead Molecule, RP-6530, that has an excellent pre-clinical profile. RP-6530 is currently in non-GLP Tox studies and is expected to enter Clinical Development in H2 2013.
In December 2013, Rhizen announced the start of a Phase I clinical trial. The study entitled A Phase-I, Dose Escalation Study to Evaluate Safety and Efficacy of RP6530, a dual PI3K delta /gamma inhibitor, in patients with Relapsed or Refractory Hematologic Malignancies is designed primarily to establish the safety and tolerability of RP6530. Secondary objectives include clinical efficacy assessment and biomarker response to allow dose determination and potential patient stratification in subsequent expansion studies.
Partners by Region
Rhizen’s pipeline consists of internally discovered (with 100% IP ownership) novel small molecule programs aimed at high value markets of Oncology, Immuno-inflammtion and Metabolic Disorders. Rhizen has been successful in securing critical IP space in these areas and efforts are on for further expansion in to several indications. Rhizen seeks partnerships to unlock the potential of these valuable assets for further development from global pharmaceutical partners. At present global rights on all programs are available and Rhizen is flexible to consider suitable business models for licensing/collaboration.
In 2012, Rhizen announced a joint venture collaboration with TG Therapeutics for global development and commercialization of Rhizen’s Novel Selective PI3K Kinase Inhibitors. The selected lead RP5264 (hereafter, to be developed as TGR-1202) is an orally available, small molecule, PI3K specific inhibitor currently being positioned for the treatment of haematological malignancies.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014195888Intermediates
Intermediate 1: 3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one: To a solution of 2-(l-bromopropyl)-3-(3-fluorophenyl)-4H-chromen-4-one1 (8.80 g, 24.36 mmol ) in DMSO (85 ml), n-butanol (5 ml) was added and heated to 120° C for 3h. The reaction mixture was cooled to room temperature (RT), quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (2.10 g, 29 %) which was used without further purification in next step.
Intermediate 2: 3-(3-fluorophenyl)-2-propionyl-4H-chromen-4-one: DMSO (1.90 ml, 26.82 mmol) was added to dichloromethane (70 ml) and cooled to -78°C. Oxalyl chloride (1.14 ml, 13.41 mmol) was then added. After 10 minutes, intermediate 1 (2.00 g, 6.70 mmol) in dichloromethane (20 ml) was added dropwise and stirred for 20 min. Triethylamine (7 ml) was added and stirred for lh. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow liquid (1.20 g, 60%) which was used as such in next step.
Intermediate 3: (+)/(-)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one :
To a solution of intermediate 2 (0.600 g, 2.02 mmol) in DMF (7.65 ml) under nitrogen purging, formic acid : trietylamine 5 : 2 azeotrope (1.80 ml) was added followed by [(S,S)tethTsDpenRuCl] (3.0 mg). The reaction mixture was heated at 80°C for 1.5 hours under continuous nitrogen purging. The reaction mixture was quenched with water, extected with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (0.450 g, 74%). Mass: 299.0 (M+).
Enantiomeric excess: 78%, enriched in the late eluting isomer (retention time: 9.72 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 4: (+)/(-)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one :
The title compound was obtained as yellow solid (0.500 g, 83%) by using a procedure similar to the one described for intermediate 3, using intermediate 2 (0.600 g, 2.02 mmol), DMF (7.65 ml), formic acid : trietylamine 5 : 2 azeotrope (1.80 ml) and [(R,R)tethTsDpenRuCl] (3.0 mg). Mass: 298.9 (M+). Enantiomeric excess: 74.8%, enriched in the fast eluting isomer (retention time: 8.52 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: (R)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one:
Step 1 : (R)-2-(l-(benzyloxy)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one: To 2-(3-fluorophenyl)-l-(2-hydroxyphenyl)ethanone (2.15 g, 9.36 mmol ), in dichloromethane ( 20 ml), HATU (4.27 g, 11.23 mmol), R-(+)2-benzyloxybutyric acid (2.00 g, 10.29 mmol) were added and stirred for lOmin, then triethylamine (14.0 ml, 101.1 mmol) was added dropwise and stirred at RT for 24h. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as yellow solid (1.65 g, 45%). JH-NMR (δ ppm, CDC13, 400 MHz): 8.24 (dd, / = 7.9,1.5 Hz, 1H), 7.74 (dt, / = 7.1,1.7 Hz, 1H), 7.58 (dd, / = 8.3,0.4 Hz, 1H), 7.44-7.06 (m, 10H), 4.51 (d, / = 7.8 Hz, 1H), 4.34 (d, / = 7.8 Hz, 1H), 4.25 (dd, / = 7.8,6.2 Hz, 1H), 2.17-1.90 (m, 2H), 0.95 (t, / = 7.5 Hz, 3H). Mass: 389.0 (M+).
Step 2: (R)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one : To (R)-2-(l-(benzyloxy)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one (1.50 g, 3.86 mmol) in dichloromethane (15 ml) cooled to 0°C and aluminium chloride (1.00 g, 7.72 mmol) was added portion wise and stirred at RT for 6h. The reaction mixture was quenched with 2N HC1 solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as yellow solid (0.552 g, 48%).‘ JH-NMR (δ ppm, CDC13, 400 MHz): ‘ 8.24 (dd, / = 8.0,1.6 Hz, 1H), 7.72 (m, , 1H), 7.52 (dd, / = 8.4,0.5 Hz, 1H), 7.44 (m, 2H), 7.12-7.01(m,3H), 4.49 (t, / = 7.0 Hz, 1H), 1.94 (m, 2H), 0.93 (t, / = 7.5 Hz, 3H). Mass: (299.0(M+). Purity: 96.93%.
25[a] D -14.73 (c = 1, CHCI3). Enantiomeric excess: 85.92%, enriched in the fast eluting isomer (retention time: 8.57 min.) as determined by HPLC on a chiralpak AS-3R column.
Compound A
(RS)- 2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
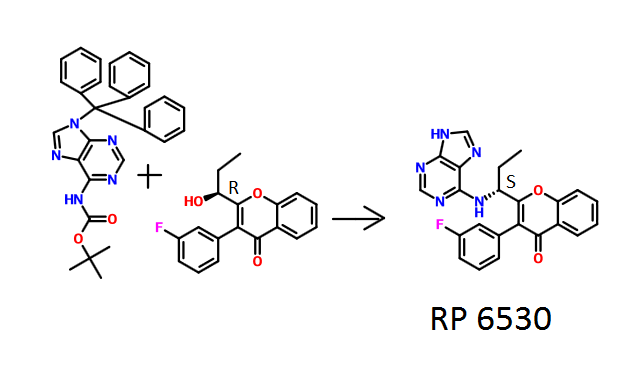
To a solution of intermediate 1 (2.50 g, 8.41 mmol) in THF (25 ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (4.81 g, 10.09 mmol) and triphenylphosphine (3.31 g, 12.62 mmol) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (2.5 ml, 12.62 mmol) was added and stirred at RT for 2h. The reaction mixture was concentrated and column chromatographed with ethyl acetate : petroleum ether to afford a yellow coloured intermediate. To the intermediate, dichloromethane (65 ml) and trifluoroacetic acid (7.9 ml) were added and the resulting mixture was stirred at RT for 12 h. The reaction mixture was then basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as pale-brown solid (1.05 g, 30 %). MP: 148-150°C. Mass: 415.6 (M+).
Compound Al
(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
Method A: To a solution of intermediate 3 (0.250 g, 0.838 mmol) in THF (5ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (0.479 g, 1.00 mmol) and triphenylphosphine (0.329 g, 1.25 mmol) were added and the resulting mixture was stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.25 mmol) was then added and stirred at RT for 12 h. The reaction mixture was concentrated and column chromatographed with ethyl acetate: pet.ether to afford the yellow coloured intermediate. To the intermediate in dichloromethane (6 ml), trifluoroacetic acid (1.2 ml) was added stirred at RT for 12 h. The reaction mixture was basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as an off-white solid (0.015 g, 4 %). MP: 137-140°C. JH-NMR (δ ppm, DMSO- , 400 MHz): 12.94 (s, 1H), 8.12-8.10 (m, 4H), 7.84-7.80 (m, 1H), 7.61 (d, / = 8.3 Hz, 1H), 7.50-7.41 (m, 2H), 7.28-7.18 (m, 3H), 5.20-5.06 (m, 1H), 2.10-1.90 (m, 2H), 0.84 (t, / = 3.7 Hz, 3H). Enantiomeric excess: 77.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 7.90 min.).
Method B : To a solution of intermediate 5 (2.60 g, 8.68 mmol) in THF (52 ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (4.96 g, 10.42 mmol) and triphenylphosphine (2.76 g, 13.03 mmol) were added and the resulting mixture was stirred at RT for 5 min. Dusopropylazodicarboxylate (0.25 ml, 1.25 mmol) was then added and stirred at RT for 12 h. The reaction mixture was concentrated and column chromatographed with ethyl acetate: petroleum ether to afford the yellow coloured intermediate. To the intermediate in dichloromethane (55 ml), trifluoroacetic acid (14.2 ml) was added and stirred at RT for 12 h. The reaction mixture was basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as pale-yellow solid (1.00 g, 27 %). MP: 168-170°C. Mass: 416.5(M++1) Enantiomeric excess: 86.5% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 7.90 min.).
Method C : The title compound was separated by preparative SFC conditions from Compound A (1.090 g) on a CHIRALPAK AY-H column (250 x 30 mm; 5μπι) using methanol : C(¾ (35:65) as the mobile phase at a flow rate of 80 g / min. Off-white solid (0.378 g). e.e. 100%. Rt: 2.37 min. Mass: 416.1(M++1). MP: 149-152°C.
Scheme 1A

CAUTION ethyl compd below, NOT THE PRODUCT
Example 47
(S)-2-(l-(9H-purin-6-yIamino) ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one
[428] To a solution of intermediate 65 (2.0g, 8.68 mmoles) in dichloromethane (20ml), triethylamine (3.6ml, 26.06 mmoles) was added followed by N-Boc-Alanine (1.97g, 10.42 mmoles). To this mixture HATU (6.6g, 17.37 mmoles) was added and stirred at RT for 12h. The reaction mixture was quenched by the addition of water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the isoflavone intermediate (1.70g). To a solution of this intermediate (1.7g) in dichloromethane (20ml), trifluoroacetic acid (3 ml) was added and stirred at RT for 2h. The reaction mixture was concentrated, basified with sodium bicarbonate solution, extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the amine intermediate (0.641 g). To a solution of this amine intermediate (0.30g, 1.05 mmoles) in tert-butanol (6ml), N, N- diisopropylethylamine (0.36ml, 2.17 mmoles) and 6-bromopurine (0.168g, 0.847 mmoles) were added and refluxed for 24h. The reaction mixture was concentrated, diluted with water, extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: ethyl acetate to afford the title compound as off-white solid (0.041g, 10% yield). MP: 135-138 °C. Ή-NMR (δ ppm, DMSO-D6, 400 MHz): δ 12.95(s,lH), 8.15(t, / = 6.8Hz, 1H), 8.11(s, 1H), 8.08(s, 1H), 8.03(d, J = 7.8 Hz, 1H), 7.81(t ,J = 7.3Hz, 1H), 7.60 (d, J = 8.3Hz, 1H), 7.49 (t, J = 7.3Hz, 2H), 7.25(m,3H), 5.19(br m, 1H), 1.56(d, J = 6.9Hz,3H). Mass: 402.18(M+ +1).
Scheme 1
Base

This scheme provides a synthetic route for the preparation of compound of formula wherein all the variables are as described herein in above

15 14 10 12 12a
Abstract 2704: RP6530, a dual PI3K δ/γ inhibitor, potentiates ruxolitinib activity in the JAK2-V617F mutant erythroleukemia cell lines
– Author Affiliations
- 1Rhizen Pharmaceuticals SA, Fritz-Courvoisier 40, Switzerland;
- 2Incozen Therapeutics Pvt. Ltd., Hyderabad, India.
Abstract
Background: Myelofibrosis (MF) represents a life-threatening neoplasm that manifests particularly in the elderly population and is characterized by bone marrow fibrosis and extramedullary hematopoeisis. While ruxolitinib, a JAK1/2 inhibitor, has recently been approved by the USFDA for its disease modifying potential in MF patients, it is still not considered as a curative option. Targeting another kinase such as PI3K, downstream of JAK, could therefore be a more efficient way of treating myelofibrotic neoplasms. RP6530 is a novel, potent, and selective PI3K δ/γ inhibitor that demonstrated high potency against PI3Kδ (IC50 = 25 nM) and γ (IC50 = 33 nM) enzymes with selectivity over α (>300-fold) and β (>100-fold) isoforms. The objective of this study was to evaluate the effect of a combination of ruxolitinib and RP6530 in the JAK2-V617F mutant Human Erythroleukemia (HEL) cell line.
Methods: Passive resistance was conferred by incubating HEL cells with increasing concentrations of ruxolitinib over an 8-10-week period. Endogenous JAK2, PI3Kδ, PI3Kδ, and pAKT were estimated by Western Blotting. RP6530, ruxolitinib, and the combination of RP6530 + Ruxolitinib were tested for their effect on viability and apoptosis. Cell viability was assessed by a MTT assay. Induction of apoptosis was analyzed by Annexin V/PI staining.
Results: Resistance to ruxolitinib was confirmed by a right-ward shift in EC50 of ruxolitinib in a HEL cell proliferation assay (0.82 μM Vs. 12.2 μM). Endogeous pAKT expression was 3.7-fold higher in HEL-RR compared to HEL-RS cells indicating activation of the AKT signaling pathway. While single-agent activity of RP6530 was modest (33-46% inhibition @ 10 μM) in both HEL-RS and HEL-RR cells, addition of 10 μM RP6530 to ruxolitinib was synergistic resulting in a near-complete inhibition of proliferation (>90% for HEL-RS and >70% for HEL-RR). While the order of addition did not affect the potency of RP6530, addition of 5 μM RP6530, 4 h prior to the addition of ruxolitinib resulted in a significant reduction in EC50 of ruxolitinib (5.8 μM) in HEL-RR cells. On lines with cell proliferation data, incubation of 10 μM RP6530 with ruxolitinib for 72 h increased the percent of apoptotic cells (55% in HEL-RS and 37% in HEL-RR) compared to either agent alone (16-27% in HEL-RS and 17-21% in HEL-RR).
Conclusions: Ruxolitinib resistance in the V617F JAK-2 mutant HEL cells is accompanied by an increase in pAKT expression. Inhibition of pAKT via the addition of RP6530, a dual PI3K δ/γ inhibitor, resulted in a reversal of ruxolitinib resistance. Complementary activity was also observed in HEL-RS cells indicating that a combination of ruxolitinib and RP6530 could have a positive bearing on the clinical outcome in MF patients.
Citation Format: Swaroop Vakkalanka, Seeta Nyayapathy, Srikant Viswanadha. RP6530, a dual PI3K δ/γ inhibitor, potentiates ruxolitinib activity in the JAK2-V617F mutant erythroleukemia cell lines. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 2704. doi:10.1158/1538-7445.AM2015-2704
- 01 Sep 2015 Phase-I clinical trials in Haematological malignancies (Second-line therapy or greater) in USA (PO) (NCT02567656)
- 18 Nov 2014 Preclinical trials in Multiple myeloma in Switzerland (PO) prior to November 2014
- 18 Nov 2014 Early research in Multiple myeloma in Switzerland (PO) prior to November 2014
| WO2011055215A2 | Nov 3, 2010 | May 12, 2011 | Incozen Therapeutics Pvt. Ltd. | Novel kinase modulators |
| WO2012151525A1 | May 4, 2012 | Nov 8, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
| WO2013164801A1 | May 3, 2013 | Nov 7, 2013 | Rhizen Pharmaceuticals Sa | Process for preparation of optically pure and optionally substituted 2- (1 -hydroxy- alkyl) – chromen – 4 – one derivatives and their use in preparing pharmaceuticals |
| US20110118257 | May 19, 2011 | Rhizen Pharmaceuticals Sa | Novel kinase modulators | |
| US20120289496 | May 4, 2012 | Nov 15, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
| WO 2011055215 |
WO2015175966
WO2015051252
-
The Distillery: Therapeutics 09/04/2014
BC Innovations, TherapeuticsIndication Target/marker/pathway Summary Licensing status Publication and contact information Cardiovascular disease Intimal hyperplasia Phosphoinositide 3-kinase-g (PI3Kg) Rodent studies suggest inhibiting … -
BC Innovations, Targets & MechanismsTargets & Mechanisms: PI3K inhibition: solid immunotherapy Table 1. A peek at PI3K inhibitors. According to a study in Nature by Ali et al., inhibition of phosphoinositide 3-kinase-d (PI3Kd) or the PI3K catalytic …
-
RP6530: Phase I started 12/23/2013
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland Product: RP6530 Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) delta; Phosphoinositide 3-kinase (PI3K) gamma Description: Dual … -
Rhizen preclinical data 07/01/2013
Week in Review, Preclinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland Product: RP6530 Business: Cancer Indication: Treat B cell lymphoma In vitro, 2-7 M RP6530 led to a >50% dose-dependent inhibition in growth of immortalized …
/////
c1cccc4c1C(/C(=C(/[C@H](CC)Nc3c2c(ncn2)ncn3)O4)c5cc(ccc5)F)=O
CCC(C1=C(C(=O)C2=CC=CC=C2O1)C3=CC(=CC=C3)F)NC4=NC=NC5=C4NC=N5


Reblogged this on New Drug Approvals.
LikeLike