PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Icenticaftor (QBW251) is an orally active CFTR channel potentiator, with EC50s of 79 nM and 497 nM for F508del and G551D CFTR, respectively. Icenticaftor can be used for chronic obstructive pulmonary disease (COPD) and cystic fibrosis research.
Cystic fibrosis (CF) is the most prevalent life-threatening Mendelian disorder in Caucasian populations. CF arises from mutations of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The CFTR ion channel orchestrates gating of chloride and bicarbonate ions across epithelial cell membranes in various tissues, including the lung, pancreas, intestine, reproductive tract, and sweat glands. While CF is a systemic disorder, the primary mortality derives from reduced CFTR activity in the airways. Subsequent acidification3 and dehydration leads to accumulation of a viscous mucus layer, occluding the airways and trapping bacteria, leading to infections, reduced lung function, and ultimately, respiratory failure. The most common CFTR mutation, F508del (Class II, found in 90% of CF patients), impairs folding of the CFTR protein (a Class II trafficking defect), resulting in a reduced amount of channel present at the plasma membrane. With the G551D mutation (class III), theamount of protein at the membrane is unaffected, but its open probability (Po) is reduced, also resulting in a reduced channel gating. Thus, to address the underlying causes of CF, two distinct CFTR modulators are required: correctors to increase CFTR levels at the plasma membrane and potentiators to enable effective opening of the channel
Chronic obstructive pulmonary disease (COPD) is anticipated to shortly become the third leading cause of death globally. COPD is characterized by persistent airflow obstruction with cigarette smoke exposure recognized as the primary risk factor. Airflow limitation is associated with all COPD patients; however, the disease is heterogeneous, with variable phenotypes ranging from chronic bronchitis (CB) to emphysema. Small airway disease exhibits increased numbers of goblet cells and mucus plugging with associated smooth muscle hyperplasia, airway fibrosis, and increased inflammation. Excess mucus secretion is believed to play an important role in COPD pathogenesis and is associated with progression of the disease.
Cystic fibrosis (CF) is a fatal genetic disease caused by mutations in the gene encoding the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), a protein kinase A activated epithelial anion channel involved in salt and fluid transport in multiple organs, including the lung. Most CF mutations either reduce the number of CFTR channels at the cell surface (e.g. synthesis or processing mutations) or impair channel function (e.g. gating or conductance mutations) or both.
PCT publication No. WO 2011/113894 describes compounds which restore or enhance the function of mutant and/or wild type CFTR for the treatment of cystic fibrosis, primary ciliary dyskinesia, chronic bronchitis, chronic obstructive pulmonary disease, asthma and other CFTR related diseases. The compounds described therein include (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (Example 5 of WO 2011/113894).
The synthesis described in WO 2011/113894 to make (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide is long, uses expensive starting materials and toxic reagents. Schemes 1 and 2 outline a synthesis from WO 2011/113894 used to make(S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
In Scheme 1, the intermediate ethyl 3-amino-5-(trifluoromethyl)picolinate (B4) is made via a Buchwald-Hartwig coupling reaction which requires the use of an expensive starting material (B1) and an expensive palladium catalyst which has to be controlled in the final product. Also, the conversion of B4 to B5 requires the use of NBS, a mutagenic reagent which has to be controlled in the API.
Moreover, the conversion of B5 to B8 is accomplished through the addition of 2,5-hexanedione, a well-known neurotoxin, as shown in Scheme 2. Transformation of the pyrrole in B8 to the amine B9 uses hydroxylamine which is a mutagenic and thermally unstable compound that is dangerous to use in large quantities. The overall process described in WO 2011/113894 requires many protecting group manipulations that lead to a low atom economy and afford a lot of waste. Thus there is a need for an improved synthetic process for making (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (1.4 kg, 5.47 mol), tetramethyl ethylene diamine (TMEDA) (1.75 kg, 15 mol) and tetrahydrofuran (THF) (10kg) were charged to a dry and inert reactor. At -25°C a solution of 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithiumchloride complex, 1 M in THF/toluene (TMPMgCl.LiCl)(14.5 kg, 15 mol) was slowly added. After stirring the reaction mixture for 30 min., CO2 gas was carefully bubbled into the reactor so that the temperature of the exothermic reaction did not exceed -20°C. The reaction mixture was then quenched onto a mixture of t-butyl methyl ether (TBME) and 5% aq. H2SO4 (50 kg). The biphasic mixture was separated and the organic phase was extracted with 2M NaOH solution. The aqueous phase was acidified to pH 1-2 with 5% aq. H2SO4 and extracted with TBME. After a distillative solvent change to cyclohexane the product was crystallized from cyclohexane to yield 1.1 kg 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (65% yield).
HRMS: [M-H]- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9337
Example 2: Methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (5.0 g, 19.53 mmol) was added to a 100 ml reactor followed by toluene (20 ml) and dimethylcarbonate (17.59 g, 195.30 mmol). To the stirred solution at 20 °C was slowly added 2,2,6, 6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (27.34 ml, 27.34 mmol) within 45 minutes. A sample was taken and diluted in acetic acid for HPLC analysis in order to confirm full conversion of II to the methylester. Within the same vessel 5% aq. H2SO4 (36 ml) was slowly added to the reaction mixture until a pH below 2 was obtained (caution, exothermic). The biphasic mixture was separated and the lower aqueous phase back-extracted with toluene (10 ml).
In order to isolate the methylester the organic phases were combined and concentrated by rotary evaporation to yield a residue which was chromatographed on reverse-phase silica to yield the final product: methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate as a yellow solid, 5.3 g, 86 % yield. The solid was optionally recrystallized from methanol and water to further increase purity.
HRMS: MH+ expected C9H8BrF3NO3, 313.9561 ; found C9H8BrF3NO3, 313.9634
HPLC Conditions:
HPLC: Column : Agilent Zorbax SB-C18 (150 mm x 3.0 mm, particle size 3.5 urn)
Eluent A : Water / TFA = 1000/1 (v/v)
Eluent B: Acetonitrile / TFA = 1000/1 (v/v)
Wavelength : 230 nm
Flow-rate : 0.8 ml/min
Gradient: eluent B: 45% to 90% over 9 mins
Retention time 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate: 5.80 min
Alternative synthesis for 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid:
Isolation of Example 1
In order to proceed to Example 1 without the isolation of VII, the work-up continues from the combined toluene phases post-H2SO4 quench as follows:
To the combined organic phases was slowly added 50% aq. sodium hydroxide (30 ml) until a pH of above 10 was obtained. The reaction mixture was heated to 35 °C and after 15 mins addition of water (30 ml) followed by 30 mins further stirring preceded sample-taking to ensure full hydrolysis of the methylester to Example 1 by HPLC. Water was added (130 ml), followed by TBME (60 ml) and the phases separated. To the aqueous phase was cautiously added concentrated H2SO4 (30 g) until a pH of below 2.5 was obtained (caution, exothermic and release of CO2 causes foaming). TBME (100 ml) was added and the phases separated. The organic phase contained the C2, and could be evaporated to dryness by rotary evaporation to confirm the yield, 5.4 g C2, 92 % yield.
HRMS: M-H- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9333
For HPLC method details see above. Retention time C2: 2.94 min
Alternative synthesis for ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate:
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (0.5 g, 1.95 mmol) was added to a reactor followed by THF (2 ml) and the solution cooled to 0 °C. To the mixture was added 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (4.88 ml, 3.91 mmol), and the mixture was left to stir for 15 minutes at 0 °C. An aliquot of the solution (50 ul) was then added to a reactor containing diethylcarbonate (20 ul, 19.5 mmol). A second aliquot (50 ul) was taken of the metallated II and added to a reactor containing ethyl chloroformate (14 ul, 19.5 mmol). After 2 minutes both reactors were quenched with a 1 :1 mixture of acetonitrile/HCl (1 M). The reaction with diethylcarbonate gave 56 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate and the reaction with ethyl chloroformate gave 68 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate product according to the HPLC method described above.
Example 3: Synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1: 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (1.3 kg, 4.33 mol) and
copper(II)sulfate pentahydrate (0.108 kg, 0.433 mol) were charged into an inert autoclave
followed by aqueous ammonia 25% (12 kg). The mixture was stirred and heated up to 100 °C, whereby a pressure of 7 bar resulted. The solution was stirred for 2 hr and then cooled down to
5 °C. Sulfuric acid (8 M) was dosed upon cooling, so that a temperature range of 5 °C to 30 °C was held until a pH of about 5 was reached. Isopropylacetate was added and the pH was
further adjusted to 1-2. The phases were separated and the organic phase was azeotropically dried by partial distillation. n-Heptane was added and the mixture stirred for 15 hr at 20 °C
during which the product crystallized out. After filtration and drying 3-amino-6-methoxy-5-(trifluoromethyl)picolinic acid was obtained as a yellow solid (0.92 kg, 90%).
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Step 2: 3-amino-6-methoxy-5-(trifluoromethyl) picolinic acid (20 g, 84.7 mmol) and HATU (38.6 g, 101.6 mmol) were charged to a reactor followed by a solution of (S)-3-amino-1 ,1 ,1-trifluoro-2- methylpropan-2-ol in isopropylacetate (7 %, 188 g, 93 mmol). The solution was stirred at room temperature, diisopropyl ethyl amine (21.9 g, 169 mmol) was added and stirring was continued for at least 16h at 25 °C. Water (250 ml) was then added dropwise within 15 min. keeping the temperature below 25 °C. The water phase was separated and the organic phase was extracted with 5% aqueous HCl , 5% potassium carbonate solution, and water. The organic layer was concentrated to about 60% solution. At 50 °C n-heptane (41 g) was added and the solution was cooled by a linear ramp to 5 °C while adding more n-heptane (131 g). The precipitate was filtered off and dried at 50 °C resulting in a yellow to beige product (S)-3-amino-6-methoxy-N- (3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (21.1 g, 69 % yield).
13C NMR (101 MHz, DMSO-d6): δ ppm 18.92, 42.15, 53.52, 72.40, 115.5-116.5 m, 118-126 m, 122-130.7 m, 124.82, 128.3 m, 140.95, 148.49, 166.27
Example 4: Telescoped process for the synthesis of the HCl salt of 3-amino-6-methoxy- 5-(trifluoromethyl)picolinic acid (V)
1 Equivalent* of (III) and 6 equivalents of dimethyl carbonate (DMC) were dissolved in 3.5 parts** of toluene at room temperature. To this solution 1.5 equivalent of TMPMgCl.LiCl solution in THF was added at 15-25°C within ca. 1 h. Tert butyl methyl ether (MTBE, 5.9 parts) was added and the mixture was quenched in 7.3 parts of 10% sulfuric acid at 25-40°C. The water phase was discarded and to the organic phase 6.2 parts of 30% sodium hydroxide solution were added. The mixture was stirred well at 40°C for 1-2h. After the successful conversion of (VIII) to (IV), 2.5 parts of water were added to dissolve the partially precipitated sodium carbonate. The water phase was discarded and the organic phase was cooled to 20°C and extracted with 4.8 parts of 25% aqueous ammonia. The aqueous phase was transferred in an autoclave and 0.0979 parts (10mol%) of copper sulfate pentahydrate were added. The autoclave was well inertized by a pressure method and heated up to 100°C, while the pressure raises up to ca. 8 bar absolute pressure. After the successful conversion of (IV) to (V), the green solution was added to a mixture of 3.7 parts of MTBE and 6.8 parts of 50% sulfuric acid resulting in a biphasic solution of pH 1-2. The water phase was separated and the organic phase washed two times with 2.5 parts of water each. The organic phase was dried by distillation at JT 50°C/400mbar while 3.7 parts of MTBE were added/replaced. To the dried organic solution 0.41 parts of HCl gas was dosed at 0-5°C under or over solvent level. The suspension was stirred for ca.1 h, then filtered off and washed with 48 parts of TBME. The product was dried at 40°C/20 mbar for ca. 12h. (yield from (III): 72%, slightly beige solid).
*equivalents are based on the molar amount of the starting material (III) = 1 equivalent
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Example 5: Alternative synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1 : (VIII) (1.0 g), (S)-3-amino-1 ,1 ,1-trifluoro-2-methylpropan-2-ol as mandellic acid salt (1.128 g, 1.2 eq.) and 2,3,4,6, 7, 8-hexahydro-1H-pyrimido[1,2-a]pyrimidine (TBD, 0.588 g, 1.3 eq.) were added to a pre-dried flask as solids. To this was added the anhydrous THF (10 ml) and the cloudy solution heated to 55 °C. Sampling and analytical determination of purity at 2.5 hrs confirmed 88 A% product upon which water (10 ml) was added and the phases separated. The organic phase was distilled to a concentrated mixture upon which toluene (20 ml) was added. The organic layer was extracted with 10% aq. citric acid (10 ml) followed by three consecutive extractions with 1 M aq. NaOH. The organic phase was then dried with magnesium sulfate and evaporated to dryness to give 1.196 g of (S)-3-bromo-6-methoxy-N-(3,3,3-trifluoro- 2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (IX) as a white solid (95 A%, 88% yield).
HRMS: MH+ expected C12H12BrF6N2O3, 424.9857; found C12H12BrF6N2O3, 424.9931
HPLC (method described above): retention time = 4.94 min
Step 2: IX (79 mg, 0.186 mmol) was combined with copper(II)sulfate pentahydrate (4.6 mg, 0.019 mmol), methanol (0.6 ml) and 23% aqueous ammonium hydroxide solution (559 ul) within a glass microwave vial. The headspace was inertized with nitrogen, then the vial sealed and placed in the microwave unit for heating to 105 °C for 7.5 hrs. Isopropylacetate (5 ml) was added to the deep green reaction mixture and a solvent-switch brought about by rotary evaporation. To the mixture now in water and isopropyl acetate was added 8M H2SO4 (5 ml), the phases mixed and then left to separate. The aqueous phase was further extracted with isopropylacetate and the combined organic phases washed with aq. NaCl (5 ml). The organic phase was dried over MgSO4 and evaporated to yield of a yellow residue, 66 mg.
A portion of the residue (16 mg) was re-dissolved in heptane / ethyl acetate and submitted for combiflash purification (n-heptane / ethyl acetate gradient, elution at 20% ethyl acetate) providing (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (VII) as a residue on evaporation in 91 A% purity containing trace residual solvents (17 mg, corrected to 13 mg by 1H NMR, 80 % yield back-calculated).
Examples 4, 5 and 6: 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide and its enantiomers
Example 4: 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide,
was prepared according to the following procedure:
A solution comprising 3-amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (Intermediate D)(4 g, 16.94 mmol) and 3-amino-1,1,1-trifluoro-2-methylpropan-2-ol hydrochloride (Intermediate R) (3.04 g, 16.94 mmol) in NMP (188 ml) was treated with HATU (7.73 g, 20.33 mmol) followed by dropwise addition (2 ml portions) of DIPEA (8.88 ml, 50.8 mmol) over 1 hour. After stirring for a further hour, the reaction mixture was poured into water (450 ml) and EtOAc (450 ml). The aqueous phase was acidified with 5M HCl (50 ml) and the layers were separated. The organic portion was washed with 2M NaOH (200 ml), water (4×200 ml), brine (2×100 ml), dried over MgSO 4, filtered and concentrated in vacuo to afford a brown solid. Purification of the solid by chromatography on silica (220 g pre-packed silica cartridge) eluting with 0-50% EtOAc in iso-hexane afforded the racemate, 3-amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide (Ex. 4) as a yellow solid;
Chiral separation of the racemate by Supercritical Fluid Chromatography was carried out using the following conditions to afford the compounds listed hereinafter:
Mobile Phase: 12% 2-propanol+0.1% DEA/50% CO 2
Column: Chiralcel OD-H, 250×10 mm id, 5 μm (2 columns linked in series)
Detection: UV @ 220 nm
Flow rate: 10 ml/min
Sample concentration: 3.5 g in 30 ml EtOH
Injection volume: 100 μl
Examples 5 and 6 are Entantiomers
Example 5: First eluted peak Rt=7.30 minutes. 3-Amino-6-methoxy-5-trifluoromethyl-pyridin e-2-carboxylic acid ((S)-3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide (“Compound A”):
Optical rotation [α] 21D at 589 nm −20.83° (c=0.513, MeOH).
The stereochemistry of this compound was confirmed by X-ray crystallography.
Example 6: Second eluted peak Rt=8.29 minutes. 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid ((R)-3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide
LC-MS Rt=1.15 mins [M+H]+ 362.4 (Method 2 minLC_v003).
Alternatively, Example 5 may be prepared according to the following method: To a solution of 3-amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (Intermediate D) (10 g, 42.3 mmol) and (S)-3-amino-1,1,1-trifluoro-2-methylpropan-2-ol hydrochloride (Intermediate RA)(7.60 g, 42.3 mmol) in NMP (400 ml) was added HATU (19.3 g, 50.8 mmol) followed by dropwise addition of DIPEA (22.19 ml, 127 mmol) over ˜1 hr. After stirring at room temperature for 30 min, the mixture was added to EtOAc (2 L), washed with 1M NaOH (2×1 L), water (1 L), brine (1 L), dried (MgSO 4) and evaporated under reduced pressure to give the crude product as a dark brown oil. Purification by chromatography on silica eluting with a gradient of 1 to-25% of EtOAc in iso-hexane afforded a yellow oil. Recrystallisation of the oil from iso-hexane/DCM afforded 3-amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid ((S)-3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide as a crystalline solid;
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) ion channel are established as the primary causative factor in the devastating lung disease cystic fibrosis (CF). More recently, cigarette smoke exposure has been shown to be associated with dysfunctional airway epithelial ion transport, suggesting a role for CFTR in the pathogenesis of chronic obstructive pulmonary disease (COPD). Here, the identification and characterization of a high throughput screening hit 6 as a potentiator of mutant human F508del and wild-type CFTR channels is reported. The design, synthesis, and biological evaluation of compounds 7–33 to establish structure–activity relationships of the scaffold are described, leading to the identification of clinical development compound icenticaftor (QBW251) 33, which has subsequently progressed to deliver two positive clinical proofs of concept in patients with CF and COPD and is now being further developed as a novel therapeutic approach for COPD patients.
a Reagents and conditions: (i) aq NaOH, THF, RT, 97%; (ii) aq Me2NH or MeNH2, THF, RT, 56−92%; (iii) 41, HATU, Et3N, NMP, RT, 52− 78%; (iv) NH2OH·HCl, Et3N, EtOH−water, reflux, then chiral HPLC, 34−36%; (v) aq NaOH, MeOH, 60°C, 97%; (vi) cat H2SO4, MeOH, reflux, 75%; (vii) TMSCl, KI, MeCN, reflux, 54%; (viii) EtOH, DEAD, Ph3P, dioxane, RT, 61%; (ix) aq NaOH, THF, reflux, 26%; (x) (S)-41, HATU, DIPEA, DMF, RT, 89%; (xi) NH2OH·HCl, Et3N, EtOH−water, reflux, 37−53%; (xii) (S)-41, HATU, DIPEA, NMP, RT, 59%.
Anti-inflammatory, Farnesoid X receptor (FXR) agonist
Comment
Treatment of non-alcoholic steatohepatitis
Novartis is developing tropifexor, a non-bile acid farnesoid X receptor agonist, and its analog LJP-305, for treating NASH, PBC, liver fibrosis, bile acid diarrhea and non-alcoholic fatty liver disease. In June 2021, this drug was reported to be in phase 2 clinical development.
Nonalcoholic steatohepatitis (NASH) is a liver disease that is becoming more prevalent as worldwide obesity and type 2 diabetes increase. It is characterized by accumulation of fat in the liver, inflammation, hepatocyte ballooning, and fibrosis.
Another liver disease, primary biliary cholangitis (PBC), is a cholestatic condition in which bile flow from the liver to the intestine is reduced or interrupted. It is thought to be autoimmune.
PBC is associated with decreased expression of the farnesoid X receptor (FXR), a ligand-activated nuclear receptor that is highly expressed in the liver and other organs. FXR is a key regulator of bile acid production, conjugation, and transport. FXR activation also suppresses lipogenesis; thus, it has been proposed as a treatment for NASH.
Recently, David C. Tully and colleagues at the Genomics Institute of the Novartis Research Foundation (San Diego) and the Novartis Institutes for Biomedical Research (Emeryville, CA) discovered tropifexor, a highly potent FXR agonist. They began by replacing an indole group in an existing partial FXR agonist with a 2-substituted benzothiazole-6-carboxylic acid, a change that resulted in a dramatic increase in potency. Further changes, including optimization of the benzothiazole substituent, resulted in more potent, orally bioavailable tropifexor.
Rats treated orally with tropifexor (0.03 to 1 mg/kg) showed an upregulation of the FXR target genes, BSEP and SHP, and a down-regulation of CYP8B1. Its EC50 for FXR is between 0.2 and 0.26 nM depending on the biochemical assay.
The patent which covers tropifexor and related compounds was published in 2010.[3]
Novel, stable crystalline polymorphic form II of tropifexor , useful for treating non-alcoholic steatohepatitis (NASH), fatty liver and primary biliary cholangitis (PBC).Tropifexor was originally developed by Novartis and then licensed to Pfizer for cooperative development. It is a non-steroidal FXR (farnesoid receptor) agonist, currently in clinical phase II of indications for NASH (non-alcoholic steatohepatitis), fatty liver and primary biliary cholangitis. The structure of Tropifexor is shown in the following formula (1):
Drug polymorphism is a common phenomenon in drug development and an important factor affecting drug quality. Different crystal forms of the same drug may have significant differences in physical and chemical properties such as appearance, fluidity, solubility, storage stability, bioavailability, etc., and there may be great differences, which will affect the storage transfer, application, stability, and efficacy of the drug In order to obtain an effective crystal form that is conducive to production or pharmaceutical preparations, it is necessary to conduct a comprehensive investigation of the crystallization behavior of the drug to obtain a crystal form that meets the production requirements. At present, there is no literature that discloses the crystal form of Tropifexor, and there is no related literature report. The present invention obtains a new crystal form of the compound through a large number of experimental studies on the Tropifexor compound. The new crystal form has the advantages of high solubility, good stability, low moisture absorption, simple preparation process and easy operation, etc., and has excellent properties in industrial production. Superiority.Example 1 Preparation method of Tropifexor crystal form II[0049]After mixing 60.3 mg of Tropifexor and p-aminobenzoic acid (13.7 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 51.3 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0050]Example 2 Preparation method of Tropifexor crystal form II[0051]After mixing 60.3 mg of Tropifexor and p-hydroxybenzoic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 48.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0052]Example 3 Preparation method of Tropifexor crystal form II[0053]After mixing 60.3 mg of Tropifexor and salicylic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. Filter with suction and place in a drying box at 50°C and vacuum dry to constant weight to obtain 50.0 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0054]Example 4 Preparation method of Tropifexor crystal form II[0055]After mixing 60.3 mg of Tropifexor and 2,4-dihydroxybenzoic acid (15.4 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 49.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.
claiming crystalline polymorphic form I of tropifexor,Example 1 Preparation method of Tropifexor crystal form I 50.0 mg of Tropifexor was added to ethanol (1.0 ml), heated to 60° C. and stirred to obtain a clear solution, and then water (3 ml) was added dropwise to the Tropifexor solution. Stir and precipitate solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 38.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form I; its X-ray powder diffraction pattern was basically consistent with Figure 1, its DSC pattern was basically consistent with Figure 2, and its TGA pattern was basically consistent with Figure 3
Methyl 2-[(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4- yl}methoxy)-8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1 -1 A). Into a 25-mL round-bottom flask equipped with a stir bar was added sequentially 4-(((1 R,3r,5S)- 8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethoxy)phenyl)isoxazole (1 .29 mmol), N,N-dimethylacetamide (3.6 mL), cesium carbonate (3.31 mmol), and methyl 2- bromo-4-fluorobenzo[d]thiazole-6-carboxylate (3.87 mmol). After stirring the resulting slurry at room temperature for 10 minutes, the mixture was then warmed to 60 °C and stirred for 1 h. The reaction slurry was allowed to cool to room temperature, and was diluted with 200 mL of ethyl acetate and washed with water (3 χ 30 mL). The organic extracts were concentrated under vacuum and directly purified using normal phase silica gel chromatography (40 g silica column) with a 15 min gradient of 10 % to 60 % ethyl acetate/hexanes. Desired fractions were concentrated in vacuo, and the resulting residue crystallized upon standing to give methyl 2- [(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8- azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1-1 A) as a white crystalline solid. MS (m/z) : 618.2 (M+1 ).
2-r(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4-yl}methoxy)- 8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). To a 25-mL round-bottom flask equipped with a stir bar was added the ester (0.89 mmol), THF (4 mL),
MeOH (2 mL), and 3 N aqueous KOH solution (1 mL, 3 mmol). The resulting homogenous solution was stirred for 1 hour at 70 °C, cooled to room temperature, and then quenched with AcOH (roughly 0.2 mL of glacial acetic, 3 mmol) until pH=6 was achieved (Whatman class pH strip paper). At this time the reaction was diluted with ethyl acetate (40 mL) and washed with water (3 5 mL). The ethyl acetate fraction was concentrated under vacuum to give to an oily residue. To the resulting oil was then added MeOH (6 mL). The oil quickly dissolved, then immediately began to crystallize. Upon standing for 2.5 hrs, the mother liquor was withdrawn and crystals washed (3 x 2 mL of ice cold MeOH). The crystals were dried via vacuum (10 mm Hg pressure at 45 °C overnight) and then recrystallized from acetonitrile, filtered, and dried under vacuum to give 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethyl)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -2B).
Examples 1 -2A and the corresponding acid 1 -2B can be prepared following the same procedures, from the reaction of intermediate 4-((8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethyl)phenyl)isoxazole.
PAPER
European journal of medicinal chemistry (2021), 209, 112910
Farnesoid X receptor (FXR) agonists are emerging as potential therapeutics for the treatment of various metabolic diseases, as they display multiple effects on bile acid, lipid, and glucose homeostasis. Although the steroidal obeticholic acid, a full FXR agonist, was recently approved, several side effects probably due to insufficient pharmacological selectivity impede its further clinical application. Activating FXR in a partial manner is therefore crucial in the development of novel FXR modulators. Our efforts focusing on isoxazole-type FXR agonists, common nonsteroidal agonists for FXR, led to the discovery a series of novel FXR agonists bearing aryl urea moieties through structural simplification of LJN452 (phase 2). Encouragingly, compound 11k was discovered as a potent FXR agonist which exhibited similar FXR agonism potency but lower maximum efficacy compared to full agonists GW4064 and LJN452 in cell-based FXR transactivation assay. Extensive in vitro evaluation further confirmed partial efficacy of 11k in cellular FXR-dependent gene modulation, and revealed its lipid-reducing activity. More importantly, orally administration of 11k in mice exhibited desirable pharmacokinetic characters resulting in promising in vivo FXR agonistic activity.
^ Clinical trial number NCT03517540 for “Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and Cenicriviroc (CVC) in Adult Patients With Nonalcoholic Steatohepatitis (NASH) and Liver Fibrosis. (TANDEM)” at ClinicalTrials.gov
^WO Application Filing 2012087519, Alper PB, Chianelli D, Mutnick D, Vincent P, Tully DC, “Compositions and methods for modulating fxr”, published 2012-06-28, assigned to Genomics Institute of the Novartis Research Foundation. Retrieved 17 May 2019.
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015); $1,902 Million (Y2014); $1,558 Million (Y2013); $1,007 Million (Y2012); $630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex. Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
Advanced kidney cancer (US FDA approved in March 2009)[3]
Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
Prevention of organ rejection after liver transplant(Feb 2013)
Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2
Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4
Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6
Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:
See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.
(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):
According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.
(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).
Only 8% yield of the corresponding compound (4B)
and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),
It is a derivative of sirolimus of formula III),
and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),
wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),
was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche
Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),
as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),
wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Clip
References
a WO 9 409 010 (Sandoz-Erfindungen; 28.4.1994; GB-prior. 9.10.1992).
b US 6 277 983 (American Home Products; 21.8.2001; USA-prior. 27.9.2000).
US 6 384 046 (Novartis; 7.5.2002; GB-prior. 27.3.1996).
US 20 040 115 (Univ. of Pennsylvania; 15.1.2004; USA-prior. 9.7.2002).
fermentation of rapamycin (sirolimus):
Chen, Y. et al.: Process Biochemistry (Oxford, U. K.) (PBCHE5) 34, 4, 383 (1999).
The Merck Index, 14th Ed., 666 (3907) (Rahway 2006).
US 3 929 992 (Ayerst McKenna & Harrison Ltd.; 30.12.1975; USA-prior. 29.9.1972).
WO 9 418 207 (Sandoz-Erfindungen; 18.8.1994; GB-prior. 2.2.1993).
EP 638 125 (Pfizer; 17.4.1996; J-prior. 27.4.1992).
US 6 313 264 (American Home Products; 6.11.2001; USA-prior. 8.3.1994).
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]
Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.
[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.
“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.
When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID12944570.
^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID24767339.
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015); $1,902 Million (Y2014); $1,558 Million (Y2013); $1,007 Million (Y2012); $630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex. Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
Advanced kidney cancer (US FDA approved in March 2009)[3]
Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
Prevention of organ rejection after liver transplant(Feb 2013)
Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2
Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4
Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6
Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:
See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.
(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):
According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.
(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).
Only 8% yield of the corresponding compound (4B)
and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),
It is a derivative of sirolimus of formula III),
and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),
wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),
was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche
Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),
as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),
wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]
Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.
[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.
“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.
When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID12944570.
^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID24767339.
Vildagliptin was approved by the European Medicines Agency (EMA) on Sep 26, 2007, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 20, 2010, following by China Food and Drug Administration (CFDA) on Aug 15, 2011. It was developed and marketed as Galvus® by Novartis in EU.
Vildagliptin is a potent selective inhibitor of dipeptidyl peptidase-4 (DPP-4) that improves glycaemic control by increasing islet α-cell and β-cell responsiveness to glucose. It is used to reduce hyperglycemia in type 2 diabete.
Galvus®is available as film-coated tablet for oral use, containing 50 mg free Vildagliptin. The recommended dose of vildagliptin is 100 mg, administered as one dose of 50 mg in the morning and one dose of 50 mg in the evening.Drug Name:VildagliptinResearch Code:LAF-237; DSP-7238; NVP-LAF-237Trade Name:Galvus® / Jalra® / Xiliarx® / Equa®MOA:Dipeptidyl peptidase-4 (DPP-4) inhibitorIndication:Type 2 diabetesStatus:ApprovedCompany:Novartis (Originator)Sales:$1,140 Million (Y2015); $1,224 Million (Y2014); $1,200 Million (Y2013); $910 Million (Y2012); $677 Million (Y2011);ATC Code:A10BH02
Vildagliptin, previously identified as LAF237, is a new oral anti-hyperglycemic agent (anti-diabetic drug) of the new dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs. Vidagliptin subsequently acts by inhibiting the inactivation of glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP) by DPP-4. This inhibitory activity ultimately results in a two-fold action where GLP-1 and GIP are present to potentiate the secretion of insulin by beta cells and suppress glucagon secretion by alpha cells in the islets of Langerhans in the pancreas. It is currently in clinical trials in the U.S. and has been shown to reduce hyperglycemia in type 2 diabetes mellitus. While the drug is still not approved for use in the US, it was approved in Feb 2008 by European Medicines Agency for use within the EU and is listed on the Australian PBS with certain restrictions.
Vildagliptin, sold under the brand name Galvus among others, is an oral anti-hyperglycemic agent (anti-diabetic drug) of the dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs. Vildagliptin inhibits the inactivation of GLP-1[2][3] and GIP[3] by DPP-4, allowing GLP-1 and GIP to potentiate the secretion of insulin in the beta cells and suppress glucagon release by the alpha cells of the islets of Langerhans in the pancreas.
Adverse effects observed in clinical trials include nausea, hypoglycemia, tremor, headache and dizziness. Rare cases of hepatoxicity have been reported.[5]
There have been case reports of pancreatitis associated with DPP-4 inhibitors. A group at UCLA reported increased pre-cancerous pancreatic changes in rats and in human organ donors who had been treated with DPP-4 inhibitors.[6][7] In response to these reports, the United States FDA and the European Medicines Agency each undertook independent reviews of all clinical and preclinical data related to the possible association of DPP-4 inhibitors with pancreatic cancer. In a joint letter to the New England Journal of Medicines, the agencies stated that “Both agencies agree that assertions concerning a causal association between incretin-based drugs and pancreatitis or pancreatic cancer, as expressed recently in the scientific literature and in the media, are inconsistent with the current data. The FDA and the EMA have not reached a final conclusion at this time regarding such a causal relationship. Although the totality of the data that have been reviewed provides reassurance, pancreatitis will continue to be considered a risk associated with these drugs until more data are available; both agencies continue to investigate this safety signal.”[8]
Vildagliptin is an active pharmaceutical substance with an empirical formula of C17H25N3O2 and a molecular weight of 303.40 g/mol. Vildagliptin is the international common accepted name for (2S)-1-[[(3-hydroxytricyclo[3.3.1.13,7]dec-1-yl)amino]acetyl]-2-pyrrolidine carbonitrile and has the structure of formula (I).
[0003]Vildagliptin is a dipeptidyl peptidase IV (DPP-IV) inhibitor and is disclosed in U.S. Pat. No. 6,166,063 (“the ‘063 patent”), the disclosure of which is incorporated herein by reference. The ‘063 patent discloses a synthesis of vildagliptin using the synthetic process represented in Scheme 1.
[0004]Vildagliptin can exist as the (2S) and (2R) enantiomers. The stereoisomer with the desired biological activity is the (2S) enantiomer. Accordingly, it is desirable to synthesize (2S)-vildagliptin with high stereochemical purity. A process that yields vildagliptin with a high enantiomeric purity is disclosed in International Patent Publication WO 2004/092127, the disclosure of which is incorporated herein by reference. This reference discloses compositions containing from 95% to 99.99% of (2S)-vildagliptin.
[0069]This example illustrates the synthesis of the compound of formula (I) in accordance with embodiments of the invention.
[0070]Into a 100 mL rounded reaction vessel were charged 3 g (17.37 mmol) of 1-chloroacetyl-2-cyanopyrrolidine, 3.22 g (19.82 mmol) of 1-amino-3-adamantanol, 2.78 g (20.1 mmol) of potassium carbonate, and 30 mL isopropyl acetate. The mixture was refluxed for 4 h, cooled to room temperature, and the salts were filtered and washed with acetonitrile. The mother liquors were evaporated to dryness to obtain an oil which was aged in MEK from which a white solid crystallizes at 0-5° C. The solid was filtered washing the cake with MEK and dried at 40° C. in a vacuum oven until constant weight.
[0071]Yield: 36%. Assay: 99.21%. HPLC purity: 97.55% of vildagliptin (measured according to Example 2). HPLC chiral purity: more than 99.99% of vildagliptin (measured according to Example 7).
[0072]These results demonstrate that a compound of formula (I) comprising less than 0.01% of (2R)-1-[N-(3-hydroxytricyclo[3.3.1.13,7]dec-1-yl)glycyl]-2-pyrrolidinecarbonitrile (i.e., (2R)-vildagliptin).
Vildagliptin is chemically known as (S)-l-[2-(3-Hydroxyadamantan-l-ylamino) acetyl] pyrrolidine-2-carbonitrile and exist as (2S) and (2R) enantiomers. The stereoisomer with the desired biological activity is the (2S) enantiomer, represented by the following structure:
U.S. Patent No. 6,166,063 (“the Ό63 patent”) discloses new class of Dipeptidyl peptidase 4 (DPP-4) inhibitors such as vildagliptin. The ‘063 patent further discloses a process for the preparation of vildagliptin by acylation of L-prolinamide with chloroacetyl chloride in the presence of a base in dichloromethane or tetrahydrofuran as solvent, filtration and subsequent dehydration with trifluoroacetic anhydride (TFAA) to provide (S) -1- (2- chloroacetyl) pyrrolidin-2-carbonitrile. The carbonitrile intermediate is isolated by distilling out the solvent, co-distillation with ethyl acetate, partitioning between water and ethyl acetate, extraction of the resulting aqueous layer with ethyl acetate followed by aqueous washings of the organic layer and concentrating to obtain carbonitrile intermediate as yellow solid. This is later reacted with about 2 moles of l-aminoadamantane-3-ol in the presence of about 4 moles of potassium carbonate in dichloromethane (DCM) or tetrahydrofuran (THF) for 6 days. Finally, the obtained crude vildagliptin is subjected to chromatography employing SIMS/Biotage Flash chromatography system providing vildagliptin with melting point of 138°C-140°C. The disclosed process is schematically represented as follows:
Amide Carbonitrile
A similar process is described in J. Med. Chem. 2003, 46, 2774-2789, where acylation of L-prolinamide with chloroacetyl chloride is carried out in the presence of potassium carbonate in tetrahydrofuran as solvent and subsequent dehydration with TFAA to provide (S) -1- (2-chloroacetyl) pyrrolidin-2 -carbonitrile. The carbonitrile intermediate was isolated by adding ethyl acetate, distillation of the solvent, partitioning between water and aqueous sodium bicarbonate, extraction of the resulting aqueous layer with ethyl acetate followed by aqueous washings of the organic layer and concentrating to obtain carbonitrile intermediate as yellow- white solid which was reacted with about 2-3 moles of 1- aminoadamantane-3-ol in the presence of about 3 moles of potassium carbonate in DCM or THF for 1-3 days followed by purification from a mixture of ethyl acetate and isopropanol provided Vildagliptin as a white solid.
U.S. Patent No. 6,011,155 discloses a process for the preparation of (S) -1- (2- bromooacetyl) pyrrolidin-2-carbonitrile by acylation of L-prolinamide with bromoacetyl bromide in the presence of triethyl amine and catalytic amount of DMAP in DCM as solvent wherein the resulting (S)-l -(2 -bromoacetyl) pyrrolidin-2-carboxamide is isolated and subsequently dehydrated with TFAA to obtain the carbonitrile intermediate as dark yellow solid.
U.S. Patent application No. 2008/0167479 discloses preparation of Vildagliptin with high chemical and enantiomeric purities wherein (S) -1- (2-chloroacetyl) pyrrolidin-2- carbonitrile is prepared in one step process by acylation of prolinamide with chloroacetyl chloride in a mixture of isopropyl acetate and DMF followed by dehydration with cyanuric chloride to obtain the carbonitrile intermediate as an oil which was crystallized from isopropanol. The resulting carbonitrile intermediate is reacted with l-aminoadamantane-3- ol in the presence of alkali metal carbonates such as potassium carbonate and an optional additive such as I in a solvent comprising at least an ester or ether or nitrile solvent and purification of vildagliptin from methyl ethyl ketone or from a mixture of isopropanol and methyl t-butyl ether.
PCT Publication No. 2010/022690 discloses a process for the preparation of vildagliptin wherein (S)-l -(2-chloroacetyl) pyrrolidin-2-carboxamide intermediate is isolated as a trialkylamine hydrohalide salt in two fractions and. dehydrated with TFAA to obtain (S)-l- (2-chloroacetyl) pyrrolidin-2-carbonitrile as light yellow powder after crystallization from heptane. The resulting carbonitrile intermediate is then reacted with 3-amino-l- adamantanol in the presence of alkali metal carbonate base and an alkali metal iodide as a catalyst in a mixture of organic ketones, ester and polar aprotic solvents. The crude product was subjected to multiple crystallizations in order to achieve high chemical purity of vildagliptin. This publication also disclosed final crystallization of vildagliptin from 2- butanone, toluene, 2-methyl tetrahydrofuran, isopropyl acetate, dimethyl carbonate, isopropanol. This process adds an extra step of isolation of the said carboxamide intermediate, uses mixture of solvents in the preparation of vildagliptin and to multiple crystallizations which makes the process uneconomical on large scale.
PCT Publication No. 2011/101861 discloses a process for the preparation of vildagliptin wherein (S)-l-(2-chloroacetyl) pyrrolidin-2-carboxamide and (S)-l-(2-chloroacetyl) pyrrolidin-2-carbonitrile intermediates are isolated as solids after purification and drying. Further, (S)-l-(2-chloroacetyl) pyrrolidin-2-carbonitrile is then converted to vildagliptin by reacting it with l-aminoadamantane-3-ol in the presence of potassium carbonate and KI in a suitable ether solvent like THF and purifying the obtained vildagliptin from a mixture of ethyl acetate and methanol. This publication also provided an alternate process for the preparation of vildagliptin by reacting 2-(3-hydroxyadamantan-l-yl amino) acid or derivative thereof with pyrrolidine-2-carbonitrile and various solvents from which vildagliptin may be crystallized such as ethyl acetate, 2-butanone, or mixture of ethyl acetate-methanol, ethyl acetate-isopropanol, methanol-DCM, ethyl acetate-cyclohexane and 2-butanone-methyl t-butyl ether.
U.S. Patent No. 7,375,238 discloses a one-pot process for the preparation of vildagliptin without isolation of the carboxamide and carbonitrile intermediates and further involves preparation of Vildagliptin by using potassium carbonate and potassium iodide (KI) as catalysts in 2-butanone solvent. Purification of the crude vildagliptin was carried out from a mixture of isopropanol and methyl t-butyl ether in the presence of 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU) base and final recrystallization from 2-butanone afforded pure vildagliptin. This process suffers from certain draw backs such as use of mixture of solvents for the acylation and condensation reactions; use of base and expensive additive such as KI in the condensation reaction.
PCT Publication No. 2011/012322 discloses a process wherein the (S) -1- (2-chloroacetyl) pyrrolidin-2-carbonitrile intermediate is isolated, purified and reacted with 1- aminoadamantane-3-ol in the presence of a phase transfer catalyst, optionally an inorganic base and a solvent selected from nitrile, ketone, ether, ester and mixtures thereof in a two phase reaction system wherein the first phase consist of a liquid phase and the second phase consists of an inorganic base. The final purification of vildagliptin was carried out in 2- butanone solvent.
PCT Publication No. 2013/179300 discloses preparation of vildagliptin from organic solvents such as aromatic hydrocarbons, aliphatic hydrocarbons, halogenated hydrocarbons, ethers, nitrile, dialkyl formamides, dialkylacetamides, dialkyl sulfoxides in the presence of organic or inorganic base. The resulting crude vildagliptin was purified by acid-base treatment and crystallization from a solvent selected from aliphatic hydrocarbons, aromatic hydrocarbons, ketones, esters, nitrile, ether, cyclic ether and alcohol or mixtures thereof.
PCT Publication No. 2012/022994 involves conversion of racemic vildagliptin to (S)- enantiomer via formation of vildagliptin adducts and final purification from ethyl acetate or mixture of ethyl acetate with 1% water.
U.S. Application No. 2006/0210627 discloses crystalline Form A of vildagliptin and its preparation from 2-butanone, isopropanol, acetone or a mixture of isopropanol-ethyl acetate in the presence of DBU base. This publication also discloses amorphous vildagliptin and its preparation by lyophilization from a water solution.
PCT Publication No. 2014/102815 disclosed a process for the preparation of vildagliptin by isolating the carboxamide and carbonitrile intermediates after crystallization and drying. The resulting carbonitrile intermediate is reacted with l-aminoadamantane-3-ol in the presence of organic base or inorganic base in nitrile, ester or alcohol solvent.
IN 3965 MUM/2013 publication discloses a process for the preparation of vildagliptin by preparing and crystallizing (S) -1- (2-chloroacetyl) pyrrolidin-2-carbonitrile intermediate and reacting it with l-aminoadamantane-3-ol in the presence of a potassium carbonate, optionally in presence of suitable catalyst such as KI in ketone solvent or in mixture of ketone with non polar solvents.
C.N. publication No. 102617434 discloses a one pot process for the preparation of Vildagliptin by reacting salt of pyrrolidine carbonitrile such as TFA salt with haloacetyl halide in the presence of a base followed by insiru reaction with l-aminoadamantane-3-ol in the presence of tertrabutyl ammonium iodide in halogenated hydrocarbon or ether as solvent to get vildagliptin which is further crystallized from ethyl acetate-petroleum ether.
C.N. publication No. 103804267 discloses a process for the preparation of vildagliptin by reacting (S)-l -(2 -haloacetyl) pyrrolidin-2-carbonitrile with l-aminoadamantane-3-ol in a mixed system of an organic solvent and water in the presence of a base and phase transfer catalyst followed by crystallization of the obtained crude vildagliptin.
C.N. publication No. 103787944 disclosed dehydration of-1- (2-chloroacetyl) -2- (S) – pyrrolidine carboxamide in the presence of a dehydrating agent and an acid-binding agent in an organic solvent followed by crystallization from mixture of isopropyl ether and ethyl acetate to provide l-(2-chloroacetyl)-2-(S)-pyrrolidine carbonitrile as white or pale yellow solid powder.
Furthermore, several techniques are known in the art for the purification of vildagliptin such as chromatography (US 6,166,063); or acid-base purification (IN 61 /MUM/2012 publication) or via formation of inorganic salt complexes (WO 2011/042765); or by solvent crystallizations such as mixture of ethyl acetate and isopropanol (J. Med. Chem. 2003, 46, 2774-2789); isopropanol and MTBE in the presence of DBU base and final recrystallization from 2-butanone (US 7,375,238); methyl ethyl ketone or from a mixture of isopropanol and MTBE (US 2008/0167479); acetone, 2-butanone, cyclohexanone, ethyl acetate, isopropyl acetate or dimethyl carbonate (IN 61 /MUM/2012 publication); 2- butanone (WO 2011/012322); aliphatic hydrocarbons, aromatic hydrocarbons, ketones, esters, nitrile, ether, cyclic ether and alcohol or mixtures thereof (WO 2013/179300); or from ethyl acetate or mixture of ethyl acetate with 1% water (WO 2012/022994).
Most of the processes known in the art for synthesizing vildagliptin are associated with one or more of the following disadvantages:
a) use of toxic TFAA for dehydration which is costly and environmentally harmful, b) lengthy and time consuming condensation process,
c) conventional solvents used in the condensation stage are costly, volatile, flammable, toxic, causing adverse health effects, in, addition to this potentially unsafe peroxide forming solvents such as THF were used, which process is more costlier than the process not having such elements,
d) purification of vildagliptin by chromatographic purification or by formation of inorganic salt complexes or by multiple crystallizations which are tedious, labor intensive, uses high amounts of solvents, require precise monitoring and time consuming and hence not viable for commercial scale operations.
Therefore, the present invention fulfills the need in the art and provides simple, industrially feasible and scalable processes for the preparation and purification of vildagliptin that circumvent disadvantages associated with the prior art process, proved to be advantageous from environmental and industrial point of view and also fulfill purity criteria. These processes allow the final product to be produced in a higher yield and purity by minimizing number of processing steps and reducing the number of solvent usage which is very practical for scale-up production, especially in terms of operating efficiency.
The new processes has a further advantage in recovering the expensive 1- aminoadamantane-3-ol from the reaction mixture and recycling in a simple manner that avoids use of inorganic salt complexes, which is economical and applicable on an industrial scale.
EXAMPLE 1: Preparation of (2S)- 1 -(Chloroacetyl)-2-pyrrolidinecarbonitrile.
To a solution of L-Prolinamide (100 gms) dissolved in DCM (1000 mL) was added triethyl amine (88.6 gms) and DMAP (1.07 gms) at 25-30°C under N2 atmosphere and stirred for 15 min at 25-30°C. This solution was added to a solution of chloroacetyl chloride (98.9 gms) in DCM (500 mL) under N2 atmosphere at -5 to 0°C over 2-3 hr. Raised the reaction mass temperature to 0-5°C and stirred for lhr. After reaction completion, charged phosphorus oxy chloride (201.5 gms) to the reaction mass at 0-5 °C, heated the reaction mass temperature to reflux and stirred for 6hr at same temperature. After reaction completion, allowed to cool to 10-20°C and added DM water (500 mL). Aqueous layer was separated and the organic layer was washed with DM water. To the organic layer DM water (300 mL) was added at 25-30°C and adjusted the reaction mass pH to 6.5-7.5 with -500 mL of sodium bicarbonate solution (-40 g of NaHC03 dissolved in 500 mL of DM Water). Separated the aqueous layer and concentrated the organic layer under vacuum at temperature of 30-40°C to get residual mass. Charged isopropanol (100 mL) and distilled out solvent completely under vacuum at <50°C. The resulting residue was allowed to cool to 30-40°C and charged isopropanol (500 mL). Heated the reaction mass temperature to 40- 45°C, stirred for 30 min at 40-45°C, allowed to cool to 0-5°C, stirred for 2 hr, filtered and washed wet cake with chilled isopropanol (100 mL), dried at 40-45°C for 6 hr to provide 115 gms of (2S)-l-(CMoroace1yl)-2-pyrrolidinecarbonitrile.
HPLC Purity: 99.86%.
Example 2: Preparation of Vildagliptin
To (2S)-l-(Chloroacetyl)-2 -Pyrrolidine carbonitrile (100 gms) dissolved in DM Water (500 mL), charged l-aminoadamantane-3-ol (242.2 g) at 25-35°C. Heated the reaction mass temperature to 40-45°C and stirred for 8-10 hr at 40-45°C. After reaction completion, allowed to cool to 25-30°C and charged DM water (700 mL) and DCM (600 mL). Separated the organic layer and extracted the aqueous layer with DCM. The total organic layer was concentrated under vacuum at temperature 30-40°C to get residual mass. Ethyl acetate (100 mL) was added to the residual mass and distilled completely under vacuum at <50°C. Charged ethyl acetate (500 mL) and refluxed for 1 hr. Allowed to cool to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (100 mL) then dried at 50-55°C for 6 hr to provide 130 gms of crude vildagliptin.
HPLC Purity: 99.56%.
Dimer impurity content: <0.32%;
R-isomer content (by chiral HPLC): <0.2%;
l-aminoadamantane-3-ol content (by GC): 0.56%.
EXAMPLE 3: Preparation of Vildagliptin (using K2C03 and KI)
To l-aminoadamantane-3-ol (19.4 g) taken in DM Water (50 mL), added potassium carbonate (8.0 gms), potassium iodide (0.1 gm) and stirred for 15 mins at 25-35°C. (2S)-1- (Chloroacetyl)-2-Pyrrolidine carbonitrile (10 gms) was added at 25-35°C and stirred for 15 mins at 25-35°C. Raised the reaction mass temperature to 40-45°C and stirred for 4 hr at 40-45°C. After reaction completion, cooled to 25-30°C and charged DCM (50 mL). Separated the organic layer and extracted the aqueous layer with DCM. The total organic layer was washed with DM water and the resulting organic layer was concentrated under vacuum at temperature <40°C to get residual mass. Charged ethyl acetate (70 mL) to above residual mass and refluxed for 1 hr. Cooled to 25-30°C and stirred for 2 hr. Filtered the reaction mass and wash wet cake with ethyl acetate (10 mL). Suck dried for 30 min, dried initially at 25-35°C for 1 hr and then at 50-55°C for 6 hr to provide 12 gms of crude vildagliptin.
EXAMPLE 4; Preparation of Vildagliptin (using K2HP04 buffer and KI)
·
To l-aminoadamantane-3-ol (19.4 g) taken in DM Water (100 mL), added K2HP04 (10.1 gms), potassium iodide (0.1 gm) and stirred for 15 rnins at 25-35°C. (2S)-l-(Chloroacetyl)-
2- Pyrrolidine carbonitrile (10 gms) was added at 25-35°C and stirred for 15 mins at 25- 35°C. Raised the reaction mass temperature to 40-45°C and stirred for 8-10 hr at 40-45°C. After reaction completion, cooled to 25-30°C and filtered the reaction mass to remove salts. The resulting filtrate was extracted with DCM, and the resulting organic layer was concentrated initially by atmospheric distillation and later under vacuum at temperature 30- 40°C to get residual mass. Charged ethyl acetate (50 mL) to above residual mass and refluxed for 1 hr. Cooled to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed the wet cake with ethyl acetate (10 mL). Suck dried for 30 min, dried initially at 25-35°C for 1 hr and then at 50-55°C for 6 hr to provide 12 gms of crude vildagliptin.
HPLC Purity: 96.54%
Dimer impurity content: 2.55%;
R-isomer content (by chiral HPLC): not detected
l-aminoadamantane-3-ol content (by GC): 0.86%.
Example 5: Purification of Vildagliptin.
Vildagliptin crude (100 gms) was dissolved in isopropanol (900 mL) by heating to 50-55°C and stirred for 30 min. Filtered the reaction mass over hyflo bed (10 gms) at 50-55°C and washed the hyflo bed with hot isopropanol (100 mL). Distilled out solvent under vacuum at
35-40°C up to 4 volumes remains and allowed to cool to 20-25°C and stirred for 1 hr at same temperature. Further, allowed to cool to 5-10°C, stirred for 2 hrs, filtered and washed with isopropanol (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to provide 80 gms of pure vildagliptin.
HPLC Purity: 99.89%;
Dimer impurity content: <0.1 %;
R-isomer content (by chiral HPLC): not detected
l-aminoadarnantane-3-ol content (by GC): 0.06%.
The purified vildagliptin (I) was analyzed by powder X-ray diffraction (PXRD) and is set forth in Figure. 01.
EXAMPLE 6: Preparation of Vildagliptin To a solution of L-Prolinamide (100 gms) dissolved in DCM (1000 mL) was added triethyl amine (88.6 gms) and DMAP (1.07 gms) at 25-30°C under N2 atmosphere and stirred for 15 min at 25-30°C. This solution was added to a solution of chloroacetyl chloride (118.7 gms) in DCM (500 mL) under N2 atmosphere at -5 to 0°C over 2-4 hr. Heated the reaction mass temperature to 10-15°C and stirred until reaction completion, charged phosphorus oxychloride (201.5 gms) to the reaction mass at 0-5°C, heated the reaction mass temperature to reflux and stirred for 6hr at same temperature. After reaction completion, allowed to cool to 5-15°C and slowly added DM water (500 mL). Aqueous layer was separated and the organic layer was washed with DM water. To the organic layer, DM water (300 mL) was added at 25-30°C and adjusted the reaction mass pH to 6.5-7.5 with -200 mL of sodium bicarbonate solution (-16 g of NaHC03 dissolved in 200 mL of DM Water). Separated the aqueous layer and concentrated the organic layer under vacuum at temperature of 30-40°C to get residual mass. The residual mass was dissolved in DM Water (640 mL), charged l-aminoadamantane-3-ol (310.6 g) at 25-35°C. Heated the reaction mass temperature to 40-45 °C and stirred for 9 hr at the same temperature. After reaction completion, allowed to cool to 25-30°C and charged DM water (900 mL) and DCM (1280 mL). Separated the organic layer and extracted the aqueous layer with DCM. The aqueous layer was separated and kept aside for l-aminoadamantane-3-ol recovery. The total organic layer was treated with P.S. 133 carbon, stirred for 30 rnins at 25-30°C and filtered over hyflo bed. The resulting filtrate was concentrated under, vacuum at temperature 30-40°C to get residual mass. To the residual mass, charged ethyl acetate (128 mL) and distilled completely under vacuum at 30-40°C to get semi solid mass. Charged ethyl acetate (640 mL) to the obtained semi solid and refluxed for 1 hr. The reaction mass was allowed to cool to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (128 mL) to obtain wet cake. Again charged ethyl acetate (512 mL) to the obtained wet cake and refluxed for 1 hr. The reaction mass was allowed to cool to 25- 30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (128 mL) and then dried at 50-55°C for 6 hr to provide 175 gms of crude vildagliptin.
HPLC Purity: 99.66%.
Dimer impurity content: <0.2%;
R-isomer content (by chiral HPLC) : <0.1 %;
l-aminoadamantane-3-ol content (by GC): <0.7%.
DSC: 150.12°C.
EXAMPLE 7: Purification of Vildagliptin. Vildagliptin crude (100 gms) was dissolved in isopropanol (1100 mL) by heating to 50- 55°C and stirred for 30 min. Filtered the solution over hyflo bed at 50-55°C and wash with hot isopropanol (100 mL). Distilled out solvent under vacuum at <55°C up to 5 volumes remains and allowed to cool to 20-25 °C and stirred for 1 hr at same temperature. Further allowed to cool to 10-15 °C, stirred for 2 hrs, filtered and washed with chilled isopropanol (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to provide 80 gms of pure vildagliptin. HPLC Purity: >99.8%;
Dimer impurity content: <0.1%;
R-isomeri content (by chir‘al HPLC) : <0.1%;
l-aminoadamantane-3-ol content (by GC): <0.1%.
DSC: 151.92°C.
Example 8: Recovery of l-aminoadamantane-3-ol of formula (IV).
To aqueous layer (1700 mL) from example 1, 50% C.S.lye (435 mL) was added to adjust the pH to 13.0-14.0 at 25-35°C and stirred for 15 mins at 25-35°C. Raised the reaction mass temperature to 60-70°C and stirred for 3 hrs. Cooled to 25-35°C and added DCM (1700 mL), stirred for 15 min and separated the organic layer. The aqueous layer was extracted with DCM and the total organic layer was distilled out completely under vacuum at <40°C to get semisolid mass. Charged ethyl acetate (150 mL) and distilled out solvent completely under vacuum at <50°C to get semisolid material. Charged ethyl acetate (400 mL), stirred for 30 min at 40-45°C and cooled to 25-35°C. Further allowed to cool to 0- 5°C, stirred for 2hr, filtered the reaction mass at 5-10°C and washed with ethyl acetate (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to obtain 140 gms of 1- aminoadamantane-3-ol.
An original synthesis of vildagliptin ((S)-1-[2-(3-hydroxyadamantan-1-ylamino)acetyl]pyrrolidine-2-carbonitrile), a powerful DPP-4 inhibitor, was developed. Vildagliptin was assembled from 3-amino-1-adamantanol, glyoxylic acid and l-prolinamide in a 4-step reaction sequence with the isolation of only two intermediates. The procedure is competitive with existing protocols, leading to vildagliptin in 63% overall yield.
PAPER
A Facile and Economical Method to Synthesize Vildagliptin
A mild and economical method to prepare vildagliptin had been reported with a good yield. In this paper, vildagliptin was synthesized from L-proline and 3-amino-1-adamantanol through chloride acetylation, amination, dehydration and substitution. The total yield of the target compound was 59%.
Elemental Analysis calcd for C27H22ClN3O4: C 66.46, H 4.54, N 8.61; found: C 66.43, H 4.56, N 8.62.
TRIS Salt Formation. Methanol (400 mL) was added to a mixture of 1 (4.0 g, 8.2 mmol) and 2-amino-2-hydroxymethylpropane-1,3-diol (TRIS, 1.0 g, 8.2 mmol). The mixture was heated to 70 °C for 0.5 h. After cooling to room temperature, the solvent was removed in vacuum. The residue was sonicated in dichloromethane (10 mL) and concentrated again. The resulting white solid was dried under vacuum overnight. The crude material was crystallized by slurring the solid residue in a 4:1 mixture of acetonitrile and methanol (5 mL). The mixture was stirred at room temperature for 24 h to give 4-((N-benzyl-8-chloro-1-methyl-1,4-dihydrochromeno- [4,3-c]pyrazole-3-carboxamido)methyl)benzoic acid TRIS salt as a white salt (3.7 g, 73% yield). Melting point: 195.6 °C. 1 H NMR (400 MHz, DMSO): δ 7.92−7.80 (m, 2H), 7.78−7.64 (m, 1H), 7.41− 7.19 (m, 8H), 7.13−7.00 (m, 1H), 5.44 (s, 2H), 5.25−5.14 (m, 2H), 4.61−4.48 (m, 2H), 4.18−4.03 (m, 3H), 3.39 (s, 7H). TRIS OH masked by water peak. LC-MS m/z: 488.0/490.0 (M+H)+ ; chlorine pattern, method 3. RT = 1.58 min. Elemental Analysis calc for C31H33ClN4O7: C 61.00, H 5.36, N 9.15; found: C 60.84, H 5.34, N 9.13.
BOS-228 (LYS-228) is a monobactam discovered at Novartis and currently in phase II clinical development at Boston Pharmaceuticals for the treatment of complicated urinary tract infection and complicated intraabdominal infections in adult patients.
The compound has been granted fast track and Qualified Infectious Disease Product (QIDP) designation from the FDA.
In October 2018, Novartis licensed to Boston Pharmaceuticals worldwide rights to the product.
Step 1: Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate. To a solution of (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetic acid (854 mg, 1.59 mmol) prepared according to published patent application US2011/0190254, Intermediate B (324 mg, 1.75 mmol) and HATU (785 mg, 2.07 mmol) in DMF (7.9 mL) , DIPEA was added (832 μL, 4.77 mmol) . After 1 h of stirring, it was poured into water and extracted with EtOAc. Brine was added to the aqueous layer, and it was further extracted with ethyl acetate (EtOAc) (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo. The crude residue was purified via silica gel chromatography (0-10%MeOH-DCM) to afford the title compound (1.09 g, 97%) as a beige foam. LCMS: R t = 0.97 min, m/z =705.3 (M+1) Method 2m_acidic.
[0127]
Instead of HATU, a variety of other coupling reagents can be used, such as any of the typical carbodiimides, or CDMT (2-chloro-4, 6-dimethoxy-1, 3, 5-triazine) and N-methylmorpholine to form the amide bond generated in Step 1.
[0128]
Step 2: (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid. Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate (1.00 g, 1.42 mmol) in DMF (7.0 mL) at 0 ℃ was treated with SO 3·DMF (448 mg, 2.84 mmol) . After 2 h of stirring at rt, the solution was poured into ice-cold brine and extracted with EtOAc (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo, affording the title compound (assumed quantitative) as a white solid. LCMS: Rt =0.90 min, m/z = 785.2 (M+1) Method 2m_acidic.
To a solution of (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid (1.10 g, 1.40 mmol) in DCM (1.5 mL) at 0℃, TFA (5.39 mL, 70.0 mmol) was added, and after 10 minutes, the ice bath was removed. Additional TFA (3.24 mL, 42.0 mmol) was added after 1 hr at rt and the solution was diluted with DCM and concentrated in vacuo after an additional 30 min. Optionally, anisole may be added to the TFA reaction to help reduce by-product formation, which may increase the yield of desired product in this step. The crude residue was purified by reverse phase prep HPLC (XSelect CSH, 30 x 100 mm, 5 μm, C18 column; ACN-water with 0.1%formic acid modifier, 60 mL/min) , affording the title compound (178 mg, 23%) as a white powder. LCMS: R t = 0.30 min, m/z = 518.9 (M+1) Method 2m_acidic; 1H NMR (400 MHz, DMSO-d 6) δ 9.27 (d, J = 9.0 Hz, 1H) 6.92 (s, 1H) 5.23 (dd, J = 9.1, 5.7 Hz, 1H) 4.12-4.23 (m, 3H) 3.72-3.62 (m, 2H assumed; obscured by water) 3.61-3.52 (m, 1H assumed; obscured by water) 3.26 (dd, J = 14.5, 5.9 Hz, 1H) 1.36 (s, 4H) . 1H NMR (400 MHz, D 2O) δ 7.23 (s, 1H) , 5.48 (d, J = 5.8 Hz, 1H) , 4.71-4.65 (m, 1H) , 4.44 (t, J = 8.2 Hz, 2H) , 3.89-3.73 (m, 3H) , 3.54 (dd, J = 14.9, 4.9 Hz, 1H) , 1.65-1.56 (m, 2H) , 1.56-1.46 (m, 2H) . The product of this process is amorphous. Compound X can be crystallized from acetone, ethanol, citrate buffer at pH 3 (50 mM) , or acetate buffer at pH 4.5 (50 mM) , in addition to solvents discussed below.
Over the past several decades, the frequency of antimicrobial resistance and its association with serious infectious diseases have increased at alarming rates. The increasing prevalence of resistance among nosocomial pathogens is particularly disconcerting. Of the over 2 million (hospital-acquired) infections occurring each year in the United States, 50 to 60% are caused by antimicrobial-resistant strains of bacteria. The high rate of resistance to commonly used antibacterial agents increases the morbidity, mortality, and costs associated with nosocomial infections. In the United States, nosocomial infections are thought to contribute to or cause more than 77,000 deaths per year and cost approximately $5 to $10 billion annually.
Important causes of Gram-negative resistance include extended-spectrum 13- lactamases (ESBLs), serine carbapenemases (KPCs) and metallo-13-lactamases (for example NDM-1 ) in Klebsiella pneumoniae, Escherichia coli, and Proteus mirabilis, high-level third-generation cephalosporin (AmpC) 13-lactamase resistance among Enterobacter species and Citrobacter freundii, and multidrug-resistance genes observed in Pseudomonas, Acinetobacter, and Stenotrophomonas. The problem of antibacterial resistance is compounded by the existence of bacterial strains resistant to multiple antibacterials. For example, Klebsiella pneumonia harboring NDM-1 metallo-13- lactamase carries frequently additional serine-13-lactamases on the same plasmid that carries the NDM-1 .
Thus there is a need for new antibacterials, particularly antibacterial compounds that are effective against existing drug-resistant microbes, or are less susceptible to development of new bacterial resistance. Monobactam antibiotic, which is referred to herein as Compound X, is primarily effective against Gram-negative bacteria, including strains that show resistance to other monobactams.
The present invention relates to a process for the preparation of monobactam antibiotic Compound X and intermediates thereof.
More particularly, the present invention relates to a process for the preparation of Compound X
Compound X
also referred to as 1 -(((Z)-(1 -(2-aminothiazol-4-yl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)-1 -sulfoazetidin-3-yl)amino)ethylidene)amino)oxy)cyclopropanecarboxylic acid, or a salt thereof, or a solvate including hydrate thereof.
Patent application number PCT/US2015/02201 1 describes certain monobactam antibiotics. Compound X may be prepared using the method disclosed in PCT/US2015/02201 1 , in particular example 22, and in PCT/CN2016/099482.
A drawback from these processes is that they exhibit a large number of process steps and intermediate nitrogen protection/deprotection steps, reducing the overall yield and efficiency. Furthermore, these processes require several chromatographic purification steps to be carried out in course of the processes. We have found that the preparation of Compound X, as previously prepared on a manufacturing scale, possesses a number of disadvantages, in particular poor handling characteristics.
It would thus be beneficial to develop alternative or improved processes for the production of Compound X that do not suffer from some or all of these disadvantages.
Compound x Compound x
Scheme 1
Preparation of Compound X from Intermediates 22 and 2A
Scheme 3
Examples
The Following examples are merely illustrative of the present disclosure and they should not be considered as limiting the scope of the disclosure in any way, as these examples and other equivalents thereof will become apparent to those skilled in the art in the light of the present disclosure, and the accompanying claims.
Synthesis of Compound 8 (R = benzyl)
1 .50kg oxazolidin-2-one (7b) was charged into the reactor. 7.50kg THF was charged and the stirring started. The mixture was cooled to 10~20°C. 2.18kg potassium fert-butoxide was charged intol 2.00kg THF and stirred to dissolve.
The potassium fert-butoxide solution was added dropwise into the reactor while maintaining the temperature at 10-20 °C. The reaction was stirred for 1 ~2hrs at 10-20 °C after the addition. The solution of 2.36kg methyl-2-chloroacetate (7a) in 3.00kg of THF was added to the reactor while maintaining the temperature at 10-20 °C. The reaction mixture was stirred for 16-18 h at 20-25 °C. The IPC (in process control) showed completion of the reaction. The mixture was centrifuged and the wet cake was washed with 7.50kg THF. The filtrate was concentrated and the crude 7 was provided as reddish brown liquid, which was used for the next step without further purification,
The dried reactor was exchanged with N2 three times. 3.71 kg LiHMDS solution in THF/Hep (1 M) and 1 .30kg THF were charged under nitrogen protection. The stirring was started and the solution was cooled to -70—60 °C. The solution of 0.71 kg benzyl acetate (6) in 5.20 kg THF was added dropwisely at -70— 60 °C, and the resulted mixture was stirred for 1 -1 .5 h after the addition. The solution of 0.65kg 7 in 3.90kg THF was added dropwise while maintaining the temperature at -70—60 °C, then stirred for 30-40 minutes. The reaction mixture was warmed to 20-25 °C and stirring was continued for 0.5-1 .0 h. IPC showed 6 was less than1 .0% (Otherwise, continue the reaction till IPC passes). The reaction mixture was poured into 13.65 kg aqueous citric acid below 10 °C. The mixture was stirred for 15-20 minutes after the addition. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (6.50kg * 2). The organic layer was combined, washed by 6.50 kg 28% NaCI solution and dried with 0.65
kg anhydrous MgSC . The mixture was filtered and the wet cake was washed with 1 .30kg EA. The filtrate was concentrated under vacuum to provide crude 8. The crude 8 was stirred in 2.60 kg MTBE at 20-25 °C for 1 -1 .5 h. The mixture was cooled to 0-10 °C and stirred for 1 .5-2.0 h and filtered. The filter cake was washed with 0.65kg pre-cooled MTBE and dried under vacuum (<-0.096Mpa) at 20-25 °C for 12~16hrs till a constant weight to give 513 g of 8 as a white solid, Yield: 45%, HPLC purity 96.4%,1 H NMR (400 MHz, CHLOROFORM-c δ ppm 3.48 – 3.55 (m, 1 H) 3.56 – 3.63 (m, 2 H) 3.66 – 3.74 (m, 1 H) 4.17 – 4.26 (m, 2 H) 4.31 – 4.44 (m, 2H) 5.12 – 5.24 (m, 2 H) 7.30 – 7.44 (m, 5 H).
Synthesis of Compound 9 (R = benzyl)
The dried reactor was charged with 3.75kg HOAc and 1 .50 kg 8. The stirring was started and the reaction mixture was cooled to 0-5 °C. 3.53kg aqueous NaN02 was added dropwise at 0-10 °C, and the reaction mixture was stirred for 15-30 minutes after the addition. IPC showed 8 was less than 0.2%. The reaction mixture was treated with 7.50kg EA and 7.50 kg water. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (7.50kg * 2). The organic layers were combined, washed with 7.50 kg 28% NaCI solution, and concentrated under vacuum to provide crude 9. The crude 9 was slurried with 5.25 kg water at 10-20 °C for 3~4hrs, and filtered. The wet cake was washed with 1 .50kg water. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .44 Kg of 9, yield: 86.9%, HPLC purity 92.9%,1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.60 – 3.76 (m, 2 H) 4.44 (t, J=8.07 Hz, 2 H) 4.60 (s, 2 H) 5.25 – 5.41 (m, 2 H) 7.30 – 7.43 (m, 5 H) 1 1 .62 (br s, 1 H).
Synthesis of Compound 9a (R = benzyl)
9
The dried reactor was charged with 0.58 kg Zn, 4.72kg (Βο Ο, 6.00 kg water, 1 .20 kg NH4CI and 6.00kg THF. The reaction mixture was stirred and heated to 50-55 °C. The solution of 0.60 kg 9 in 4.20kg THF
was added dropwisely while maintaining the temperature at 50-55 °C. The reaction mixture was stirred for 0.5-1 .Ohrs after the addition. IPC showed 9 was less than 0.1 %. The reaction mixture was treated withl .50 kg ethyl acetate and stirred for 15-20 minutes. Phase was separated and the water layer was extracted by1 .50 kg ethyl acetate. The organic layers were combined, washed with 6.00 kg 28% NaCI solution and concentrated under vacuum to provide crude 9a. The crude 9a was stirred with 3.60kg*2 n-heptane to remove excess (Βο Ο. The residue was purified by silica gel chromatography column eluted with ethyl acetate: Heptane= 1 :1 to provide crude 9a solution. The solution was concentrated under reduced pressure to obtain crude 9a. The crude 9a was slurried with 1 .80 kg MTBE for 2.0-3. Ohrs, filtered, and the wet cake was washed with MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 50-55 °C for 16-18 h till a constant weight to give 392 g of 9a as a white solid, Yield: 51 %, HPLC purity 98.1 %,1H NMR (400 MHz, DMSO-cfe) δ ppm 1.17 – 1 .57 (m, 9 H) 3.39 – 3.61 (m, 2 H) 4.20 – 4.45 (m, 3 H) 5.10 – 5.32 (m, 3 H) 5.75 (s, 1 H) 7.38 (br s, 5 H) 7.75 – 7.99 (m, 1 H).
Synthesis of compound (VII) (R = benzyl, X = CI)
9a VII
The dried reactor was charged with 13.0kg HCI in IPA and the stirring was started. 1 .33 kg 9a was charged in portions at 20-25 °C. The mixture was stirred at 20-25 °C for 3-4 h. IPC showed 9a was less than 0.1 %. The reaction solution was concentrated under vacuum 40-45 °C. The residue was treated with 21 .58kg MTBE at 20-25 °C for 3-4 h. The mixture was filtered and the wet cake was washed with 2.60kg MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .045 Kg of compound VII (R = benzyl, X = CI) as a yellow solid, Yield: 93.7%, HPLC purity 99.2%,1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.16 – 3.74 (m, 3 H) 4.10 – 4.35 (m, 4 H) 5.09 – 5.39 (m, 2 H) 7.27 – 7.60 (m, 5 H) 8.72 (br s, 2 H).
Synthesis of compound (Vile) (R = benzyl)
VII Vile
To an autoclave (3L) were added VII (R = benzyl, X = CI) (100 g, 304.2 mmol, 1 .0 equiv.), DCM (2650 g, 26.5 equiv., w/w) and (S-BINAP)RuCl2 (2.4 g, 3.04 mmol, 0.01 equiv.), successively. Air in the autoclave was replaced with N2 5 times. N2 in the autoclave was was replaced with H2 5 times. The solution was stirred with 250-260 r/min and H2 (2.1 ±0.1 MPa) at 40±5°C for 24 h. The reaction mixture was filtered, and the filter cake was washed with DCM (400 g, 4.0 equiv., w/w). The filter cake was slurried with IPA (785 g, 7.85 equiv., w/w) and H2O (40 g, 0.4 equiv., w/w) overnight (18-20 h). The mixture was filtered. The filter cake was washed with IPA (200 g, 2.0 equiv., w/w) and dried at 45±5°C overnight (18-20 h). Vile (R = benzyl) was obtained as off-white solid, 80.4 g, 79.9% yield, 95.5% purity, 97.6% de, >99.5% ee. 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.34-3.38 (m, 2 H) 3.50-3.52 (m, 1 H) 3.60-3.62 (m, 1 H) 4.18-4.24 (m, 4 H) 5.23 (s, 2H) 6.16 (s, 1 H) 7.32 (m, 5H) 8.74 (s, 1 H).
Alternative synthesis of compound 9a (R = benzyl)
5b
Mg(OtBu)2
To a flask was added 5a (1 .88 g, 12.93 mmol), THF (40 mL), and CDI (2.20 g, 13.58 mmol) at 25 °C. The mixture was stirred for 3 h. To the reaction mixture was added 5b (2.00 g, 6.47 mmol), and Mg(OfBu)2 (2.21 g, 12.93 mmol). The reaction mixture was stirred at 25 °C for 24 h. The reaction mixture was concentrated under vacuum to remove most of the THF solvent. To the concentrated solution was added MTBE (40 mL), followed by addition of an aqueous solution of HCI (1 M, 60mL) to adjust to pH = 2-3. Two phases were separated, and the water phase was extracted with MTBE (20 mL). The combined organic phase was washed with aqueous NaHCC (5%, 50 mL) and brine (20%, 40 mL). The organic phase was concentrated to a weight of -19 g, and a lot of white solid was obtained in the concentration process. The suspension was cooled to 0 °C, and filtered. The filter cake was washed with cold MTBE (5 mL) and dried under vacuum to obtain product 9a (1 .6g, 63% yield).
Synthesis of compound (Vile) (R = benzyl, PG = Cbz)
Vile Vile
To a flask (5 L) were added Vile (R = benzyl) (140 g, 423.2 mmol, LOequiv.), H20 (1273 g, 9.09 equiv., w/w) and toluene (2206 g, 15.76 equiv., w/w). The solution was stirred and cooled to 0-5 °C with ice bath. Then NaHCOa (78.4 g, 933 mmol, 2.22 equiv.) was added and CbzCI (89.6 g, 527 mmol, 1 .24 equiv.) was dropped into the stirring solution, respectively. The solution was stirred at 30±5 °C overnight (18-20 h). Heptane (3612 g, 25.8 equiv., w/w) was added dropwise to the stirring solution over 1 h at 20-30 °C. The mixture was filtered. The filter cake was washed with heptane (280 g, 2.00 equiv., w/w) and MTBE (377 g, 2.69 equiv., w/w), respectively. The filter cake was dried at 45±5°C overnight (18-20 h). Vile (R = benzyl, PG = Cbz) was obtained as an off-white solid, 169.4 g, 93% yield, 96.7% purity, 98% de, >99.5% ee, 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.23-3.24 (m, 1 H) 3.30 (m, 1 H) 3.51 -3.55 (m, 2 H) 3.99 (s, 1 H) 4.17-4.21 (m, 3 H) 5.02-5.03 (m, 2H) 5.12 (s, 2H) 5.46-5.48 (d, 1 H) 7.33-7.36 (m, 10H) 7.75-7.73 (d, 1 H).
Synthesis of compound (IV) (PG = Cbz)
Vile IV
Vile (R = benzyl) (220 g, 513.5 mmol, 1 .0 equiv.) was dissolved in THF (1464g, 6.65 equiv., w/w). The solution was filtered. The filter cake was washed with THF (488g, 2.22 equiv., w/w). The filtrate (Vile) was collected. To an autoclave (3L) were added the filtrate (Vile). The reactor was cooled down to -75 – -65 °C with dry-ice/EtOH bath, and bubbled with NH3 for not less than 4 h. Then the solution was stirred at 25±5 °C with NH3 (0.5-0.6 MPa) for 24 h. The autoclave was deflated to release NH3. The reaction solution was concentrated with a rotary evaporator to remove THF until the residue was around 440 g. The residue was slurried with EA (2200 g, 10 equiv., w/w) at 70±2 °C, then cooled to 25±5 °C and stirred for 16-18 h. The mixture was filtered. The filter cake was washed with EA (440 g). The filter cake was slurried with EA (1320 g, 6.00 equiv. w/w), and the temperature was raised to 70±2 °C, then cooled to 25±5 °C and stirred for 16-20 h. The mixture was filtered. The filter cake was washed with EA, and dried at 50±5 °C overnight (18-20 h). IV (PG = Cbz) was obtained as off-white solid, 141 g, 81 .5% yield, 99.1 % purity, >99.5% assay, 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.12 – 3.23 (m, 2 H) 3.31 (br s, 1 H) 3.56 (t, J=8.01 Hz, 2 H) 3.88 (quin, J=6.02 Hz, 1 H) 3.93 – 4.03 (m, 1 H) 4.20 (t, J=8.01 Hz, 2 H) 5.02 (s, 2 H) 5.27 (d, J=5.87 Hz, 1 H) 7.12 (s, 1 H) 7.22 – 7.45 (m, 5 H).
Synthesis of compound (III) (PG = Cbz, LG = S02CH3)
IV III
To a flask was added IV (PG = Cbz) (14.00 g, 41 .50 mmol, 1 .00 equiv), and dry 1 , 2-dimethoxyethane (300 mL) under N2. The mixture was stirred at -5°C ~ 0°C for 1 h to obtain a good suspension. MsCI (7.89 g, 68.89 mmol, 5.33 mL, 1 .66 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min, and Et3N (12.60 g, 124.50 mmol, 17.26 mL, 3.00 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min side to side. The reaction mixture was stirred for additional 5 min at -5°C ~ 0°C, and was quenched with water (6 mL). The reaction mixture was concentrated to remove DME. The solid was slurried in water (250 mL) and MTBE (125 mL) for 1 h. The solid was collected by filtration, and then slurried in water (250 mL) for 1 hr. The solid was collected by filtration, and washed with water (25 mL) to give white solid. The solid was slurried in EA (150 mL) and dried in vacuum at 60°C for 24 h to give III (PG = Cbz, LG = SO2CH3) (15.00 g, 36.1 1 mmol, 87.01 % yield), 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.17 (s, 3 H) 3.26 (br d, J=15.04 Hz, 1 H) 3.47 – 3.57 (m, 1 H) 3.64 (br d, J=6.36 Hz, 2 H) 4.22 (br dd, J=17.79, 8.50 Hz, 2 H) 4.50 (br s, 1 H) 4.95 – 5.17 (m, 3 H) 7.21 – 7.56 (m, 5H) 7.43 (s, 1 H) 7.63 – 7.89 (m, 2 H).
Synthesis of compound II (PG = Cbz, LG = SO2CH3, M+ = NBu4+)
O OMs o CISO3H, 2-picoline – ° O ?yO
HN Bu4NHS04< NHCbz
“Cbz
III II
To a flask was added 2-picoline (1 1 .50 g, 12.23 mL) and DMF (10 mL). The solution was cooled to 5 SC, followed by slow addition of chlorosulfonic acid (7.20 g, 4.14 mL). The temperature was increased to 20 SC. Ill (PG = Cbz, LG = SO2CH3) (5.13 g, 12.35 mmol) was added to the reaction mixture. The reaction mixture was heated to 42 SC for 18h. IPC (in process control) showed complete conversion of starting material. The reaction was cooled to 20 SC and dropwise added to a solution of tetrabutylammonium hydrogen sulfate (4.6 g, 13.6 mmol) in the mixed solvents of dichloromethane (100 mL) and water (100 mL) at 5SC. The phases were separated and the water phase was extracted with dichloromethane (2*50mL). The combined organic phase was washed with water (5*100mL). The organic phase was concentrated to dryness and purified by column chromatography (dichloromethane/methanol = 15/1 v/v) to afford II (PG = Cbz, LG = SO2CH3, M+ = NBii4+) (8.4 g, 92.30%), 1 H NMR (400 MHz, CHLOROFORM-c/) δ ppm 0.99 (t, J=7.34 Hz, 12 H) 1 .36 – 1 .50 (m, 8 H) 1 .54 – 1 .76 (m, 8 H) 3.15 (br d, J=8.31 Hz, 2 H) 3.21 – 3.35 (m, 8 H) 3.47 (br dd, J=14.73, 7.27 Hz, 1 H) 3.54 – 3.65 (m, 1 H) 3.67 – 3.81 (m, 2 H) 4.17 – 4.32 (m, 1 H) 4.39 – 4.62 (m, 1 H) 4.74 (br s, 1 H) 5.1 1 (s, 3 H) 5.32 – 5.50 (m, 1 H) 6.47 (br s, 1 H) 7.29 – 7.47 (m, 5 H) 8.69 – 8.94 (m, 1 H).
Synthesis of compound (IA)
A solution of II (PG = Cbz, LG = SO2CH3, M+ = NBu4+) (4.0 g) in dichloromethane (38 mL) was pumped to tube A at rate of 2.0844 mL/min, and a solution of KHCO3 (3.0 g) in water (100 mL) was pumped to tube B at a rate of 1 .4156 mL/min side to side. These two streams were mixed in a cross-mixer then flowed to a tube coil that was placed in an oil bath at 100 °C. The residence time of the mixed stream in the coil was 2 min. The reaction mixture flowed through a back-pressure regulator that was set at ~ 7 bars, and was collected to a beaker. After completion of the collection, two phases was separated. The organic phase was concentrated to dryness. The residue was slurried in ethyl acetate (5 mL). The solid was filtered and the filter cake was dried to give IA (2.6 g, 75%),
To a stirring solution of compound 16b (2 g, 10.14mmol, 1 .0 eq) in DMF (20 ml_) was added CS2CO3 (5.29g, 16.22 mmol, 1 .6 eq), then the resulting solution was stirred at room temperature for 10mins, then compound 16a (5.27g, 20.28mmol, 2eq) was added dropwise to the mixture for 2 minutes, then the resulting solution was stirred for another 2 hours. TLC showed the starting material was consumed completely. The mixture was added with water (60mL) and extracted with MTBE (20mL*3). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The crude was slurried in heptane to give 1 .65 g 16 as a white solid (Yield: 57%), 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.48-7.28 (m, 10 H), 5.00-4.96 (t, J=6.0 Hz, 1 H), 3.81 (s, 3H), 3.44-3.42 (m, 2H), 2.40-2.37 (m, 2H).
Compound 16 (1 g, 2.66mmol, 1 eq) was dissolved in THF (20mL) under Nitrogen, and cooled to -40 °C. NaHMDS (1 .6mL, 2.0M THF solution, 1 .2 eq) was added dropwise. The reaction was stirred for 1 h at -40 °C. HPLC indicated the reaction was finished. The reaction was quenched with 10% Citric acid, extracted with MTBE (25 ml_ x 2). The combined organic layers were washed with brine (30 ml_), dried with Na2S04, filtered and concentrated to give 17 as a yellow solid, which was used for the next step without purification (assay yield: 65%); 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.27-7.13 (m, 10 H), 3.46 (s, 3H), 1 .21 -1 .17(dd, J=7.2, 10.4 Hz, 2H ); 1 .14-1 .1 1 (dd, J=7.2, 10.4 Hz, 2H).
Step 3
Compound 17 (100 mg) was dissolved in methanol (5 mL) and 2.0 M HCI IPAC solution (5 mL). The solution was heated at 45 °C for 3 days. HPLC indicated the reaction was finished. The reaction was cooled to room temperature and was diluted with 10 mL water. The reaction mixture was washed with MTBE (10 mL x 2), organic layer was discarded and the aqueous layer was concentrated to give compound 2A HCI (32 mg, 62% yield), 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.80-3.44 (br, 4H), 1 .56 (s, 2H), 1 .38 (s, 2H).
Step 4
To a solution of 2A HCI (0.70 g, 4.57 mmol) in methanol (5 mL) was added triethylamine (1 .26 mL, 9.14 mmol) at room temperature. The solution was stirred for 20 min, and the solvent was removed under vacuum. To the residue was added IPAC (10 mL) leading to precipitation. The solid was filtered, and the filtrate was concentrated to provide 2A (0.50g, 94% yield) containing ca. 6 wt% Et3N-HCI.
Synthesis of Compound X from compound of formula (I), (IA)
Compound x
To a flask was charged 21 (1 .00 g, 68.43 wt%, 2.50 mmol) and DMF (10 mL). The suspension was cooled to -20 °C, to which was added diphenylphosphinic chloride (0.52 mL, 2.75 mmol). The solution was stirred at -20 °C for 30 min, followed by addition of a mixed solution of (IA) (1 .52g, 3.00 mmol) and triethylamine (0.52 mL, 3.76 mmol) in DMF (2mL). The reaction mixture was stirred at 20 °C for 20 h, followed by addition of MTBE (20 mL). The reaction mixture was adjusted to pH = 2-3 using aqueous HCI solution (37%). To the mixture was added isopropanol (100 mL). The resulting mixture was stirred for 4 h to obtain a suspension. The suspension was filtered and the filter cake was dried under vacuum to afford crude 22 (1 .17 g). The crude 22 was slurried in a combined solvent of THF/H2O (= 12 mL / 3mL), and filtered to afford 22 (0.744 g, 75 wt% by Q-NMR, 53.3% yield). 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.47 – 3.55 (m, 2 H) 3.59 – 3.63 (m, 2 H) 4.13 – 4.21 (m, 3 H ) 5.05 (dd, J=8.8, 5.6 Hz, 1 H) 8.22 (s, 1 H) 9.73 (d, J=8.7 Hz, 1 H).
To a suspension of 22 (580 mg, 75 wt%, 1 .037 mmol) in DMAC (1 .5 mL) was added 2A (214.3 mg, 85 wt%, 1 .556 mmol). The reaction was stirred at 25 °C for 3 days, and in process control showed 22, Compound X = 4/96, and Z/E = 91 /9. the mixture was slowly added into 15ml acetone to precipitate yellowish solid. The reaction mixture was filtered to afford Compound X (0.7 g, 34 wt% by QNMR, 44% yield).
Synthesis of compound 3 (R2 = CH(Ph)2)
R2 = CH(Ph)2
2-(2-aminothiazol-4-yl)-2-oxoacetic acid (Y) (10.00 g, 47.93 mmol) and compound W (R2 = CH(Ph)2) (13.31 g, 46.98 mmol) were suspended in DMAC (40 mL), followed by addition of triethylamine (5.01 mL, 35.95 mmol). The reaction mixture was stirred at 20 °C for 5 h. HPLC showed completion of the reaction, and Z/E
= 97/3. To the reaction mixture was added water (120 mL) with stirring. The mixture was stirred for 20 min to obtain a suspension. The suspension was filtered and the filter cake was washed with water (50 mL).
The filter cake was slurried in a combined solvent of THF/ethyl acetate (50 mL / 50 mL) at 60 °C and cooled to 20 °C. The solid was filtered and dried at 50 °C for 3 h to get 3 (R2 = CH(Ph)2) (19.5 g, 88% yield). 1H
Alternative Synthesis of Compound X from compound of formula (I), (IA)
Compound x
IA (40.14 g, 62.63 mmol) was dissolved in methanol (200 ml_), followed by addition of Pd/C (10%, 1 .1 g). The reaction mixture was maintained under hydrogen atmosphere (1 -2 bar) at 20 °C for 24 h. In process control showed completion of the reaction. The reaction mixture was filtered. The filtrate was concentrated to give an oil of IB (M+ = NBu4+) (58.20 g, 55 wt% by Q-NMR, 100% yield). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 0.93 (t, J=7.3 Hz, 12 H) 1 .23 – 1 .36 (m, 8 H) 1 .57 (m, 8 H) 2.99 – 3.28 (m, 8 H) 3.37 (dd, J=14.3, 7.5 Hz, 1 H) 3.65 – 3.70 (m, 3 H) 3.84 – 3.88 (m, 1 H) 4.08 (d, J=5.6 Hz, 1 H) 4.18 – 4.22 (m, 2 H).
3 (R2 = CH(Ph)2) (0.95 g, 2.17 mmol) was dissolved in THF (20 ml_). To the solution was added /V-methyl morpholine (0.77 g, 7.60 mmol) and 2-chloro-4,6-dimethoxy-1 ,3,5-triazine (0.57 g, 3.26 mmol). The reaction mixture was stirred at 20 °C for 1 h followed by addition of IB (M+ = NBu +) (2.70 g, 48.98 wt%, 2.61 mmol). The reaction was stirred at 20 °C for 5 h. In process control showed completion of the reaction. To the reaction mixture was added ethyl acetate (20 ml_). The organic phase was washed with brine (10 ml_). Solvent was removed. Acetone (40ml) was added to dissolve residue. TFA (1 .24 g, 10.86 mmol) dissolved in acetone (3 ml) was added slowly. The white solid was filtered and washed by acetone (10 ml) two times. Dried at 40 °C for 5h to get compound 4 (R2 = CH(Ph)2). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 1 .49 – 1 .55 (m, 4 H) 3.27 (dd, J=14.4, 6.2 Hz, 1 H) 3.49 – 3.65 (m, 2 H) 3.71 (dd, J=14.4, 6.2 Hz, 1 H) 4.04 – 4.10 (m, 1 H) 4.07 (dd, J=16.0, 8.6 Hz, 1 H) 4.17 (dd, J=1 1 .8, 6.0 Hz, 1 H) 5.28 (dd, J=9.0, 5.7 Hz, 1 H) 6.88 (s, 1 H) 7.03 (s, 1 H) 7.18 – 7.32 (m, 6 H) 7.43 (m, 4 H) 9.45 (d, J=9.0 Hz, 1 H).
Crude 4 (R2 = CH(Ph)2) (2.13 g) was dissolved in dichloromethane (20 ml_). The solution was cooled to 0 °C. To the solution was added anisole (0.68 ml_, 6.24 mmol) and trifluoroacetic acid (2.16 ml_, 28.08 mmol). The reaction was warmed to 20 °C, and stirred for 15 h. In process control showed completion of the
reaction. The aqueous phase was separated and added to acetone (40 mL) to obtain a suspension. The suspension was filtered to afford Compound X (0.98 g, 54.5% yield over two steps). 1 H NMR (400 MHz, DMSO-c/e) δ ppm 1.40 (m, 4 H) 3.26 (dd, J=14.4, 6.0 Hz, 1 H) 3.54 – 3.69 (m, 3 H) 4.14 – 4.21 (m, 3 H) 5.25 (dd, J= 8.9, 5.7 Hz, 1 H) 7.02 (s, 1 H) 9.38 (d, J=9.0 Hz, 1 H).
REF
Synthesis and optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae – Identification of LYS228
57th Intersci Conf Antimicrob Agents Chemother (ICAAC) (June 1-5, New Orleans) 2017, Abst SATURDAY-297
//////////////LYS228, LYS 228, BOS-228, LYS-228, monobactam, Novartis, phase II, Boston Pharmaceuticals, complicated urinary tract infection, complicated intraabdominal infections, fast track, Qualified Infectious Disease Product, QIDP,
Sundaram Venkataraman, Srinivasulu Gudipati, Brahmeshwararao Mandava Venkata Naga, Goverdhan Banda, Radhakrishna Singamsetty, “Process for preparing form I of tegaserod maleate.” U.S. Patent US20050272802, issued December 08, 2005.US20050272802
Tegaserod maleate [USAN]
189188-57-6
Tegaserod
CAS Registry Number: 145158-71-0
CAS Name: 2-[(5-Methoxy-1H-indol-3-yl)methylene]-N-pentylhydrazinecarboximidamide
Molecular Formula: C16H23N5O
Molecular Weight: 301.39
Percent Composition: C 63.76%, H 7.69%, N 23.24%, O 5.31%
Literature References: Selective serotonin 5HT4-receptor partial agonist. Prepn: R. K. A. Giger, H. Mattes, EP505322; eidem,US5510353 (1992, 1996 both to Sandoz); K.-H. Buchheit et al.,J. Med. Chem.38, 2331 (1995). Clinical pharmacology: S. Appel et al.,Clin. Pharmacol. Ther.62, 546 (1997); and pharmacokinetics: idemet al.,J. Clin. Pharmacol.37, 229 (1997). Clinical trial in irritable bowel syndrome: S. A. Müller-Lissner et al.,Aliment. Pharmacol. Ther.15, 1655 (2001); in female patients: J. Novick et al.,ibid.16, 1877 (2002). Review of clinical efficacy: B. W. Jones et al.,J. Clin. Pharm. Ther.27, 343-352 (2002); of mechanism of action, efficacy and safety: M. Corsetti, J. Tack, Expert Opin. Pharmacother.3, 1211-1218 (2002).
Properties: mp 155°.
Melting point: mp 155°
Derivative Type: Maleate
CAS Registry Number: 189188-57-6
Manufacturers’ Codes: SDZ-HTF-919
Trademarks: Zelmac (Novartis); Zelnorm (Novartis)
Molecular Formula: C16H23N5O.C4H4O4
Molecular Weight: 417.46
Percent Composition: C 57.54%, H 6.52%, N 16.78%, O 19.16%
Therap-Cat: Gastroprokinetic; in treatment of irritable bowel syndrome.
Tegaserod is a 5-HT4agonist manufactured by Novartis and sold under the names Zelnorm and Zelmac for the management of irritable bowel syndrome and constipation.[1] Approved by the FDA in 2002, it was subsequently removed from the market in 2007 due to FDA concerns about possible adverse cardiovascular effects. Before then, it was the only drug approved by the United StatesFood and Drug Administration to help relieve the abdominal discomfort, bloating, and constipation associated with irritable bowel syndrome. Its use was also approved to treat chronic idiopathic constipation.[2]
In 2000, originator Novartis established an alliance with Bristol-Myers Squibb for the codevelopment and copromotion of tegaserod maleate, which is now available in more than 55 countries worldwide for the treatment of IBS with constipation. In 2015, Zelnorm was acquired by Sloan Pharma from Novartis.
Novartis’ brand name Zelnorm (tegaserod) had originally received approval from the US FDA in 2002 for the treatment of irritable bowel syndrome with constipation (IBS-C) [5, 8]. It was, however, voluntarily withdrawn from widespread use in the US market in 2007 after concerns arose over the possibility that tegaserod could potentially cause dangerous cardiovascular events in patients [5,8]. Since then, closer evaluations of the original data suggesting such cardiovascular risk have resulted in the limited reintroduction or ‘re-approval’ of tegaserod for treatment of IBS-C specifically in female patients less than 65 years of age and whom are considered to be at a lower risk of a cardiovascular event than the broader population . Zelnorm (tegaserod) by Sloan Pharma subsequently gained re-approval in April of 2019 [5]. Nevertheless, tegaserod remains un-approved in certain regions [7].
Despite the relative complications involved in its history of regulatory approval, ever since its first introduction in 2002 tegaserod remains the only therapy for IBS-C that possesses the unique mechanism of action of acting on serotonin-4 (5-HT(4)) receptors in smooth muscle cells and in the gastrointestinal wall to facilitate actions like esophageal relaxation, peristaltic gut movement, and natural secretions in the gut, among others
Mechanism of action
The drug functions as a motility stimulant, achieving its desired therapeutic effects through activation of the 5-HT4 receptors of the enteric nervous system in the gastrointestinal tract. It also stimulates gastrointestinal motility and the peristaltic reflex, and allegedly reduces abdominal pain.[3] Additionally, tegaserod is a 5-HT2B receptor antagonist.[4]
Withdrawal from market
On 30 March 2007, the United States Food and Drug Administration requested that Novartis withdraw Zelnorm from shelves.[5] The FDA alleges a relationship between prescriptions of the drug and increased risks of heart attack or stroke. An analysis of data collected on over 18,000 patients demonstrated adverse cardiovascular events in 13 of 11,614 patients treated with Zelnorm (a rate of 0.11%) as compared with 1 of 7,031 patients treated with placebo (a rate of 0.01%). Novartis alleges all of the affected patients had preexisting cardiovascular disease or risk factors for such, and further alleges that no causal relationship between tegaserod use and cardiovascular events has been demonstrated.[6] On the same day as the FDA announcement, Novartis Pharmaceuticals Canada announced that it was suspending marketing and sales of the drug in Canada in response to a request from Health Canada.[7] In a large cohort study based on a US health insurance database, no increase in the risk of cardiovascular events were found under tegaserod treatment.[8] Currently, tegaserod may only be used in emergency situations only with prior authorization from the FDA.[9]
Paper
The serotonin 5-HT4 receptor. 2. Structure-activity studies of the indole carbazimidamide class of agonists
J Med Chem 1995, 38(13): 2331
In a preferred embodiment of the first aspect of the present invention, the process of preparing tegaserod or a salt thereof comprises the steps of:
(a) coupling S-methyl-isothiosemicarbazide or a salt thereof and 5-methoxy-indole-3-carboxaldehyde to form 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide:
and
(b) reacting the 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide with n-pentyl amine to form tegaserod:
[0013]
The skilled person will appreciate that:
S-methyl-isothiosemicarbazide and salts thereof exist in two tautomeric forms:
1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide exists in four tautomeric forms:
tegaserod exists in four tautomeric forms:
[0017]
It is to be understood that where tautomeric forms occur, the present invention embraces all tautomeric forms and their mixtures, i.e. although S-methyl-isothio-semicarbazide and 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemi-carbazide are mostly defined for convenience by reference to one isothiosemicarbazide form only, and although tegaserod is mostly defined for convenience by reference to one guanidino form only, the invention is not to be understood as being in any way limited by the particular nomenclature or graphical representation employed.
[0018]
When an S-methyl-isothiosemicarbazide salt is used in the process of the present invention, this may be an acid addition salt with acids, including but not limited to inorganic acids such as hydrohalogenic acids (for example, hydrofluoric, hydrochloric, hydrobromic or hydroiodic acid) or other inorganic acids (for example, nitric, perchloric, sulfuric or phosphoric acid), or organic acids such as organic carboxylic acids (for example, propionic, butyric, glycolic, lactic, mandelic, citric, acetic, benzoic, salicylic, succinic, malic or hydroxysuccinic, tartaric, fumaric, maleic, hydroxymaleic, mucic or galactaric, gluconic, pantothenic or pamoic acid), organic sulfonic acids (for example, methanesulfonic, trifluoromethanesulfonic, ethanesulfonic, 2-hydroxyethanesulfonic, benzenesulfonic, p-toluenesulfonic, naphthalene-2-sulfonic or camphorsulfonic acid) or amino acids (for example, ornithinic, glutamic or aspartic acid). Preferably the S-methyl-isothiosemicarbazide salt is a hydrohalide (such as the hydrofluoride, hydrochloride, hydrobromide, or hydroiodide) or a sulfonate (such as the methanesulfonate, benzenesulfonate, or p-toluenesulfonate). Preferably the S-methyl-isothiosemicarbazide salt is S-methyl-isothiosemicarbazide hydroiodide.
The following synthetic scheme demonstrates a preferred process of the present invention.
[0032]
The invention is now demonstrated by the following non-limiting illustrative example.
EXAMPLE Step 1: Schiff’s Base Formation of 5-methoxy-indole-3-carboxaldehyde and S-methyl-isothiosemi-carbazide hydroiodide
[0033]
5-Methoxy-indole-3-carboxaldehyde (1.5 g, 1 eq) and S-methyl-isothiosemicarbazide hydroiodide (3.99 g, 2 eq) in methanol (15 ml, 10 vol) were stirred in the presence of triethylamine (3 ml, 2 vol) at 25-30° C. for 2 hours. After completion of the reaction, the methanol was removed by distillation under reduced pressure at 45-50° C. and ethyl acetate (10.5 ml, 7 vol) was added to the residue to precipitate out the product. The product, 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemi-carbazide, was separated by filtration, washed with ethyl acetate (3 ml, 2 vol) and dried under vacuum at 45-50° C. The yield was almost quantitative (˜100%).
Step 2: Conversion of 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide to 1-((5-methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (Tegaserod)
[0034]
A solution of 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide (8.0 g, 1 eq) and n-pentyl amine (2.65 g, 1 eq) was refluxed in methanol (8 ml, 1 vol) at 66° C. for 4 hours. After completion of the reaction, the methanol was removed by distillation under reduced pressure at 45-50° C. to obtain tegaserod free base as a yellowish brown solid. Yield=97%. HPLC purity=95%.
Step 3: Conversion of 1-((5-methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (Tegaserod) to Tegaserod Maleate
[0035]
1-((5-Methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (55 g, 1 eq) was taken in methanol (357.5 ml, 6.5 vol) and stirred. To this reaction mixture was added at room temperature a solution of maleic acid (74.15 g, 3.5 eq) in water (137.5 ml, 2.5 vol) and the reaction mixture stirred for one hour at room temperature. The solid obtained was then filtered through a Buchner funnel and dried at 700 mmHg and 500° C. Yield=36.8 g, 48.42%. HPLC purity=99.45%.
EV 320 251 655 US Powder X-ray diffraction (“PXRD”) analysis using a SCINTAG powder X-ray diffϊactometer model X’TRA equipped with a solid-state detector. Copper radiation of λ=1.5418 A was used. The sample was introduced using a round standard aluminum sample holder with round zero background quartz plate in the bottom.
Thermal Gravimetric Analysis TTGA):
TGA/SDTA 85 r, Mettler Toledo , Sample weight 7-15 mg.
Heating rate: 100C/ min., in N2 stream: flow rate: 50 ml/min
Example 1 : Preparation of Tegaserod maleate Form B
To a mixture of 90 g MICHO and 63 g NaOH [47 %] was added a solution of 212 g AGPΗI dissolved in 566 mL of water at room temperature. The resultant reaction mixture was heated to 400C. After 3 hours, 522 mL of ethyl acetate was added and the reaction mixture was stirred for an additional hour. The organic phase was washed with water (3 x 450 mL), and vacuum filtered. After addition of 211 mL ethyl acetate and 870 mL of n-propanol, the mixture was heated to 600C and a solution of maleic acid (86.5 g in 180 mL water), at the same temperature, was added to the reaction mixture and stirred at the same temperature. After 2 hours the reaction mixture was cooled to about 100C and stirred for an additional hour. The resulting solid was filtered off, washed with n-propanol, and dried in a vacuum oven over night to give 195.8 g of tegaserod maleate Form B.
Tegaserod maleate is an aminoguanidine indole 5HT4 agonist for the treatment of irritable bowel syndrome (IBS). Tegaserod maleate has the following structure:
According to the prescribing information (Physician’s Desk Reference, 57th Ed., at Page 2339), tegaserod as the maleate salt is a white to off-white crystalline powder and is slightly soluble in ethanol and very slightly soluble in water. Tegaserod maleate is available commercially as ZELNORM®, in which it is present as crystalline form.
Tegaserod maleate is disclosed in US patent No. 5,510,353 and in its equivalent EP 0 505 322 (example 13), and is reported to have a melting point of 1900C (table 1 example 13).
The literature (Buchheit K.H, et al., J.Med.Chem., 1995, 38, 2331) describes a general method for the condensation of amino guanidines with indole-3-carbadehydes in methanol in the presence of HCl (pH 3-4). The product obtained after solvent evaporation maybe converted to its hydrochloride salt by treatment of the methanolic solution with diethylether/HCl followed by recrystallization from
methanol/diethylether. Tegaserod base prepared according to this general method is characterized solely by a melting point of 155 0C (table 3 compound 5b). Additional Tegaserod maleate characterization was done by 1H and 13C-NMR according to the literature (Jing J. et. al., Guangdong Weiliang Yuansu Kexue, 2002, 9/2, 51).
WO 04/085393 discloses four crystalline forms of tegaserod maleate. The search report for WO 04/085393 further identifies WO 00/10526, and Drugs Fut. 1999, 24(1) which provides an overview for tegaserod maleate. Additional crystalline forms of tegaserod maleate are provided in WO 2005/058819, one of which is characterized by an X-ray Diffraction pattern having peaks at 15.7, 16.9, 17.2, 24.1, 24.6 and 25.2±0.2 two theta (designated as Form B in that PCT publication).
The solid state physical properties of tegaserod salt may be influenced by controlling the conditions under which tegaserod salt is obtained in solid Form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must take that fact into account in developing a tablet or capsule formulation, which may necessitate the use of glidants such as colloidal silicon dioxide, talc, starch or tribasic calcium phosphate.
Another important solid state property of a pharmaceutical compound is its rate of dissolution in aqueous fluid. The rate of dissolution of an active ingredient in a patient’s stomach fluid may have therapeutic consequences since it imposes an upper limit on the rate at which an orally- administered active ingredient may reach the patient’s bloodstream. The rate of dissolution is also a consideration in
formulating syrups, elixirs and other liquid medicaments. The solid state Form of a compound may also affect its behavior on compaction and its storage stability.
These practical physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which defines a particular polymorphic Form of a substance. The polymorphic form may give rise to thermal behavior different from that of the amorphous material or another polymorphic Form. Thermal behavior is measured in the laboratory by such techniques as capillary melting point, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) and may be used to distinguish some polymorphic forms from others. A particular polymorphic Form may also give rise to distinct spectroscopic properties that may be detectable by powder X-ray crystallography, solid state C NMR spectrometry and infrared spectrometry.
The discovery of new polymorphic forms of a pharmaceutically useful compound provides a new opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic.
The polymorphic forms may further help in purification of tegaserod, particularly if they possess high crystallinity. In the event of metastability, a metastable polymorphic form may be used to prepare a more stable polymorph.
Hence, discovery of new polymorphic forms and new processes help in advancing a formulation scientist in preparation of tegaserod as an active pharmaceutical ingredient in a formulation.
The present invention provides an additional polymorphic form of a maleate salt of tegaserod.
Example 1 : Preparation of sesqui-tefiaserod maleate Foπn H2 through tegaserod base
To a mixture of AGPΗI (112.7 g) in 283 mL of water was added 5-MICHO (45 g) followed by NaOH (52.8 g, 47%) and stirred at room temperature. After three hours, 522 mL of ethyl acetate were added and the mixture stirred for an additional four hours. After phase separation at 400C the organic phase was washed with water (3 x 218 ml), and filtrated under vacuum. The resulting solution was heated to 60 0C and a solution of maleic acid (14.4 g) in 45 mL water was dropped during half hour, and the reaction mixture stirred at the same temperature for an additional two hours. The mixture was cooled to 100C during one hour, kept under stirring at the same temperature for 12 hrs and then filtered under vacuum. The wet product was washed twice with 65 ml of ethyl acetate and dried in a vacuum oven at 45°C for 16 hours to give 85% of the product.
Example 2: Preparation of sesqui-tegaserod maleate Form H2
45 gr MICHO were added to a 1 L reactor at RT. A solution of 112.7 gr of AGP HI and 283 ml water was added to the reactor. 52.8 gr of NaOH 47% were added to the mixture while stirring. The mixture was heated to 400C and stirred for 12 hrs. 522 ml of Ethyl Acetate were added and the mixture was stirred for 4 hrs.
After phase separation at 400C the organic phase was washed with water (3 x 218 ml), and filtrated under vacuum.
The mixture was heated to 600C and a mixture o 14.4 gr of Maleic Acid in 45 ml water was dropped during 5 min.
The mixture was stirred at 600C for 2 hrs.
The mixture was cooled to 100C during 1 hour, stirred at 100C for 13 hrs and then filtered under vacuum. The wet product was washed twice with 65 ml of n-Propanol. The wet product was dried in a vacuum oven at 45°C.
Yield: 71.2%
Example 3: Preparation of Tegaserod maleate Form B from Sesqui-tegaserod maleate Form H2
6.9 g of maleic acid were added to a slurry of Sesqui-Tegaserod maleate Form H2 (41.5 g) in 208 ml n-propanol at room temperature. The mixture was stirred for 5 hours at the same temperature, filtered and washed with n-propanol. After drying on vacuum oven at 450C for 15 hours the product was analyzed by XRD and found to be Form B (89% yield).
The formation of hydrazones is catalyzed by both general acids and general bases. General base catalysis of dehydration of the tetrahedral intermediate involves nitrogen deprotonation concerted with elimination of hydroxide ion as shown in the Scheme (Sayer J.M., et al. J. Am. Chem. Soc. 1973, 95, 4277). R fast O I H h° NH2R’ R- -NHR’ R R
In many cases, the equilibrium constant for their formation in aqueous solution is high. The additional stability may be attributed to the participation of the atom adjacent to the nitrogen in delocalized bonding. – + RRC = N – NH2 ~*→- RRC – N = NH2
In order to obtain only the maleic salt, the product when using an acid halide (HA) or other acids has to first be converted into the free base, before the addition of maleic acid (Path a), which results in an additional step to the synthesis. On the other hand, the reaction of the present invention in the presence of organic or inorganic base results in the formation of tegaserod free base which gives only the maleate salt after the addition of maleic acid (Path b).
TGS
TGS-MA
EXAMPLES
HPLC method for detecting the level of the impurities:
Column: Atlantis dcl8(150*4.6),
Mobile phase: A.80% KH2PO4(0.02M) pH=5, 20% acetonitrile(ACN), B.100% ACN. Gradient: time 0= A: 100 B: 0, time 25 min= A:50%, B:50%, time 30 min= A:50%, B:50%, + 10 minutes of equilibration time. Wavelength= 225 nm
Sample concentration: 0.5 mg/mL
Temperature = 25°C
Example 1- Preparation of Tegaserod maleate in water with HCl.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by HCl (37%) until pH 4. The mixture was heated to reflux for 1 hour and then cooled to room temperature. To the resulting slurry was added a solution of NaHCO3 (10%) until pH 9, and heated to 65°C for 20 minutes. After cooling, 100 mL of EtOAc were added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.27 g of crude tegaserod maleate with a purity of 99.70% (by HPLC).
Example 2- Preparation of Tegaserod maleate in water with HCl in two steps. a. Preparation of Tegaserod free base.
To a mixture of AGP-HI (163.3 g, 0.6 mol) in 375 mL water was added 5-MICHO (52.5 g, 0.3 mol) followed by HCl (37%) until pH 4. The mixture was heated to reflux for 1 hour and then cooled to room temperature. To the resulting slurry was added a liter of a solution of NaHCO (10%) until pH 9, and heated to 65 °C for one hour. After cooling, 1500 mL of EtOAc were added, and the organic phase washed with water. The remaining organic phase was evaporated to dryness to give tegaserod free base with a purity of 87.42 % (by HPLC). b. Preparation of Tegaserod maleate. To a solution of 2 g of tegaserod free base in MeOH was added a solution of maleic acid (1.28 g, 0.011 mol) in 10 mL MeOH. The resulting solid was filtered off and washed with MeOH to give 1.09 g of crude tegaserod maleate with a purity of 96.81 % (by HPLC).
Example 3- Preparation of Tegaserod maleate in water with TEA.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by TEA (11.0 mL, 0.08 mol) and stirred at room temperature. After one hour, 25 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 7.92 g of crude tegaserod maleate with a purity of 94 % (by HPLC).
Example 4- Preparation of Tegaserod maleate in water with NaHCO3. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaHCO3 (6.72 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, 50 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.71 g of crude tegaserod maleate with a purity of 98 % (by HPLC) .
Example 5- Preparation of Tegaserod maleate in water with NaHCO3 in two steps. a. Preparation of Tegaserod free base. To a mixture of AGP-HI (32.66 g, 0.12 mol) in 300 mL water was added 5-MICHO (10.51 g, 0.06 mol) followed by NaHCO3(20.16 g, 0.24 mol) and heated to reflux for 1 hour. After cooling, 150 mL of EtOAc was added, and the organic phase washed with water and evaporated to dryness to give 20.4 g of tegaserod free base (91.55%) purity by HPLC). b. Preparation of Tegaserod maleate.
To a solution of 2g of the resulting tegaserod free base in 8 mL MeOH was added a solution of maleic acid (1.28 g, 0.011 mol) in 5 mL MeOH. The resulting solid was filtered off and washed with MeOH to give 2.1 g of crude tegaserod maleate with a purity of 99.63 % (by HPLC).
Example 6- Preparation of Tegaserod maleate in water with Na2CO3. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by Na2CO3 (4.24 g, 0.04 mol) and heated to reflux for 1 hour. After cooling, 50 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.48 g of crude tegaserod maleate with a purity of 98.2 % (by HPLC).
Example 7- Preparation of Tegaserod maleate in MeOH with TEA in two steps. a. Preparation of tegaserod free base
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL MeOH was added 5-MICHO (3.50 g, 0.02 mol) followed by triethylamine (11.0 mL, 0.08 mol). After 1 h at room temperature the mixture was evaporated to dryness, and washed with water, giving 5.79 g of tegaserod free base (86.90 % purity by HPLC). b. Preparation of tegaserod maleate
To a solution of 2 g of the resulting tegaserod free base in 10 mL MeOH was added a solution of maleic acid (1.16 g, 0.01 mol) in water. The resulting solid was filtrated and washed with water to give 1.45 g of crude tegaserod maleate as a white solid (94.60 % purity by HPLC). Crystallization in MeOH improved the purity to 98.94% by HPLC.
Example 8- Preparation of Tegaserod maleate in IPA with K2CO3.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL IPA was added 5-MICHO (3.50 g, 0.02 mol) followed by K2CO3 (5.53g, 0.04 mol). After 22 h at room temperature the mixture was washed with brine. The organic phase was treated with a solution of maleic acid (3.48 g, 0.03 mol) in IPA. The resulting solid was filtrated and washed with IPA to give 3.26 g of a white solid (98.97% purity by HPLC).
Example 9- Preparation of Tegaserod maleate in TEA.
To a mixture of AGP-HI (10.88 g, 0.04 mol) and 5-MICHO (3.50 g, 0.02 mol) was added 11 mL of TEA (0.08 mol). After 2 h at room temperature 25 mL of EtOAc were added and the mixture was stirred for 1 h. The resulting solid was filtrated and washed with 25 mL EtOAc, to give 5.7 g of crude.
2 g of the residue was dissolved in 13 mL MeOH and treated with 7 mL of a solution of maleic acid (2.7 g, 0.023 mol) in water. The resulting solid was filtered and washed with water to give 1.5 g of tegaserod maleate (99.26 % purity by HPLC). Crystallization of the solid in MeOH improved the purity to 99.89%) by HPLC.
Example 10- Preparation of Tegaserod maleate in toluene/water with NaHCO3. a. Preparation of tegaserod free base To a mixture of AGP-HI (10.88 g, 0.04 mol) in 200 mL of water/toluene 1:1 was added 5-MICHO (3.50 g, 0.02 mol) followed by NaHCO3 (6.72 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.25 g of tegaserod free base was obtained (93.8 % purity by HPLC). b. Preparation of tegaserod maleate To a solution of 3 g of the product in 10 mL MeOH was added a solution of maleic acid (2.31 g, 0.02 mol) in 10 mL water. The resulting solid was filtered off and washed with a solution of MeOH / water to give 2.50 g of crude tegaserod maleate with a purity of 96.6 % (by HPLC).
Example 11- Preparation of Tegaserod maleate in water with NaOH. a. Preparation of tegaserod free base
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaOH (2 g, 0.05 mol) and stirred at room temperature. After 3 hours 50 mL of EtOAc was added, and the organic phase washed with water and evaporated to dryness to give 5.6 g of tegaserod free base (98.80% purity by HPLC). b. Preparation of Tegaserod maleate.
To a solution of 1.6 g of tegaserod free base in 15 mL ethyl acetate was added a solution of maleic acid (0.7 g, 0.006 mol) in 5 mL ethyl acetate. The resulting solid was filtered off and washed with ethyl acetate to give 1.65 g of crude tegaserod maleate, with a purity of 99.87 % (by HPLC)
Example 12- Preparation of Tegaserod maleate in water with maleic acid. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by maleic acid (9.3 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.92 g of tegaserod maleate crude was obtained (92.4 % purity by HPLC).
Example 13- Preparation of Tegaserod maleate in methanol with maleic acid.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of methanol was added 5- MICHO (3.50 g, 0.02 mol) followed by maleic acid (9.29 g, 0.08 mol) and heated to reflux for 2 hours. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.51 g of tegaserod maleate crude was obtained (97.4 % purity by HPLC).
Example 14- Preparation of Tegaserod maleate in water with NaOH in one pot. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaOH (2 g, 0.05 mol) and stirred at room temperature. After 4 hours a solution of maleic acid (4.35 g, 0.0375 mol) in 25 mL water was added, and the reaction mixture was stirred overnight. The resulting solid was filtered off and washed with water to give 7.87 g of crude tegaserod maleate (99.16% purity by HPLC).
Example 15- Preparation of Tegaserod maleate in water with NaOH in one pot.
To a mixture of AGP-HI (174.2 g, 0.64 mol) in 362 mL of water was added 5-MICHO (56.2 g, 0.32 mol) followed by NaOH (68.1 g, 47%) and stirred at room temperature. After 4.5 hours, 640 mL of EtOAc was added, and the organic phase washed with water, treated with active carbon and filtrated through hyper flow bed. A solution of maleic acid (44.57 g, 0.38 mol) in 415 mL ethyl acetate / water 97:3 was added, and the reaction mixture was heating to 65 °C and stirrer overnight. The resulting solid was filtered off and washed with water and ethyl acetate to give 121.4 g of crude tegaserod maleate (up to 99.88 % purity by HPLC).
Example 16- Preparation of Tegaserod maleate (from Tegaserod acetate).
To a solution of 8.2 g of tegaserod acetate in 15 mL ethyl acetate heated to 65 °C was added a solution of 3.3 g maleic acid in 5 ml ethyl acetate/water 95:5, and the mixture was stirred at the same temperature for an additional 2 hours, followed by cooling to room temperature and stirring overnight. The resulting solid was filtered off and washed with ethyl acetate/water 95:5. After drying on vacuum oven at 45 °C for 15 hours, 9.18 g of tegaserod maleate were obtained. Tegaserod acetate is prepared according to Examples 19, 20 and 21 of U.S. Appl. No. 11/015,875 and PCT/US04/42822.
Example 19 of U.S. Appl. No. 11/015,875 reads as follows: A slurry of tegaserod base amorphous (6 g) in 50 mL ethyl acetate was stirred at 20- 30 °C for 24 hours. The solid was filtrated and washed with 15 mL of same solvent and dried in a vacuum oven at 40 °C for 16 hours.
Example 20 of U.S. Appl. No. 11/015,875 reads as follows:
A slurry of tegaserod base amorphous (6 g) in 50 mL ethyl acetate was stirred at reflux for 24 hours. The solid was filtrated and washed with 15 mL of same solvent and dried in a vacuum oven at 40 °C for 16 hours.
Example 21 of U.S. Appl. No. 11/015,875 reads as follows:
To a slurry of tegaserod maleate Form A (15 g) in EtOAc (210 mL) and water (210 mL) was added 38.4 g of NaOH 47%. The mixture was stirred overnight and the resulting white solid was isolated by filtration and washed with 100 mL of water. Drying in vacuum oven at 40 °C for 16 hours gives 12.38 g (90% yield). Tegaserod acetate was characterized by H and C-NMR.
Example 17: General method for the preparation of Tegaserod maleate Form A from crystallization.
Tegaserod maleate (1 g) was combined with the appropriate solvent (5 mL), and heated to reflux. Then, additional solvent was added until complete dissolution. After the compound was dissolved, the oil bath was removed and the solution was cooled to room temperature. The solid was filtrated and washed with 5 mL of the same solvent and dried in a vacuum oven at 40 C for 16 hours.
Example 18: Preparation of Tegaserod maleate in water with p-TSOH.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by para-toluenesulfonic acid monohydrate (0.45 g, 0.0024 mol). The mixture was heated to reflux for 4 hour and then cooled to room temperature. The resulting solid was filtered off and washed with water to give 8.32 g of a white solid (84.74 % purity by HPLC).
Example 19: Preparation of Tegaserod maleate from Tegaserod Hemi-maleate hemihydrate
To a solution of 1.72 g of Tegaserod Hemi-maleate hemihydrate in 20 mL ethyl acetate at room temperature was added a solution of 0.134 g maleic acid in 5 ml ethyl acetate/water 95:5, and the mixture was stirred at the same temperature for overnight. The resulting solid was filtered off and washed with ethyl acetate/water 95:5. After drying on vacuum oven at 45°C for 15 hours, 1.68 g of tegaserod maleate were obtained. Tegaserod Hemi-maleate hemihydrate was prepared according to Example 23 of U.S. Appl. No. 11/015,875 and PCT/US04/42822. Example 23 of U.S. Appl. No. 11/015,875 and PCT/US04/42822 reads as follows: A solution of maleic acid (2.32 g in 22 mL ethyl acetate/water 97:3) was added to a mixture of tegaserod base in ethyl acetate, and the reaction mixture was heated to 65 °C and stirrer overnight. The resulting solid was filtered off and washed with water and ethyl acetate. Drying in vacuum oven at 40 °C for 16 hours gives 12.19 g of Tegaserod hemi-maleate hemihydrate. Depending on the base polymorph used a solution or slurry is obtained. When using amorphous tegaserod base, a solution is obtained, while when using any other base polymorph of tegaserod, a slurry is obtained.
Tegaserod, chemically named 2-[(5-methoxy-liϊ-indol-3-yl)methylene]-IV-pentylhydrazine- carboximidamide, is a selective serotonin 4 (5-HT4) receptor agonist, which can be used to treat gastrointestinal disorders such as heartburn, bloating, postoperative ileus, abdominal pain and discomfort, epigastric pain, nausea, vomiting, regurgitation, intestinal pseudoobstruction, irritable bowel syndrome and gastro-oesophageal reflux. Tegaserod as the maleate salt is marketed for the short-term treatment of irritable bowel syndrome in women whose primary bowel symptom is constipation.
Tegaserod, represented by the formula (I), was first described in US 5 510 353 as well as processes for its preparation. The maleate salt of tegaserod is also disclosed, but interestingly a method of manufacturing tegaserod maleate is not disclosed. The only characterizing data is the melting point which is disclosed as 1900C for the maleate salt and 124°C for the tegaserod base.
WO 2006/116953 describes crystalline forms of the hydrobromide, dihydrogen phosphate and oxalate salts of tegaserod. Also claimed is a process for preparing the hydrochloride, hydrobromide, dihydrogen phosphate, tartrate, citrate, lactate, mesylate, oxalate, succinate, glutarate, adipate, salicylate, sulfate, mandelate, camphor sulfonate and hydrogen sulfate salts of tegaserod from a specific crystalline form of tegaserod base. Another process described is a method of preparing the dihydrogen phosphate, maleate, tartrate, citrate, mesylate, lactate, succinate, oxalate, hydrochloride, salicylate, glutarate, adipate, hydrobromide, sulfate and hydrogen sulfate from a hydrogen halide salt of tegaserod.
There are often major hurdles to overcome before an active pharmaceutical ingredient (API) can be formulated into a composition that can be marketed. For example, the rate of dissolution of an API that has poor aqueous solubility is often problematic. The aqueous solubility is a major influence on the bioavailability of the API such that a poorly soluble API can mean the API is not available to have a pharmaceutical effect on the body. The API can also cause problems during manufacture of a pharmaceutical composition. For example, flowability, compactability and stickiness are all factors affected by the solid state properties of an API.
It has thus always been an aim of the pharmaceutical industry to provide many forms of an API in order to mitigate the problems described above. Different salts, crystalline forms also known as polymorphs, solvates and amorphous forms are all forms of an API that can have different physiochemical and biological characteristics. Indeed, it has been discovered that the tegaserod maleate product on the market, Zelnorm , has been linked to an increase in heart problems in a proportion of individuals. One possible reason is that the maleate moiety reacts with the tegaserod, resulting over time in the production of a toxic impurity.
This impurity could be a contributor to the heart problems seen in some patients.
Figure 1 is a x-ray powder diffraction pattern of tegaserod maleate Form I. Figure 2 is a x-ray powder diffraction pattern of tegaserod maleate Form II. Figure 3 is a x-ray powder diffraction pattern of tegaserod maleate Form III. Figure 4 is a x-ray powder diffraction pattern of tegaserod maleate Form IV. x-Ray powder diffraction spectrum was measured on a Siemens D5000 x- ray powder diffractometer having a copper-Kα radiation.
The following examples further illustrate the invention.
Example 1 Tegaserod free base (10 gm) is dissolved in acetone (100 ml). Maleic acid (4 gm) is added to the solution and the contents are maintained for 1 hour at 25°C. The separated solid is filtered to give 12.5 gm of tegaserod maleate Form I.
Example 2 Tegaserod maleate Form II (5 gm) and acetone (70 ml) are mixed and refluxed for 1 hour and cooled to 25°C and filtered to give 4.8 gm of tegaserod maleate Form I.
Example 3 Tegaserod maleate Form I (10 gm) is dissolved in methanol (100 ml). Acetonitrile (150 ml) is added to the solution and the contents are heated to reflux. The contents are then cooled to 25°C and maintained for 30 minutes. The separated crystals are collected by filtration to give 9 gm of tegaserod maleate Form II.
Example 4 Tegaserod free base (10 gm) is dissolved in methanol (100 ml) and maleic acid (4 gm) is added to the solution. Then the contents are maintained for 30 minutes at 25°C. Then the separated solid is filtered to give 13 gm of tegaserod maleate Form III.
Example 5
Tegaserod maleate (5 gm) is dissolved in methanol (50 ml) and the solution is maintained at 25°C for 30 minutes. The separated crystals are collected by filtration to give 4.8 gm of tegaserod maleate Form III. Example 6 Tegaserod free base (10 gm) is dissolved in methanol (50 ml), maleic acid (4 gm) is added and the contents are refluxed for 30 minutes and then the resulting solution is cooled to 25°C. Methylene dichloride (200 ml) is added and the contents are maintained for 30 minutes at 25°C. The separated solid is collected by filtration to give 13 gm of tegaserod maleate Form IV.
Example 7 Maleic acid (4 gm) is added to a solution of tegaserod free base (10 gm) in methanol (50 ml). The contents are maintained for 30 minutes at 25°C and isopropyl alcohol (150 ml) is mixed and contents are maintained for 30 minutes at 25°C. The separated solid is collected by filtration to give 12.5 gm of tegaserod maleate Form IV
Beattie DT, Smith JA, Marquess D, Vickery RG, Armstrong SR, Pulido-Rios T, McCullough JL, Sandlund C, Richardson C, Mai N, Humphrey PP: The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo. Br J Pharmacol. 2004 Nov;143(5):549-60. Epub 2004 Oct 4. [PubMed:15466450]
Talley NJ: Irritable bowel syndrome. Intern Med J. 2006 Nov;36(11):724-8. doi: 10.1111/j.1445-5994.2006.01217.x. [PubMed:17040359]
Borman RA, Tilford NS, Harmer DW, Day N, Ellis ES, Sheldrick RL, Carey J, Coleman RA, Baxter GS: 5-HT(2B) receptors play a key role in mediating the excitatory effects of 5-HT in human colon in vitro. Br J Pharmacol. 2002 Mar;135(5):1144-51. doi: 10.1038/sj.bjp.0704571. [PubMed:11877320]
Vickers AE, Zollinger M, Dannecker R, Tynes R, Heitz F, Fischer V: In vitro metabolism of tegaserod in human liver and intestine: assessment of drug interactions. Drug Metab Dispos. 2001 Oct;29(10):1269-76. [PubMed:11560869]
FDA approves the reintroduction of Zelnorm™ (tegaserod) for Irritable Bowel Syndrome with Constipation (IBS-C) in women under 65 [Link]
EMA Refusal Assessment Report for Zelnorm (Tegaserod) [File]
FDA Joint Meeting of the Gastrointestinal Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee Briefing Document for Zelnorm (tegaserod maleate) [File]
BUCHHEIT K H ET AL: “THE SEROTONIN 5-HT4 RECEPTOR. 2. STRUCTURE-ACTIVITY STUDIES OF THE INDOLE CARBAZIMIDAMIDE CLASS OF AGONISTS” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 38, 1995, pages 2331-2338, XP000867864 ISSN: 0022-2623 cited in the application *
GRAUL A ET AL: “TEGASEROD MALEATE” DRUGS OF THE FUTURE, BARCELONA, ES, vol. 24, no. 1, 1999, pages 38-44, XP000874672 ISSN: 0377-8282 *
LALEZARI ET AL.: “Selective synthesis of …” J. HETEROCYCL. CHEM., vol. 8, 1971, pages 689-691, XP002354978 *
WAN ET AL.: “Improved synthesis of tegaserod maleate” CHINESE J. MED. CHEM., vol. 13, no. 1, 2003, pages 40-41, XP009057178 *
WO2004085393A1 *2003-03-252004-10-07Hetero Drugs LimitedNovel crystalline forms of tegaserod maleate
WO2006116953A1 *2005-05-022006-11-09Zentiva, A.S.A method for the preparation of tegaserod and slected salts thereof
WO2007084761A1 *2006-01-182007-07-26Teva Pharmaceutical Industries Ltd.Maleate salt of tegaserod and crystalline forms thereof
WO2007084761A1 *2006-01-182007-07-26Teva Pharmaceutical Industries Ltd.Maleate salt of tegaserod and crystalline forms thereof
WO2007120924A1 *2006-04-172007-10-25Teva Pharmaceutical Industries Ltd.Preparation of tegaserod maleate free of iodide
WO2007126889A1 *2006-03-272007-11-08Teva Pharmaceutical Industries Ltd.Preparation of tegaserod acetate
WO2007146717A3 *2006-06-122008-03-27Joginder S BajwaProcess for making salts of n-hydroxy-3-[4-[[[2-(2-methyl-1h-indol-3-yl)ethyl]amino]methyl]phenyl]-2e-2-propenamide
EP1955998A1 *2007-02-072008-08-13Chemo Ibérica, S.A.New addition salt of N-amino-N’-pentylguanidine, the process for its preparation and use thereof for obtaining tegaserod
WO2010015794A1 *2008-08-072010-02-11Generics [Uk] LimitedNovel polymorphic forms of tegaserod
CL2008000070A1 *2007-01-172008-07-25Lg Life Sciences LtdMaleic acid mono (3 – [({1 – [(2-amino-9 H -purin-9-yl) methyl] cyclopropyl} oxy) methyl] -8,8-dimethyl-3,7-dioxo-2,4 , 6-trioxa-3 lambda 5 -phosphanon-1-yl pivalate; pharmaceutical composition comprising said mono, and use to treat virus h
US5510353A *1991-03-221996-04-23Sandoz Ltd.Certain aminoguanidine compounds, pharmaceutical compositions containing them and their use in treating gastrointestinal motility disorders and disorders associated with cephalic pain
US20060178519A1 *2004-12-232006-08-10Venkataraman SundaramProcess for preparing tegaserod
Family To Family Citations
WO2006116953A1 *2005-05-022006-11-09Zentiva, A.S.A method for the preparation of tegaserod and slected salts thereof
CN100412059C *2006-06-062008-08-20江苏奥赛康药业有限公司Preparation method of tegaserod
31 Jan 2018 Phase-I clinical trials in Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA, Belgium, Italy, Japan (Intratumoural) (NCT03301896)
31 Jan 2018 Phase-I clinical trials in Solid tumours (Inoperable/Unresectable, Late-stage disease, Metastatic disease, Monotherapy, Second-line therapy or greater) in USA, Japan, Italy, Belgium (Intratumoural) (NCT03301896)
10 Oct 2017 Novartis plans a phase I trial for Solid tumours (Monotherapy, Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA, Belgium, Canada, France, Germany, Italy, South Korea and Spain in November 2017 (Intratumoural) (NCT03301896)
[517] To a solution of tert-butyl 5-bromo-2-chlorophenylcarbamate (6-1) (1.0 equiv.) in acetonitrile (0.3 M) and EtOH (0.5 M) was added K2C03 (2.0 equiv.). The reaction was degassed and flushed with N , then added (E)-ethyl 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)acrylate (6-2) (1.2 equiv.) and Pd(PPh3)4 (0.1 equiv.). The reaction was flushed again with N2 and stirred at 100 °C overnight. After cooling to room temperature, hexane was added, and the mixture was filtered through a pad of silica, eluting with EA/Hex (1 : 1) until the product was completely eluted. The filtrate was concentrated and purified on Combiflash, eluting with 0-15% EA in Hex to give (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3) as a white solid.
[518] To a solution of (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3) (1.0 equiv.) in ethyl acetate/ethanol (1 : 1 , 0.3 M) was added Wilkinson’s catalyst (0.10 equiv.).
Hydrogen gas was introduced via a ballon, and the reaction was stirred at room temperature for 24 hours. The mixture was filtered through a pad of celite, washing with dichloromethane. The filtrate was concentrated in vacuo and purified by Combiflash using 0-10% ethyl acetate in hexane to give ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4) as a solid.
[519] A solution of ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4) (1 .0 equiv.), 4,4,4,,4′,5,5,5′,5′-octamethyl-2,2′-bi(l ,3,2-dioxaborolane) (2.0 equiv.), tris(dibenzylideneacetone)dipalladium(0) (0.05 equiv.), 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (0.20 equiv.), and potassium acetate (2.0 equiv.) in 1 ,4-dioxane (0.2 M) was degassed and stirred at 100 °C overnight. After cooling to ambient temperature, the reaction content was concentrated in vacuo. The crude material was purified by Combiflash using 0-50% ethyl acetate in hexane to afford ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5) as a brown oil. The product was stored at -20°C and used within a month of synthesis.
[520] To a solution of 4-bromo-3-methylphenol (6-6) (1.0 equiv.) in DMF (0.5 M) at 0 °C was added portionwise 60% wt NaH (1.5 equiv.). The addition was controlled such that internal reaction temperature never went above 10 °C. The reaction was stirred at room temperature for 45 minutes, then a solution of chloro(methoxy)methane (1.2 equiv.) in DMF (3 M) was added dropwise via additional funnel. The reaction was stirred at room temperature for 3.5 hours, and then quenched by pouring into ice. The resulting mixture was stirred at room temperature for 1 hour. Ether was added, and the two layers were separated. The aqueous layer was extracted (lx) with ether. The combined organic layers were washed with water (2x), brine, dried over MgS04, and concentrated to give 1 -bromo-4-(methoxymethoxy)-2-methylbenzene (6-7) as a colorless oil. The crude material was used in the next step without further purification.
[521] A solution of l -bromo-4-(methoxymethoxy)-2-methylbenzene (1.0 equiv.), triethylamine (5.0 equiv.) in DMF (0.5 M) was degassed and flushed with nitrogen. To the reaction was added TES-acetylene (1.05 equiv.), Cul (0.098 equiv.), and Pd(PPh3)2Cl2 (0.098 equiv.). The reaction was heated to 60 °C and stirred overnight. After cooling to room temperature, water and ether were added. The layers were separated, and the organic layer was washed with water (2x). The organic layer was separated and passed through a pad of silica (packed with hexane). The silica was eluted with 10% EA in Hex. The fractions were combined and concentrated to give triethyl((4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane as a black oil. The crude material was used in the next step without further purification.
[522] To a solution of triethyl((4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane (1.0 equiv.) at
0 °C was slowly added tetrabutylammonium fluoride (1M solution in THF, 0.20 equiv.). At this
point, the ice-bath was removed and the reaction mixture was allowed to stir at room temperature for 45 minutes. The reaction mixture was then passed through a pad of silica (packed with hexane) and eluted with 20% EtOAc in Hexanes to remove insoluble salts. The crude product was then purified by Combiflash using 0-10% EtOAc in Hexanes to give 1 -ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8) as a slightly brown liquid.
[523] A solution of l -ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8) (1 .0 equiv.), 3,5-dichloropicolinonitrile (6-9) (0.90 equiv.), Cul (0.10 equiv.), and Pd(PPh3)2CI2 (0.10 equiv.), and triethylamine (5.0 equiv.) in DMF (0.25 M) was degassed and flushed with nitrogen. The reaction mixture was then heated to 60 °C and stirred overnight. After cooling to room temperature, water was added. The mixture was extracted with EA (2x). The combined organic layers were washed with 10% aq NH4OH (2x), brine, and concentrated. The crude material was filtered through a pad of silica (wetted with hexane). The silica was eluted with 10% EA in Hex. The fractions were combined and concentrated. The resulting solids were washed in hot ether and filtered to give a yellow solid, which was used in the next step without further purification. The filtrate was concentrated and purified by Combiflash using 0- 10% EtOAc in Hexanes to give 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10) as a yellow solid.
[524] A solution of 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10) (1 .0 equiv.), ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5) (1.25 equiv.), tris(dibenzylideneacetone)dipalladium(0) (0.10 equiv.), dicyclohexyl(2′,6′-dimethoxybiphenyl-2-yl)phosphine (0.20 equiv.), and sodium bicarbonate (3.0 equiv.) in «-butanol /H20 (5: 1 , 0.2 M) was degassed and stirred at 100 °C overnight. After cooling to ambient temperature, the reaction content was diluted with ethyl acetate and water. The two phases were separated, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous MgS04, and concentrated in vacuo. The crude material was purified by flash chromatography on a COMBIFLASH® system (1SCO) using 0-40% ethyl acetate in DCM first to remove the impurity, then 0-4% MeOH in DCM to give ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-benzo[f][l ,7]naphthyridin-8-yl) propanoate (6-11). Further purification was accomplished by precipitating and washing in hot ether.
[525] A solution of ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-11) (1.0 equiv.) in EtOH/THF (3: 1 , 0.16 M) was flushed with nitrogen. Then, 10% wt Pd/C (0.20 equiv. by weight) was added. The reaction was flushed with hydrogen (2x) and stirred under a hydrogen balloon. After 24 hours, the reaction was filtered through a pad of celite, washing with 5%MeOH in DCM. The filtrate was checked for the presence of starting material using LCMS. The hydrogenation reaction was repeated until no more
of the alkyne starting material or alkene intermediate was detected. The crude product was purified by Combiflash using 0-4% eOH in DCM to give ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-12) as a white solid.
[526] Ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[fJ[l ,7]naphthyridin-8-yl)propanoate (6-12) (1 .0 equiv.) was dissolved in EtOH (0.2 M), then added a solution of 4M HC1 in dioxane (0.2 M). The product precipitated out as a yellow salt. After stirring for 3 hours, the reaction was poured into a stirring solution of ether. The mixture was stirred for 10 minutes, then filtered and washed with ether. Ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fJ[l ,7]naphthyridin-8-yl)propanoate (6-13) was obtained as a yellow solid which was dried on vacuum overnight (bis-HCl salt). Alternatively, the crude product was purified by Combiflash using 0-5% MeOH in DCM to give the free base.
[527] To a solution of ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fJ [ l ,7]naphthyridin-8-yl)propanoate (6-13) (1.0 equiv.) dissolved in DMF (0.14 M) was added a solution of diethyl 3-(2-bromoethoxy)-l ,l -difluoropropylphosphonate (6-14: described in Example 7 – Step 1) (1 .3 equiv.) in DMF (0.7 M) and cesium carbonate (4 equiv.). The reaction was stirred at 60 °C. After 1.5 hours (or until reaction is complete by LCMS), DCM (2 volume equivalent) was added to the reaction. The solids (inorganic) were filtered, and the filtrate was concentration. The crude product was purified by Combiflash using 0-5%MeOH in DCM to give ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[fJ
[1 ,7]naphthyridin-8-yl)propanoate (6-15) as an oil which upon standing became a white solid.
[528] To a solution of ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-15) (1.0 equiv.) in DCM (0.16 M) at 0 °C was added slowly TMSBr (10 equiv.). The reaction was stirred at room temperature overnight. Additional TMSBr (5.0 equiv.) was added at 0 °C, and the reaction was again stirred at room temperature overnight. The solvent was removed by evaporation and the crude orange solids dried on hi-vac briefly. The solids were suspended in EtOH (0.5 M) and added 2.5 N
NaOH (10.0 equiv.). The reaction was stirred at 80 °C for 3 hours. After cooling to room temperature, the mixture was adjusted to pH 9 to 10 and directly purified on RP-HPLC using a CI 8 column, eluting with 10-40% 95:5 (MeCN/5mM NH4OAc) in l OmM NH4OAc (pH 9) gradient. The fractions containing the product were combined and concentrated in vacuo. The resulting white gel was dissolved in refluxing 1 :1 EtOH/water (0.04 M) with the addition of a few drops of ammonium hydroxide. While hot, the mixture was slowly poured into a stirring hot solution of acetone (0.009
M) preheated at 50 °C. The acetone suspension was slowly cooled to room temperature for 15 minutes with continued stirring, and then sat in an ice bath for 10 minutes. The solids were filtered and washed successively with acetone (2x) and ether (2x). The solids were dried on hi-vac overnight to give the 3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphonopropoxy)ethoxy)-2-methylphenethyl)benzo [fj[l ,7]naphthyridin-8-yl)propanoic acid (19) as a solid. Ή NMR (Dimethylsulfoxide-d6): δ 9.02 (s, 1 H), 8.82 (s, 1H), 8.55 (d, 1H, J = 8.4 Hz), 7.58 (s, 1H), 7.48 (d, 1 H, J = 8.4 Hz), 7.07 (d, 1H, J = 8.4 Hz), 6.75 (s, 1 H), 6.68 (d, 1H, J = 8.4 Hz), 4.03-4.00 (m, 2H), 3.72-3.68 (m, 4H), 3.16-3.12 (m, 2H), 3.03-2.96 (m, 4H), 2.67-2.64 (m, 2H), 2.33-2.32 (m, 2H), 2.26 (s, 3H). LRMS [M+H] = 604.2
Toll-like receptors (TLRs) are pattern recognition receptors which play an essential role in the innate immunity, by recognizing invasion of microbial pathogens and initiating intracellular signal transduction pathways to trigger expression of genes, the products of which can control innate immune responses. Specifically, Toll like receptor (TLR) agonists activate innate immune cells through the TLR-MyD88-NFk and IRF3/7 pathways. TLR7, TLR8, and TLR9 belong to a subfamily of TLRs based on their genomic structure, sequence similarities, and homology. TLR7, TLR8, and TLR9 are located in intracellular endolysosomal compartments and show a unique pattern of cell type-specific expression that is thought to be responsible for different pathogen response profiles.
Small molecule agonists of TLR7 and/or TLR8 have been reported and shown to activate innate immune responses by inducing selected cytokine biosynthesis, the induction of co-stimulatory molecules, and by increased antigen-presenting capacity. Such compounds include imidazoquinoline amine derivatives (U.S. Patent No. 4689338), imidazopyridine amine derivative (U.S. Patent No. 5446153), imidazonaphthyridine derivative (U.S. Patent No.
benzonaphthyridine amine derivatives (U.S. Patent Nos. 8466167 and 9045470).
The synthetic TLR7 agonist, Imiquimod (1 -(2-methylpropyl)-1 H-imidazo[ 4,5-c]quinolin-4-amine) is FDA-approved in a cream formulation for the topical treatment of cutaneous basal cell carcinoma, actinic keratosis and genital warts, and has limited activity against cutaneous melanoma and breast tumors (J. Immunol. 2014, 193(9) : 4722^1-731 ). Systemic administration of Imiquimod, and structurally similar Resiquimod, is limited by cytokine- mediated adverse effects including severe flu-like symptoms (Expert Opin. Emerging Drugs (2010), 15:544-555). Consequently, Imiquimod is used exclusively in topical applications and is not used to treat deep, non-cutaneous tumors such as melanoma or solid tumors.
An injectable lipid modified imidazoquinoline (TLR7/8 dual agonist) that forms a tissue depot with gradual, sustained release which allows for local TLR triggering activity without systemic cytokine release has been reported (J. Immunol. 2014, 193(9): 4722^731 ). However, this compound was shown to be ineffective for large tumors and in addition the serum concentration of this compound 24 hours post subcutaneous administration decreased by approximately 50% (Journal for ImmunoTherapy of Cancer, 2014, 2:12). Therefore, there remains a need for intratumor administration of a TLR7 agonist with prolonged sustained release, which may benefit the treatment of large tumors.
Presenter: Alex Cortez, senior Investigator I at the Genomics Institute of the Novartis Research Foundation
Target: Toll-like receptor 7 (TLR7)
Disease: Solid tumors
Reporter’s notes: Cortez shared another story in the realm of immuno-oncology, although the program that yielded this compound actually started in the world of vaccines. Cortez’s team had been focusing on vaccine adjuvants, small molecules that turn on the immune system to enhance a vaccine’s effect. They developed one such class of compound that activates toll-like receptor 7 (TLR7), a protein in the immune system that recognizes dangerous-looking molecules and can trigger the release of infection-clearing proteins. After observing TLR7 agonists’ ability to induce an immune response with vaccines, the researchers wondered whether the molecules could also be effective in immuno-oncology.
They found that LHC165 adsorbed to aluminum hydroxide reduced tumor growth in mice and, intriguingly, showed signs of an abscopal effect, in which untreated tumors shrink concurrently with treated tumors. The implication is that if the immune system recognizes one tumor site, it can recognize others. As with several of the candidates presented throughout the day, LHC165 bears a phosphate group and is injected into the tumor. It’s currently in Phase I trials in patients with advanced malignancies, which means they’ve already tried second and third line therapies, as a single agent and in combination with the checkpoint inhibitor PDR001.
US9618508FLOW CYTOMETRY ANALYSIS OF MATERIALS ADSORBED TO METAL SALTS2011-12-142013-12-12
US2014112950COMBINATION VACCINES WITH LOWER DOSES OF ANTIGEN AND/OR ADJUVANT2012-03-022014-04-24
Asciminib is an orally bioavailable, allosteric Bcr-Abl tyrosine kinase inhibitor with potential antineoplastic activity. Designed to overcome resistance, ABL001 binds to the Abl portion of the Bcr-Abl fusion protein at a location that is distinct from the ATP-binding domain. This binding results in the inhibition of Bcr-Abl-mediated proliferation and enhanced apoptosis of Philadelphia chromosome-positive (Ph+) hematological malignancies. The Bcr-Abl fusion protein tyrosine kinase is an abnormal enzyme produced by leukemia cells that contain the Philadelphia chromosome.
ABL001 has been used in trials studying the health services research of Chronic Myelogenous Leukemia and Philadelphia Chromosome-positive Acute Lymphoblastic Leukemia.
Class Antineoplastics; Pyrazoles; Pyrrolidines; Small molecules
Mechanism of Action Bcr-abl tyrosine kinase inhibitors
Highest Development Phases
Phase III Chronic myeloid leukaemia
No development reported Precursor cell lymphoblastic leukaemia-lymphoma
Most Recent Events
04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in Australia (PO)
04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in France (PO)
04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in Germany (PO)
The tyrosine kinase activity of the ABLl protein is normally tightly regulated, with the N-terminal cap region of the SH3 domain playing an important role. One regulatory mechanism involves the N-terminal cap glycine-2 residue being myristoylated and then interacting with a myristate binding site within the SHI catalytic domain. A hallmark of chronic myeloid leukemia (CML) is the Philadelphia chromosome (Ph), formed by the t(9,22) reciprocal chromosome translocation in a haematopoietic stem cell. This chromosome carries the BCR-ABL1 oncogene which encodes the chimeric BCR-ABL1 protein, that lacks the N-terminal cap and has a constitutively active tyrosine kinase domain.Although drugs that inhibit the tyrosine kinase activity of BCR-ABL1 via an ATP-competitive mechanism, such as Gleevec® / Glivec® (imatinib), Tasigna® (nilotinib) and Sprycel® (dasatinib), are effective in the treatment of CML, some patients relapse due to the emergence of drug-resistant clones, in which mutations in the SHI domain compromise inhibitor binding. Although Tasigna® and Sprycel® maintain efficacy towards many Gleevec-resistant mutant forms of BCR-ABLl, the mutation in which the threonine-315 residue is replaced by an isoleucine (T315I) remains insensitive to all three drugs and can result in CML patients developing resistance to therapy. Therefore, inhibiting BCR-ABLl mutations, such as T315I, remains an unmet medical need. In addition to CML, BCR-ABLl fusion proteins are causative in a percentage of acute lymphocytic leukemias, and drugs targeting ABL kinase activity also have utility in this indication.Agents targeting the myristoyl binding site (so-called allosteric inhibitors) have potential for the treatment of BCR-ABLl disorders (J. Zhang, F. J. Adrian, W. Jahnke, S. W. Cowan- Jacob, A. G. Li, R. E. Iacob4, T. Sim, J. Powers, C. Dierks, F. Sun, G.-R. Guo, Q. Ding, B. Okram, Y. Choi, A. Wojciechowski, X. Deng, G. Liu, G. Fendrich, A. Strauss, N. Vajpai, S. Grzesiek, T. Tuntland, Y. Liu, B. Bursulaya, M. Azam, P. W. Manley, J. R. Engen, G. Q. Daley, M. Warmuth., N. S. Gray. Targeting BCR-ABL by combining allosteric with ATP -binding-site inhibitors. Nature 2010;463:501-6). To prevent the emergence of drug resistance from ATP inhibitor and/or allosteric inhibitor use, a combination treatment using both types of inhibitor can be developed for the treatment of BCR-ABLl related disorders. In particular, the need exists for small molecules, or combinations thereof, that inhibit the activity of BCR-ABLl and BCR-ABLl mutations via the ATP binding site, the myristoyl binding site or a combination of both sites.Further, inhibitors of ABL 1 kinase activity have the potential to be used as therapies for the treatment of metastatic invasive carcinomas and viral infections such as pox and Ebola viruses.The compounds from the present invention also have the potential to treat or prevent diseases or disorders associated with abnormally activated kinase activity of wild-type ABL1, including non-malignant diseases or disorders, such as CNS diseases in particular neurodegenerative diseases (for example Alzheimer’s, Parkinson’s diseases), motoneuroneuron diseases (amyotophic lateral sclerosis), muscular dystrophies, autoimmune and inflammatory diseases (diabetes and pulmonary fibrosis), viral infections, prion diseases.
Asciminib is an allosteric inhibitor of BCR-ABL kinase in phase III clinical development at Novartis for the treatment of patients with chronic myelogenous leukemia (CML) in chronic phase who have been previously treated with ATP-binding site tyrosine kinase inhibitors. Early clinical trials are also under way in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) and as first-line threapy of CML.
To illustrate tautomerism with the following specific examples, (R)-N-(4- (chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)-5-(lH-pyrazol-5-yl)nicotinamide
(right structure, below) is a tautomer of (R)-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)-5-(lH-pyrazol-3-yl)nicotinamide (left structure, below) and vice versa:
[0045] Where the plural form (e.g. compounds, salts) is used, this includes the singular
[00365] A mixture of (R)-5-Bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)nicotinamide (Stage 9.2, 100 mg, 0.216 mmol) and 5-(4 ,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -((2-(trimethylsilyl)ethoxy)methyl)- IH-pyrazole (215 mg, 0.663 mmol), Pd(PPh3)2Cl2 (17 mg, 0.024 mmol), Na2C03 (115 mg, 1.081 mmol), DME (917 μί), water (262 μΕ) and EtOH (131 μί) in a MW vial was sealed, evacuated / purged 3 times with argon and subjected to MW irradiation at 125°C for 20 min. The RM was diluted with 2 mL
of DME, stirred with Si-Thiol (Silicycle 1.44 mmol/g, 90 mg, 0.130 mmol) for 3 h. The mixture was centrifuged and the supernatant was filtered through a 0.45 μηι PTFE filter and the solvent was evaporated off under reduced pressure. The crude product was purified by flash
chromatography (RediSep® Silica gel column, 12 g, cyclohexane / EtOAc from 40% to 100% EtOAc) to afford the protected intermediate as a colorless oil. Ethylene diamine (96 μί, 1.428 mmol) and TBAF 1 M in THF (1.428 mL, 1.428 mmol) were then added and the RM was stirred at 80-85°C for 5 days. The solvent was evaporated off under reduced pressure and the residue was dissolved in EtOAc (40 mL), washed 3 times with sat. aq. NaHCC and brine, dried over Na2S04 and The solvent was evaporated off under reduced pressure to give a residue which was purified by preparative SFC (Column DEAP, from 25% to 30% in 6 min) to yield the title compound as a white solid.
[00366] Alternatively, Example 9 was prepared by adding TFA (168 mL, 2182 mmol) to a solution of N-(4-(chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinamide (Stage 9.1, 31.3 g, 54.6 mmol) in DCM (600 mL). The mixture was stirred at RT for 2.5 h. The solvent was evaporated off under reduced pressure and the residue was dissolved in EtOAc (1.5 L),washed with a sat. solution of NaHC03 (3 x 500 mL) and brine (500 mL), dried over Na2S04 and the solvent was evaporated off under reduced pressure to give a residue which was suspended in DCM (300 mL), stirred at RT for 15 min, filtered, washed with DCM (200 mL), dried and purified by chromatography (Silica gel, 1 kg, DCM / MeOH 95:5). The residue was dissolved in MeOH (500 mL) and treated with Si-Thiol (Biotage, 5.0 g , 6.5 mmol) for 16 h at 25°C. The resin was filtered off, the solvent was evaporated off under reduced pressure and the residue was crystallized from MeCN to afford the title compound as a white crystalline solid.
[00367] Alternatively, Example 9 was prepared by the dropwise addition of aqueous HC1
(7.7 mL of 6M) to a solution of N-(4-(chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-( 1 -(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinamide (Stage 9.1, 3.8 g, 7.12 mmol) in MeOH (20 mL) and THF (10 mL) with cooling (below 35°C). The mixture was stirred at 22°C for 2 h and then added to cooled (10°C) 1.2 M NaOH (22 mL).
Throughout the addition the temperature was kept below 30°C and pH was kept in the range of 9-10. The RM was then stirred for 30 min at 30°C. The solvent was evaporated off under reduced pressure, until the desired compound precipitated. The precipitate was filtered and dried to give the title compound as a yellow solid.
[00368] Analytical data for Example 9: HPLC (Condition 5) tR = 5.54 min, HPLC Chiral
[00370] l-(Tetrahydro-2H-pyran-2-yl)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (29.6 g, 102 mmol), K3P04 (51.6 g, 236 mmol) and Pd(PPh3)4 (4.55 g, 3.93 mmol) were added to a suspension of (R)-5-bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)nicotinamide (Stage 9.2, 36.4 g, 79 mmol) in toluene (360 mL) under an argon atmosphere and the mixture was stirred at 110°C for 4 h. The RM was poured into brine (500 mL) and extracted with EtOAc (2 x 1 L). The combined extracts were washed with brine (500 mL), dried over Na2S04, and the solvent was evaporated off under reduced pressure to give a residue which was purified by chromatography (Silica gel column, 1.5 kg, DCM / MeOH 95:5) to afford a dark yellow foam, that was dissolved in MeOH / DCM (1 L of 3: l) and treated with Si-Thiol (Biotage, 35 g , 45.5 mmol) for 17 h at 30°C. The resin was filtered off, and solvent was evaporated off under reduced pressure, until the desired compound crystallized. The product was filtered washed with MeOH and dried to afford the title compound.
[00371] Alternatively, Stage 9.1 was prepared by adding 4-(chlorodifluoromethoxy)aniline
(16.6 g, 84.9 mmol), NMM (21.7 g, 212.1 mmol), hydroxybenzotriazole hydrate (HOBt H20, 11.9 g, 77.77 mmol) and l-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCIHCl, 20.9 g, 109.0 mmol) to a solution of 6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinic acid (Stage 9.4, 29.83 g, 70.7 mmol) in THF (271 mL). The mixture was stirred for 1.5 h at 25°C and then at 65°C for 16 h. After cooling the RM to 35 °C, further EDCIHCl (13.3 g, 69.4 mmol) was added and the RM was stirred for 1.5 h at 35°C then again at 65°C for 16 h. After cooling the RM to 35°C, water (150 mL) was added, the THF was removed under reduced pressure, EtOAc (180 mL) was added and the mixture was stirred for at 35 °C fori h. The two layers were separated and the aq. phase was then extracted with EtOAc (60 mL). The combined organic layers were washed with water (90 mL), brine (90 mL). The solvent was evaporated off under reduced pressure to give a brown solid which was purified by column chromatography (Silica gel, DCM / MeOH 40: 1 to 20: 1) to afford the title compound as a yellow solid.
[00372] Analytical data for Stage 9.1: HPLC (Condition 5) tR = 6.12 min, UPLC-MS
[00374] (R)-Pyrrolidin-3-ol (9.55 g, 109.6 mmol) and DIPEA (35.1 ml, 201.3 mmol) were added to a suspension of 5-bromo-6-chloro-N-(4-(chlorodifluoromethoxy)phenyl)nicotinamide (Stage 9.3, 37.7 g, 91.5 mmol) in iPrOH (65 mL) and stirred at 140°C for 1 h. EtOAc (700 mL) was added and the solution was washed IN HC1 (2 x 200 mL), sat. NaHCC (200 mL) and brine (2 x 200 mL), dried over Na2S04, and the solution was concentrated under reduced pressure until crystallization commenced. n-Heptane (1 L) were added and the mixture was stirred at RT for 30 min, filtered and washed with ΪΡΓ20 (500 mL) to afford the title compound as a white crystalline solid. HPLC (Condition 5) tR = 6.68 min, UPLC-MS (Condition 3) tR = 1.10 min, m/z =
[00376] DMF (2.55 mL, 33.0 mmol) and SOCl2 (24.08 ml, 330 mmol) were added to a suspension of 5-bromo-6-chloro-nicotinic acid (26 g, 110 mmol) in toluene (220 mL) and the RM was stirred at 80°C for 1 h. The solvent was evaporated off under reduced pressure and the residue was dissolved in THF (220 mL) and cooled to -16°C. DIPEA (38.4 mL, 220 mmol) was added, followed by dropwise addition of a solution of 4-(chlorodifluoromethoxy)aniline (22.35 g, 115 mmol) in THF (220 mL) over 15 min. The suspension was stirred for 1 h at RT. The solvent was evaporated off under reduced pressure and the residue was dissolved in TBME (700 mL), washed with IN HC1 (2 x 200 mL), sat. NaHC03 (200 mL) and brine (2 x 200 mL), dried over Na2S04, and the solvent was evaporated off under reduced pressure to give the product which was crystallized from EtOAc – n-heptane to afford the title compound as a white crystalline solid. HPLC (Condition 5) tR = 7.77 min, UPLC-MS (Condition 3) tR = 1.24 min, m/z =
[00378] Aq. NaOH (180 niL of 2.6 M) was added to a solution of methyl 6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-(l -(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinate (Stage 9.5, 11 lg, 299 mmol) in MeOH (270 mL) and the RM was stirred at RT for 14 h. The MeOH was evaporated off under reduced pressure and the aq. residue was treated with brine (90 mL), extracted with MeTHF twice (540 mL + 360 mL) and the combined organic layers were washed with water (90 mL). MeTHF was added to the combined aq. layers, the biphasic mixture was cooled to 0 °C and acidified (pH = 4-4.5) with aq. HC1 solution (18%) and extracted with
MeTHF. The combined organic extracts were washed with brine and the solvent was evaporated off under reduced pressure to give a residue which was recrystallized from a EtOAc / TBME (1 : 1) to afford the title compound as a white solid. HPLC (Condition 7) tR = 4.74 min, LC-MS
[00382] DIPEA (105.3 g, 142.2 mL, 814.4 mmol) was added to a solution of methyl-5-bromo-6-chroronicotinate (85 g, 339.5 mmol) and (R)-pyrrolidin-3-ol (54.2 g, 441.2 mmol) in isopropyl acetate and the RM was stirred at 70°C for 14 h . The solvent was evaporated off under reduced pressure to give a the residue which was dissolved in toluene (850 mL), washed with water (127 mL) and brine (127 mL)and concentrated under reduced pressure until precipitation commenced. n-Heptane (340 mL) was slowly added to the stirred mixture at 22 °C, which was then cooled to 0 °C and the product was filtered, washed with a toluene / n-heptane mixture
^Breccia M, Colafigli G, Scalzulli E, Martelli M (August 2021). “Asciminib: an investigational agent for the treatment of chronic myeloid leukemia”. Expert Opinion on Investigational Drugs. 30 (8): 803–811. doi:10.1080/13543784.2021.1941863. PMID34130563.
“Asciminib”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02081378 for “A Phase I Study of Oral ABL001 in Patients With CML or Ph+ ALL” at ClinicalTrials.gov
Clinical trial number NCT03106779 for “Study of Efficacy of CML-CP Patients Treated With ABL001 Versus Bosutinib, Previously Treated With 2 or More TKIs” at ClinicalTrials.gov
Uveal melanoma (UM) is the most common cancer of the eye in adults (Singh AD. et al., Ophthalmology. 201 1 ; 1 18: 1881-5). Most UM patients develop metastases for which no curative treatment has been identified so far. The majority of UM tumors have mutations in the genes GNAQ (guanine nucleotide-binding protein G(q) subunit alpha) and GNA11 (guanine nucleotide-binding protein G(q) subunit 1 1 ), which encode for small GTPases (Harbour JW. Pigment Cell Melanoma Res. 2012;25: 171-81). Both of these mutations lead to activation of the protein kinase C (PKC) pathway. The up-regulation of PKC pathway has downstream effects which leads to constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway that has been implicated in causing uncontrolled cell growth in a number of proliferative diseases.
Whilst anti-proliferative effects have been observed with certain PKC pathway inhibitors, no sustained MAPK pathway inhibition has been observed. Thus far, PKC inhibitors (PKCi) have had limited efficacy as single agents in patients (Mochly-Rosen D et al., Nat Rev Drug Discov. 2012 Dec;1 1 (12):937-57). Moreover, inhibition of PKC alone was unable to trigger cell death in vitro and/or tumor regression in vivo (Chen X, et al., Oncogene. 2014;33:4724-34).
The protein p53 is a transcription factor that controls the expression of a multitude of target genes involved in DNA damage repair, apoptosis and cell cycle arrest, which are all important phenomena counteracting the malignant growth of tumors. The TP53 gene is one of the most frequently mutated genes in human cancers, with approximately half of all cancers having inactivated p53. Furthermore, in cancers with a non-mutated TP53 gene, typically the p53 is functionally inactivated at the protein level. One of the mechanisms of p53 inactivation is through its interaction with human homolog of MDM2 (Mouse double minute 2) protein. MDM2 protein functions both as an E3 ubiquitin ligase, that leads to proteasomal degradation of p53, and an inhibitor of p53 transcriptional activation. Therefore, MDM2 is an important negative regulator of the p53 tumor suppressor. MDM2 inhibitors can prevent interaction between MDM2 and p53 and thus allow the p53 protein to exert its effector functions. Whilst TP53 mutations are not common in UM, there are reports suggesting the p53 pathway is inactivated by either high expression of MDM2 protein or downregulation of the PERP protein in UM patients.
A combination of an MDM2 inhibitor (Nutlin-3) has been shown to act synergistically with reactivation of p53 and induction of tumor cell apoptosis (RITA) and Topotecan to cause growth inhibition in UM cell lines (De Lange J. et al., Oncogene. 2012;31 :1 105-16). However, Nutlin-3 and Topotecan delayed in vivo tumor growth only in a limited manner.
Example 9: 3-amino-N-(3-(4-amino-4-methylpiperidin-l-yl)pyridin-2-yl)- 6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamide
Synthesis of tert-butyl (4-meth l-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate
To a solution of 3-fluoro-2-nitropyridine (11.2 g, 81 mmol) in dioxane (200 mL) was added tert-butyl (4-methylpiperidin-4-yl)carbamate (26 g, 121 mmol). Huenig’s Base (28.3 mL,
162 mmol) was added and the mixture was heated to 85 °C for 18 hrs. The reaction was cooled to RT and concentrated to give a brown solid. The solids were washed with 200 mL of 4: 1 heptane:EtOAc. Slurry was concentrated to half volume and filtered to collect (26.2 g, 78 mmol, 96%) brown solid. LC-MS (Acidic Method): ret.time= 1.46 min, M+H = 337.4
Step 2: Synthesis of tert-butyl (4-meth l-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate
To a solution of tert-butyl (4-methyl-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate (11.6 g, 37.2 mmol) in ethyl acetate (200 mL). 10% Pd-C (3.48 g) was added and stirred under H2 balloon pressure at RT for 4h. A small amount of MgS04 was added to the reaction and then the reaction mixture was filtered through a pad of cellite, then washed with ethyl acetate (100 mL) and the filtrate was concentrated to afford a brown solid (8.54 g, 27.9 mmol, 85%). LC-MS (Acidic Method): ret.time= 0.91 min, M+H = 307.4.
Step 3: Synthesis of tert-butyl (l-(2-(3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamido)pyridin-3-yl)-4-meth lpiperidin-4-yl)carbamate
To a solution of 3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxylic acid in dimethyl formamide (125 mL) was added ((lH-benzo[d][l,2,3]triazol-l- yl) oxy)
tris(dimethylamino) phosphonium hexafluorophosphate(V) (1.8g, 4.24 mmol) and 4-methylmorpholine (1 mL, 9.79 mmol). Reaction stirred at RT for 40 minutes. Tert-butyl (l-(2-aminopyridin-3-yl)-4-methylpiperidin-4-yl) carbamate in dimethylformamide (25 mL) was added and reaction stirred for 16 hrs at RT. The reaction mixture was diluted with EtOAc and was washed with NaHC03(aq) (3 x 200mL) and brine (lx 200mL). The organic phase was dried with Na2S04, filtered and concentrated. The crude product was taken up in acetonitrile (30 mL) and mixture was allowed to stand at RT for a period of time. Yellow solid collected by filtration (1.39g, 74%). LC-MS (Acidic Method): ret.time= 1.13 mm, M+H = 573.3.
Step 4: Synthesis of 3-amino-N-(3-(4-amino-4-methylpiperidin-l-yl)pyridin-2-yl)-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamide
A solution of tert-butyl (l-(2-(3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamido)pyridin-3-yl)-4-methylpiperidin-4-yl)carbamate (l -39g, 2.06 mmol) in dichloromethane (10 mL) was cooled to 0 °C. 2,2,2-trifluoroacetic acid (2.4 ml, 31 mmol) was added dropwise to the solution. The mixture was allowed to warm to 22 °C and stirred for 4 hrs. Reaction mixture was concentrated to remove DCM and excess TFA. A red oil was produced, which was taken up in 100 mL CHCI3/IPA 3: 1 and saturated aq. NaHCC was added to neutralize the solution. The mixture was then stirred at 22°C for 16 hrs. The mixture transfered to separatory funnel and aqueous layers were washed with CHCI3/IPA 3: 1 (3X 100 mL). Combined organic phases were dried with Na2S04, filtered and concentrated to afford a yellow solid. The crude product was recrystallized from acetonitrile. A yellow solid was collected by filtration (0.82g, 83%). LC-MS (Acidic Method ): ret.time= 0.75 mm, M+H = 473.2. 1H NMR (400 MHz, Methanol-^) δ 8.92 (dd, J = 5.1, 1.4 Hz, 1H), 8.68 (s, 1H), 8.47 – 8.27 (m, 1H), 8.12 (dd, J = 4.9, 1.6 Hz, 1H), 7.83 – 7.50 (m, 2H), 7.18 (dd, J = 7.9, 4.9 Hz, 1H), 3.02 – 2.65 (m, 4H), 1.54 – 1.24 (m, 4H), 0.74 (s, 3H).
REFERENCES
Visser, M.; Papillon, J.; Fan, J.; et al. NVP-LXS196, a novel PKC inhibitor for the treatment of uveal melanoma
253rd Am Chem Soc (ACS) Natl Meet (April 2-6, San Francisco) 2017, Abst MEDI 366

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











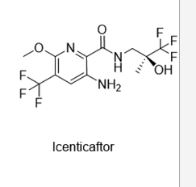
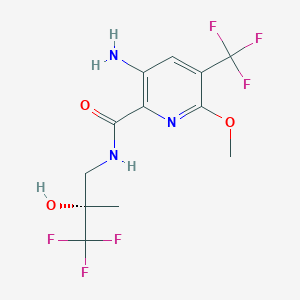





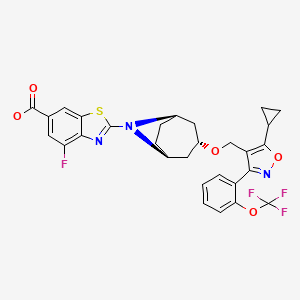

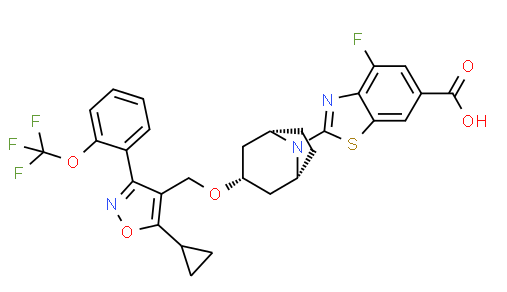


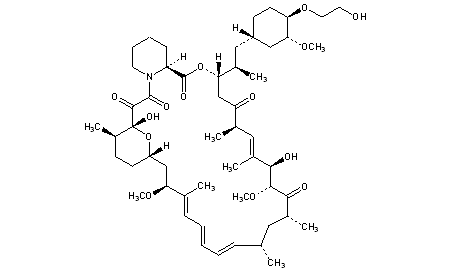

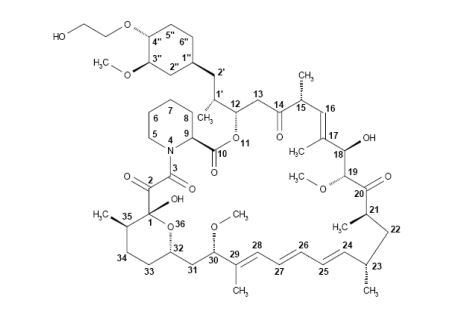





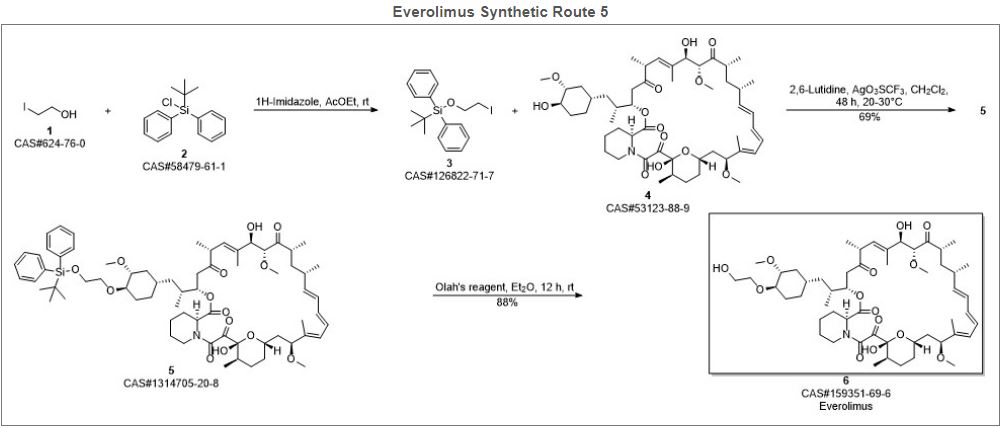

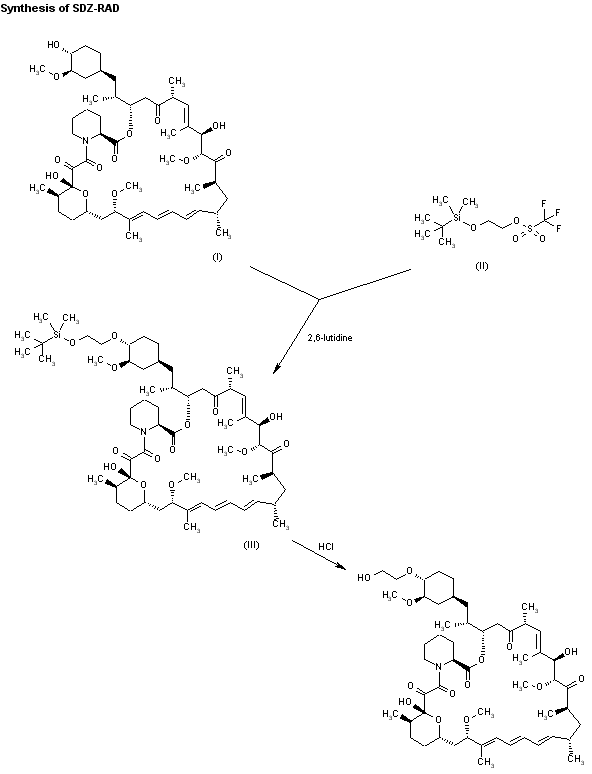
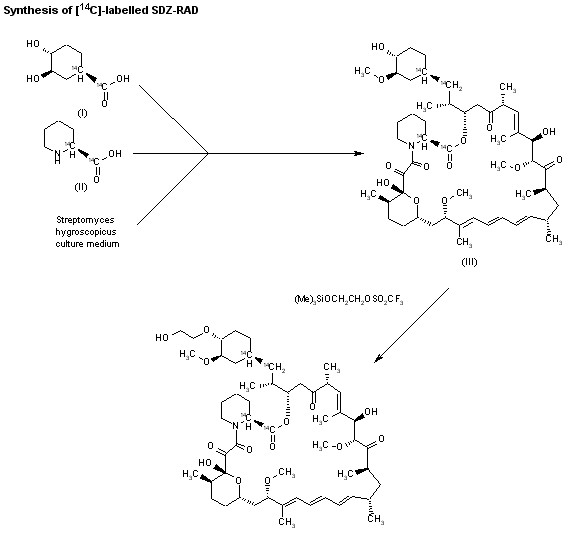
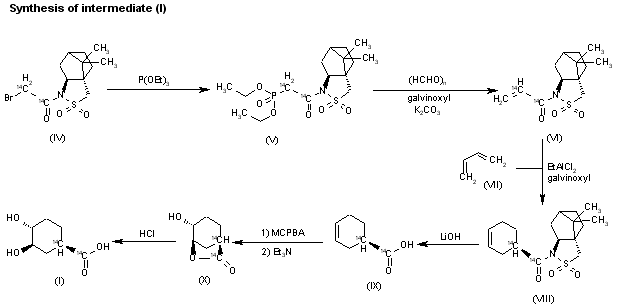
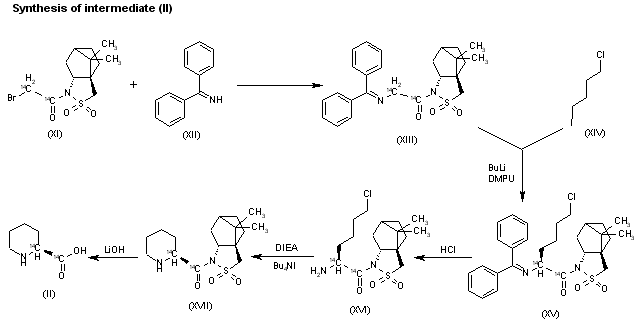

















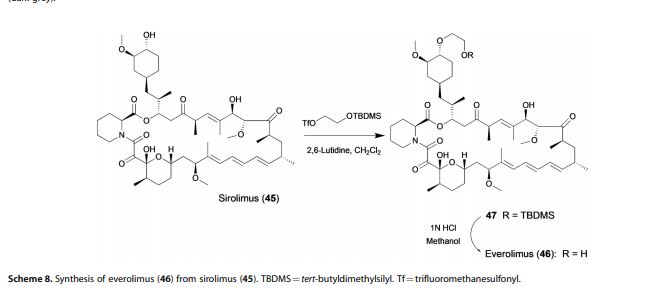




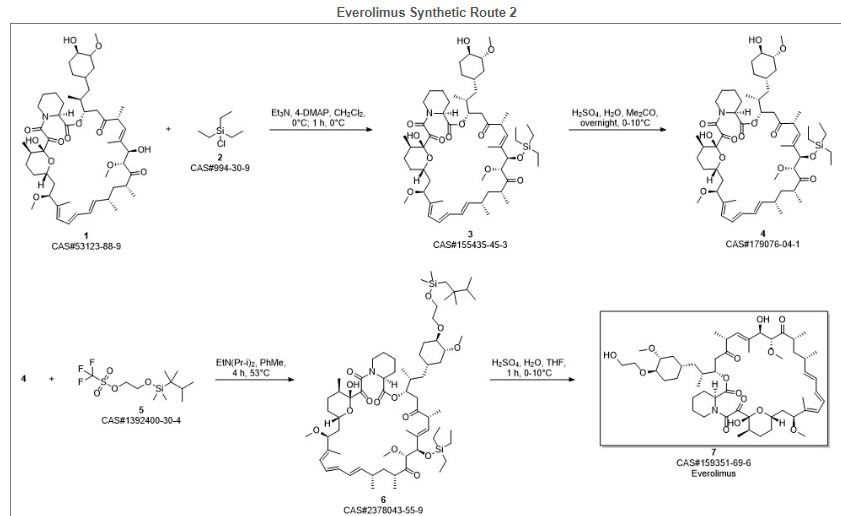
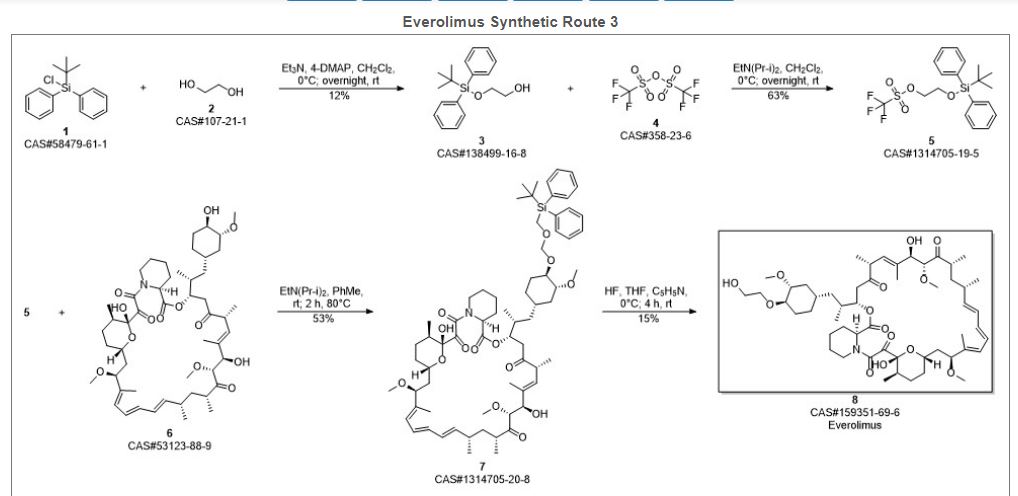
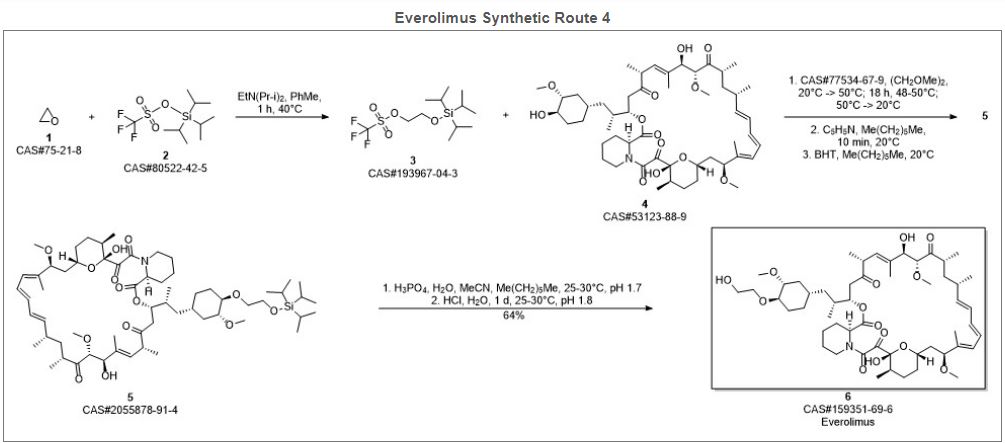
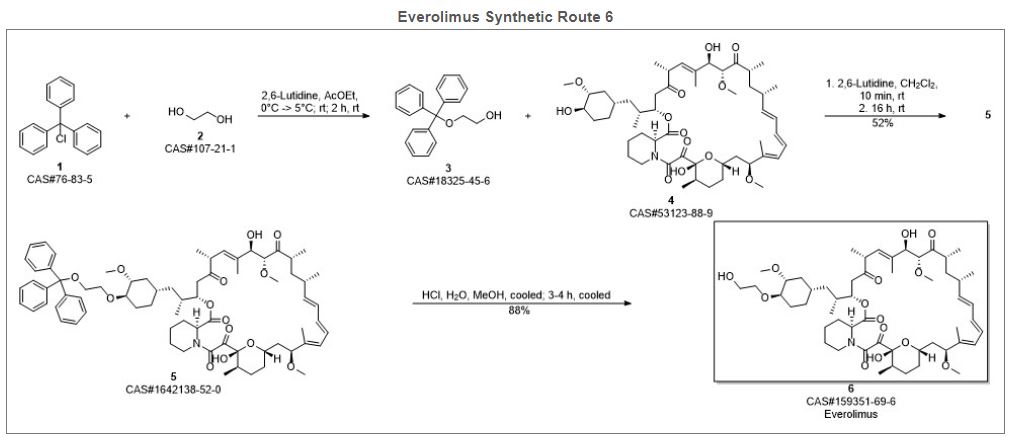


















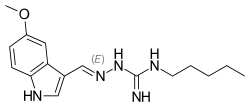

















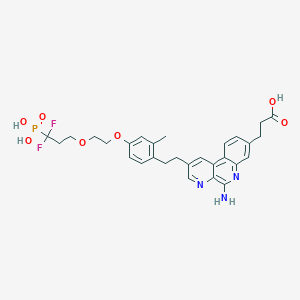

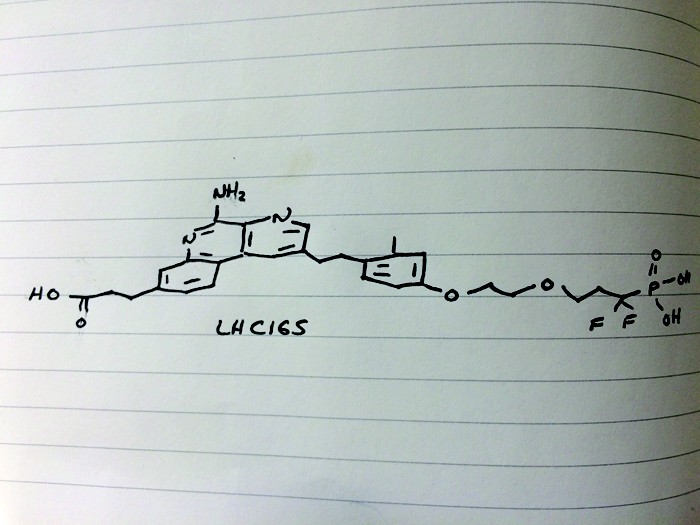

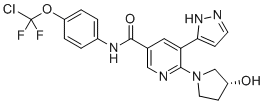
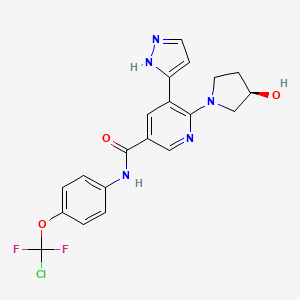












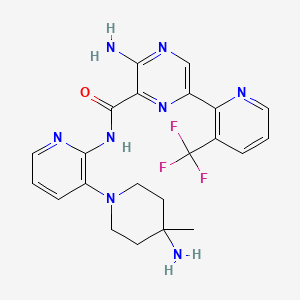

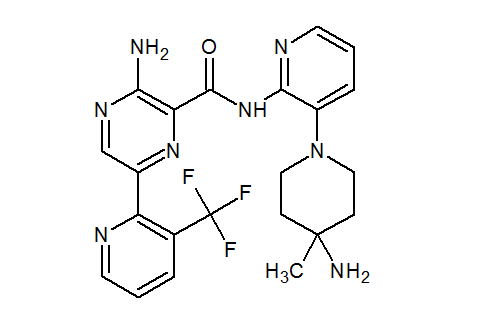








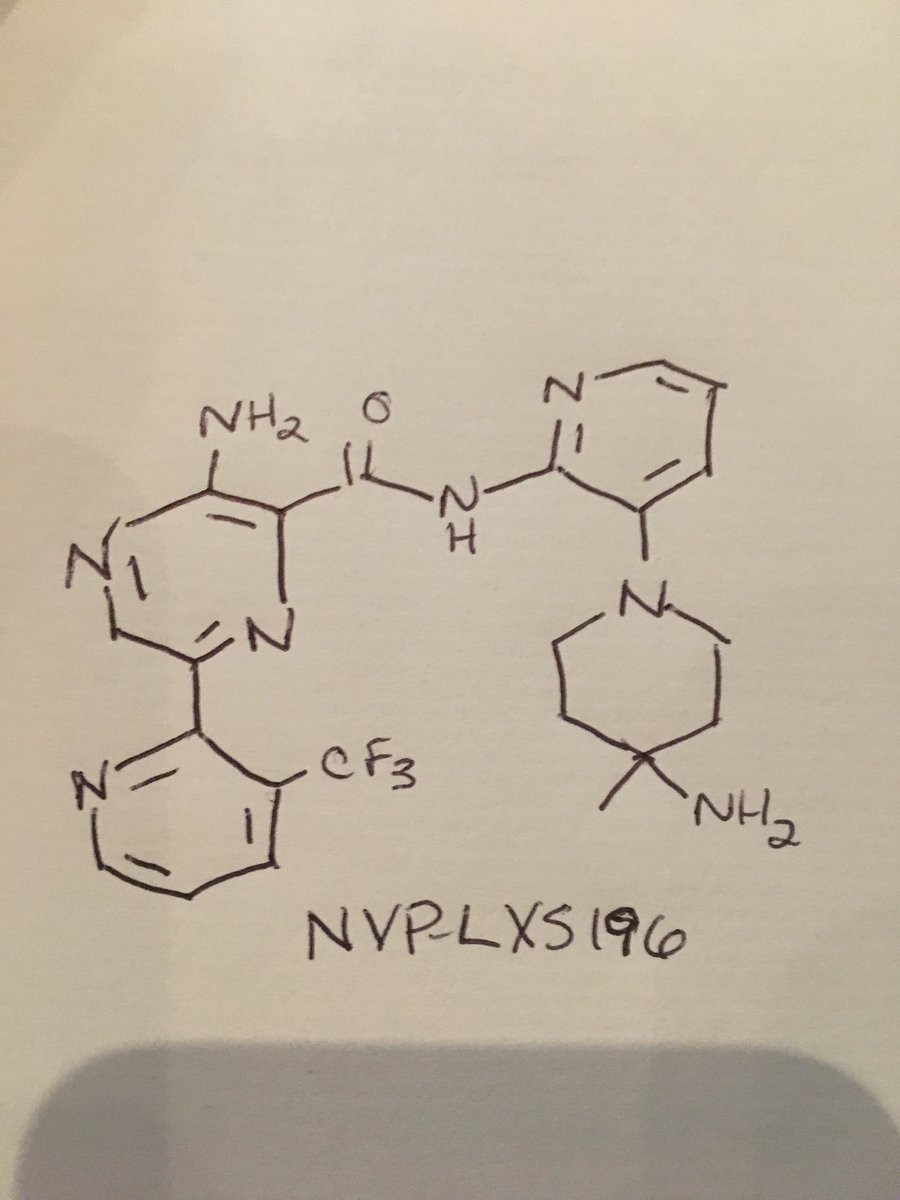

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
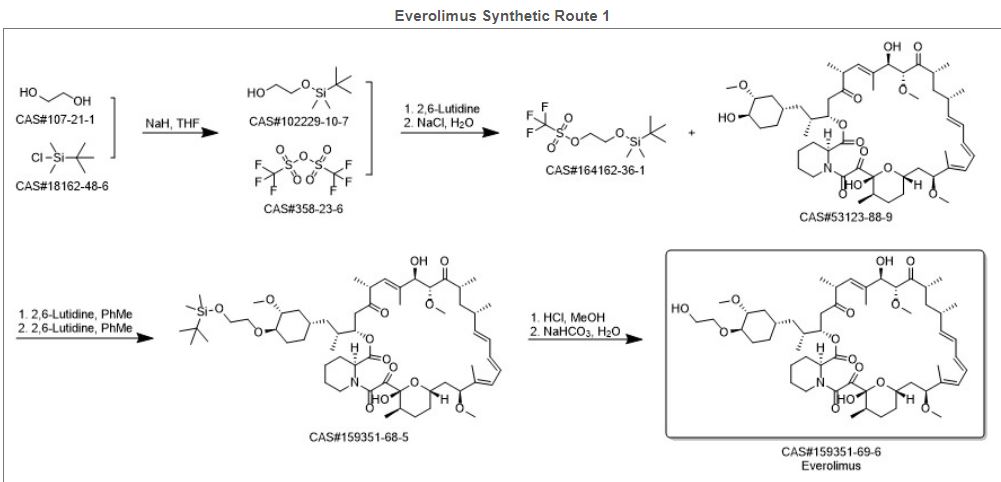{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}