

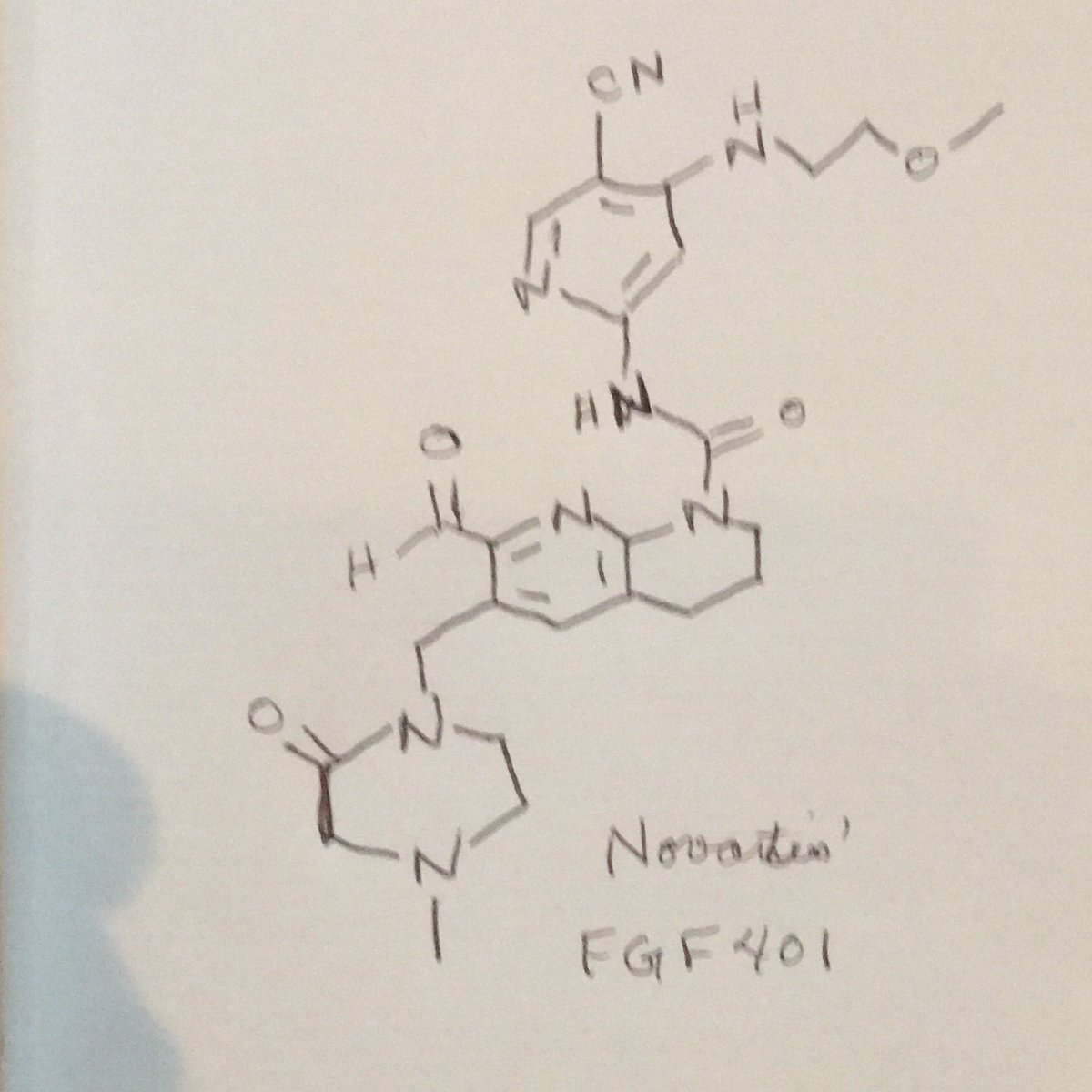
FGF 401
NVP-FGF-401
CAS 1708971-55-4
MF C25 H30 N8 O4, MW 506.56
1,8-Naphthyridine-1(2H)-carboxamide, N-[5-cyano-4-[(2-methoxyethyl)amino]-2-pyridinyl]-7-formyl-3,4-dihydro-6-[(4-methyl-2-oxo-1-piperazinyl)methyl]-
N-[5-Cyano-4-[(2-methoxyethyl)amino]-2-pyridinyl]-7-formyl-3,4-dihydro-6-[(4-methyl-2-oxo-1-piperazinyl)methyl]-1,8-naphthyridine-1(2H)-carboxamide
/V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide
Phase I/II Hepatocellular carcinoma; Solid tumours
- Originator Novartis
- Developer Novartis Oncology
- Class Antineoplastics
- Mechanism of Action Type 4 fibroblast growth factor receptor antagonists
- 26 Jan 2016 Phase-I/II clinical trials in Solid tumours and Hepatocellular carcinoma in USA, Hong Kong, Japan, Taiwan, France, Germany and Spain (PO)
- 26 Dec 2014 Phase-I/II clinical trials in Hepatocellular carcinoma in Singapore (PO)
- 26 Dec 2014 Phase-I/II clinical trials in Solid tumours in Singapore (PO)
Activation of FGFRs (fibroblast growth factor receptors) has an essential role in regulating cell survival, proliferation, migration and differentiation.1 Dysregulation of the FGFR signaling pathway has been associated with human cancer.1 FGFRs represent an important target for cancer therapeutics because a growing body of evidence indicates that they can act in an oncogenic fashion to promote multiple steps of cancer progression, including induction of mitogenic and survival signals
FGF-401 is a FGFR4 inhibitor in phase I/II clinical studies at Novartis for the treatment of positive FGFR4 and KLB expresion solid tumors and hepatocellular carcinoma
Normal growth, as well as tissue repair and remodeling, require specific and delicate control of activating growth factors and their receptors. Fibroblast Growth Factors (FGFs) constitute a family of over twenty structurally related polypeptides that are developmental^ regulated and expressed in a wide variety of tissues. FGFs stimulate proliferation, cell migration and differentiation and play a major role in skeletal and limb development, wound healing, tissue repair, hematopoiesis, angiogenesis, and tumorigenesis (reviewed in Ornitz, Novartis Found Symp 232: 63-76; discussion 76-80, 272-82 (2001)).
The biological action of FGFs is mediated by specific cell surface receptors belonging to the Receptor Protein Tyrosine Kinase (RPTK) family of protein kinases. These proteins consist of an extracellular ligand binding domain, a single transmembrane domain and an intracellular tyrosine kinase domain which undergoes phosphorylation upon binding of FGF. Four FGFRs have been identified to date: FGFR1 (also called Fig, fms-like gene, fit- 2, bFGFR, N-bFGFR or Cek1 ), FGFR2 (also called Bek-Bacterial Expressed Kinase-, KGFR, Ksam, Ksaml and Cek3), FGFR3 (also called Cek2) and FGFR4. All mature FGFRs share a common structure consisting of an amino terminal signal peptide, three extracellular immunoglobulin-like domains (Ig domain I, Ig domain II, Ig domain III), with an acidic region between Ig domains (the “acidic box” domain), a transmembrane domain, and intracellular kinase domains (Ullrich and Schlessinger, Cell 61 : 203,1990 ; Johnson and Williams (1992) Adv. Cancer Res. 60: 1 -41). The distinct FGFR isoforms have different binding affinities for the different FGF ligands.
Alterations in FGFRs have been associated with a number of human cancers including myeloma, breast, stomach, colon, bladder, pancreatic and hepatocellular carcinomas. Recently, it was reported that FGFR4 may play an important role in liver cancer in particular (PLoS One, 2012, volume 7, 36713). Other studies have also implicated FGFR4 or its ligand FGF19 in other cancer types including breast, glioblastoma, prostate, rhabdomyosarcoma, gastric, ovarian, lung, colon (Int. J. Cancer 1993; 54:378-382; Oncogene 2010; 29:1543-1552; Cancer Res 2010; 70:802-812; Cancer Res 201 1 ; 71 :4550-4561 ; Clin Cancer Res 2004; 10:6169-6178; Cancer Res 2013;
73:2551 -2562; Clin Cancer Res 2012; 18:3780-3790; J. Clin. Invest. 2009; 1 19:3395-3407; Ann Surg Oncol 2010; 17:3354-61 ; Cancer 201 1 ; 1 17:5304-13; Clin Cancer Res 2013; 19:809-820; PNAS 2013; 1 10:12426-12431 ; Oncogene 2008; 27:85-97).
Therapies involving FGFR4 blocking antibodies have been described for instance in
WO2009/009173, WO2007/136893, WO2012/138975, WO2010/026291 , WO2008/052798 and WO2010/004204. WO2014/144737 and WO2014/01 1900 also describe low molecular weight FGFR4 inhibitors.
in spite of numerous treatment options for patients with cancer, there remains a need for effective and safe therapeutic agents and a need for new combination therapies that can be administered for the effective long-term treatment of cancer.
Liver cancer or hepatic cancer is classified as primary liver cancer (i.e. cancer that forms in the tissues of the liver) and secondary liver cancer (i.e. cancer that spreads to the liver from another part of the body). According to the National Cancer Institute at the National Institutes of Health, the number of estimated new cases and deaths from liver and intrahepatic bile duct cancer in the United States in 2014 was 33,190 and 23,000, respectively. Importantly, the percent surviving five years or more after being diagnosed with liver and intrahepatic bile duct cancer is only about 16%.
It has now been found that a combination of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in free form or in pharmaceutically acceptable salt form and at least one further active ingredient, as defined herein, shows synergistic combination activity in an in vitro cell proliferation assay as shown in the experimental section and may therefore be effective for the delay of progression or treatment of a proliferative disease, such as cancer, in particular liver cancer.
| Inventors |
Nicole Buschmann, Robin Alec Fairhurst, Pascal Furet, Thomas Knöpfel, Catherine Leblanc, Robert Mah, Pierre NIMSGERN, Sebastien RIPOCHE, Lv LIAO, Jing XIONG, Xianglin ZHAO, Bo Han, Can Wang, |
| Applicant |
Novartis Ag |

Nicole Buschmann
Global Discovery Chemistry
Basel, Switzerland

Drawn by worlddrugtracker, helping millions………………..
PATENT
WO 2015059668
https://www.google.com/patents/WO2015059668A1?cl=en
PATENT
WO 2016151500
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1-yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid salt form has the following structure:

Example 1 – A/-(5-cvano-4 (2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1-yl)methyl)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid salt form (1 :1).

Step 1 : 2-(dimethoxymethyl)-1 ,8-naphthyridine.
The procedure described in J. Org. Chem., 2004, 69 (6), pp 1959-1966 was used. Into a 20 L 4-necked round-bottom flask was placed 2-aminopyridine-3-carbaldehyde (1000 g, 8.19 mol), 1 , 1-dimethoxypropan-2-one (1257 g, 10.64 mol), ethanol (10 L), and water (2 L). This was followed by the addition of a solution of sodium hydroxide (409.8 g, 10.24 mol) in water (1000 mL) drop wise with stirring at 0-15 °C. The solution was stirred for 3 h at 0-20 °C and then concentrated under vacuum. The resulting solution was extracted with 3×1200 mL of ethyl acetate and the organic layers were combined. The mixture was dried over sodium sulfate and concentrated under vacuum. The residue was washed with 3×300 mL of hexane and the solid was collected by filtration. This resulted in the title compound as a yellow solid. 1 H-NMR (400 MHz, DMSO-cf6) δ 9.1 1 (dd, 1 H), 8.53 (d, 1 H), 8.50 (dd, 1 H), 7.73 (d, 1 H), 7.67 (dd, 1 H), 5.44 (s, 1 H), 3.41 (s, 6H).
Step 2: 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 5-L pressure tank reactor (5 atm) was placed 2-(dimethoxymethyl)-1 ,8-naphthyridine (200 g, 979 mmol), ethanol (3 L), Pt02 (12 g). The reactor was evacuated and flushed three times with nitrogen, followed by flushing with hydrogen. The mixture was stirred overnight at 23 °C under an
atmosphere of hydrogen. This reaction was repeated four times. The solids were filtered out and the resulting mixture was concentrated under vacuum to give the title compound as a yellow solid. 1 H-NMR (400 MHz, DMSO-d6) δ 7.14 (d, 1 H), 6.51 (d, 1 H), 6.47 – 6.41 (m, 1 H), 4.98 (s, 1 H), 3.28 -3.19 (m, 2H), 3.23 (s, 6H), 2.64 (t, 2H), 1 .73 – 1.79 (m, 2H).
Step 3: 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
Into a 3 L 4-necked round-bottom flask was placed 7-(dimethoxymethyl)-1 ,2,3, 4-tetrahydro-1 ,8-naphthyridine (1 14.6 g, 550.3mmol) in acetonitrile (2 L). This was followed by the addition of NBS (103 g, 578 mol) in portions with stirring at 25 °C. The resulting solution was stirred for 30 min at 25 °C. The resulting mixture was concentrated under vacuum and the residue was diluted with 1000 mL of diethylether. The mixture was washed with 3×100 mL of ice/water. The aqueous phase was extracted with 2×100 mL of diethylether and the organic layers were combined. The resulting mixture was washed with 1×100 mL of brine, dried over sodium sulfate and concentrated under vacuum to give the title compound as a light yellow solid. LC-MS: (ES, m/z): 286.03 [M+H]+. 1 H-NMR: (300MHz, CDCI3) δ 1 .86 – 1 .94 (2H, m), 2.70 – 2.74 (2H, m), 3.9 – 3.43 (2H, m), 3.47 (6H, s), 5.23 (1 H, s), 5.58 (1 H, s), 7.29 (1 H, s).
Step 4: 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde.
To a solution of 6-bromo-7-(dimethoxymethyl)-1 ,2,3, 4-tetrahydro-1 ,8-naphthyridine (15.0 g, 52.2 mmol) in THF (400 mL) at -78 °C under argon, was added MeLi (1 .6 M in Et20, 32.6 mL, 52.2 mmol), the solution was stirred for 5 min, then n-BuLi (1 .6 M in hexane, 35.9 mL, 57.5 mmol) was added slowly and the solution was stirred for 20 min. THF (100 mL) was added to the reaction at -78 °C. Subsequently, n-BuLi (1 .6 M in hexane, 49.0 mL, 78 mmol) was added and the reaction mixture was stirred for 20 min, then again n-BuLi (1 .6 M in hexane, 6.53 mL, 10.45 mmol) was added and the mixture was stirred for 10 min at – 78 °C. DMF (2.10 mL, 27.2 mmol) was added and the reaction mixture was stirred at -78 °C for 45 min, then it was allowed to warm to room temperature, poured into sat. aq. NH4CI and extracted twice with DCM. The combined organic phases were dried over Na2S04, filtered and evaporated to give the title compound as an orange oil. (UPLC-MS 3) tR 0.63 min; ESI-MS 237.2 [M+H]+.
Step 5: ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate.
Ethyl bromoacetate (1.27 mL, 1 1 .48 mmol) was added to a mixture of tert-butyl (2-(methylamino)ethyl)carbamate (2.0 g, 1 1 .48 mmol), triethylamine (4.81 mL) and THF (24 mL) at 0 °C. After stirring 24 h at room temperature the reaction mixture was partitioned between saturated aqueous NaHC03 and DCM, extracted 2x with DCM, the organic layers dried over Na2S04 and
evaporated to give the title compound as a clear pale-yellow oil. 1H NMR (400 MHz, CDCI3) δ 5.20 (s, br, 1 H), 4.18 (q, 2H), 3.24 (s, 2H), 3.22 – 3.16 (m, 2H), 2.65 – 2.61 (m, 2H), 2.38 (s, 3H), 1 .42 (s, 9H), 1 .24 (t, 3H).
Step 6: ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride.
Concentrated hydrochloric acid (10 mL) was added to a solution of ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate (3.05 g, 1 1 .13 mmol) in THF (20 mL) and EtOH (100 mL) at room temperature. After stirring 1 h at room temperature the reaction mixture was evaporated, ethanol (20 mL) added, evaporated, further ethanol (50 mL) added and then stirred at 60 °C for 70 min. The cooled reaction mixture was then evaporated to give the title compound as a pale-yellow glass. 1 H NMR (400 MHz, DMSO-d6) δ 8.58 (s, br, 3H), 4.19 (q, 2H), 4.26 – 4.15 (m, 2H), 3.44 (s, br, 2H), 3.21 (s, br, 2H), 2.88 (s, 3H), 1 .21 (t, 3H).
Step 7: 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one.
Sodium triacetoxyborohydride (3.10 g, 14.61 mmol) was added to a mixture of 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde (obtained in step 4, 2.30 g, 9.74 mmol), ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride (obtained in step 6, 2.6 g, 14.61 mmol) and triethylamine (6.75 mL, 48.7 mmol) in 1 ,2-dichloroethane (20 mL) at room temperature. The reaction mixture was stirred for 21 h at room temperature and additional sodium triacetoxyborohydride (2.6 g, 9.74 mmol) was added. After a further 4 h stirring at room temperature, again additional sodium triacetoxyborohydride (1 .3 g, 4.87 mmol) was added and the reaction maintained at 4 °C for 2.5 days. The reaction mixture was then warmed to room temperature, saturated aqueous NaHC03 solution added, the mixture extracted with DCM (3x), the combined organic layers dried over Na2S04 and evaporated. The residue was applied to a 120 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 10% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1 H NMR (400 MHz, CDCI3) δ 7.08 (s, 1 H), 5.30 (s, br, 1 H), 5.20 (s, 1 H), 4.69 (s, 2H), 3.44 – 3.34 (m, 2H), 3.40 (s, 6H), 3.22 – 3.15 (m, 2H), 3.24 (s, 2H), 2.71 – 2.64 (m, 2H), 2.58 – 2.50 (m, 2H), 2.31 (s, 3H), 1 .98 – 1.82 (m, 2H). (UPLC-MS 6) tR 0.33; ESI-MS 335.3 [M+H]+.
Step 8: 4-fluoro-5-iodopyridin-2-amine.
A suspension of 4-fluoropyridin-2-amine (336 g, 2.5 mol) and NIS (745 g, 2.75 mol) in MeCN (9 L) was treated with TFA (1 14 g, 1 mol). The reaction mixture was then stirred at room temperature for 8 h. The reaction mixture was diluted with EtOAc (10 L), washed with sat. aq. Na2S203 (2 x 5 L), brine (4 x 5 L). The combined organic layers were dried over Na2S04, filtered and concentrated to get the crude product. The crude product was purified by recrystallization from EtOAc/pentane (1/10) to afford the title compound as a white solid. 1H NMR (400 MHz, DMSO-cf6) δ 8.14 (d, 1 H), 6.45 (s, 2H), 6.33 (d, 1 H).
Step 9: 6-amino-4-fluoronicotinonitrile.
4-fluoro-5-iodopyridin-2-amine (obtained in step 8, 240 g, 1 mol), zinc cyanide (125 g, 1.05 mol), zinc (13 g, 0.2 mol), Pd2(dba)3 (25 g, 25 mmol) and dppf (55 g, 0.1 mol) in DMA (800 mL) were degassed and charged into the round bottom flask under nitrogen. The mixture was stirred at 100 °C for 3 h. The reaction mixture was diluted with 5% NaHC03 (2 L), extracted with EtOAc (4 x 600 mL). The combined organic layers were washed with 5% NaOH (1 L), dried over Na2S04, concentrated to 700 mL. The resulting organic phase was eluted through silica gel column with EtOAc (1.7 L). The combined organic filtrate was washed with 2 M HCI (3 x 800 mL). The pH of the aqueous phase was adjusted to 10 with saturated NaHC03. The aqueous phase was extracted whit DCM (3 x 500 mL). The combined DCM was dried over Na2S04 and concentrated. The residue was further purified by column chromatography (eluted with pentane: EtOAc 10: 1 to 3:2) followed by recrystallization from pentane/EtOAc 3/1 to give the title compound as white solid. 1 H NMR (400 MHz, DMSO-d6) δ 8.40 (d, 1 H), 7.40 (s, 2H), 6.34 (d, 1 H).
Step 10: tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate.
A mixture of 2,4-dichloro-5-cyanopyridine (1 Og, 57.8 mmol), fe/f-butyl carbamate (8.2 g, 70.5 mmol), Pd(OAc)2 (0.26 g, 1 .1 mmol), Xantphos (1 .34 g, 2.3mmol) and K2C03 (12 g, 87 mmol) in THF (150 mL) was degassed 3x with nitrogen. The mixture was then heated at 70 °C for 4-5 h and monitored by chromatography until complete conversion. Following completion of the reaction, additional THF (100 mL) was added and heated the mixture at 70 °C for additional 1 h and then cooled to room temperature. The suspension was then filtered through a pad of celite to remove the solid. The filtrate was then concentrated and azotropically distilled with ethyl acetete before filtering to give the title compound. 1 H NMR (DMSO-d6, 400 MHz): δ 10.82 (s, 1 H), 8.79 (s, 1 H), 8.09 (s, 1 H), 1 .49 (s, 9H).
Step 1 1 : fe/f-butyl N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)carbamate.
A mixture of tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate (obtained in step 10, 9.8 g, 38.6 mmol), 2-methoxyethylamine (5.8 g, 77.3 mmol) and DIPEA (6 g, 46.4 mmol) in DMSO (80 mL) was heated at 65-70 °C for 24 h and monitored by chromatography until complete conversion. The
solution was then cooled to room temperature and a white solid precipitated gradually. Water (20 mL) was then added slowly within 1 h. The suspension was stirred for a further 1 h, filtered and dried to give the title compound as a white solid. 1 H NMR (DMSO-d6, 400 MHz): δ 9.87 (s, 1 H), 8.18 (s, 1 H), 7.20 (s, 1 H), 6.86 (s, 9H), 3.51 (t, 2H), 3.36 (t, 2H), 3.28 (s, 3H), 1.47 (s, 9H).
Step 12: 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile.
A solution of 6-amino-4-fluoronicotinonitrile (obtained in step 9, 1 .10 g, 8.02 mmol) in DMA (20 mL) was treated with 2-methoxyethylamine (2.07 mL, 24.1 mmol) and DIPEA (4.20 mL, 24.1 mmol), heated to 50 °C and stirred for 15 h. The reaction mixture was cooled to room temperature and concentrated. The crude material was purified by normal phase chromatography (24 g silica gel cartridge, heptanes/EtOAc 100:0 to 0:100). The product containing fractions were concentrated and dried under vacuum to give the title compound as an off-white solid.
An alternative synthesis of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile is outlined below:
To tert-butyl N-{5-cyano-4-[(2-methoxyethyl)amino]pyridin-2-yl}carbamate (obtained in step 1 1 , 7g) was added 30-36% aqueous HCI (40 mL), the mixture stirred at room temperature for 30 minutes and monitored by chromatography until complete conversion. The solution was then basified with 20-30% NaOH solution to pH=9-10 and filtered to give a white solid. The solid was added to ethyl acetate (15 mL) and heated to 50-55 °C to form a clear solution. The solution was then cooled to 3-6 °C, stirred for 2-3 h and filtered. The wet cake was then dried to give the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.92 (s, 1 H), 6.39 (s, 2H), 6.15 (t, 1 H), 5.61 (s, 1 H), 3.46 (t, 2H), 3.27 (s, 3H), 3.24 (q, 2H). (UPLC-MS 3) tR 0.62; ESI-MS 193.1 [M+H]+.
Step 13: N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
A solution of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile (obtained in step 12, 481 mg, 2.50 mmol) in anhydrous DMF (1.5 mL) was added drop wise over 10 minutes to a mixture of di(1 H-1 ,2,4-triazol-1 -yl)methanone (410 mg, 2.50 mmol) and DMF (1 .5 mL) cooled at 0 °C. After stirring for 45 minutes at 0 °C the reaction mixture was allowed to warm to room temperature and after a further 90 minutes at room temperature a solution of 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one (obtained in step 7, 418 mg, 1.00 mmol) in DMF (2 mL) was added. The reaction mixture was stirred for 17.5 h at room temperature, quenched by the addition of MeOH and evaporated. The residue was applied to a 80 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 2% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, DMSO-d6) δ 13.50 (s, 1 H), 8.27 (s,
1 H), 7.52 (s, 1 H), 7.39 (s, 1 H), 6.93 (t, 1 H), 5.45 (s, 1 H), 4.65 (s, 2H), 3.94 – 3.89 (m, 2H), 3.54 -3.50 (m, 2H), 3.40 – 3.35 (m, 2H), 3.38 (s, 6H), 3.29 (s, 3H), 3.20 – 3.16 (m, 2H), 3.05 (s, 2H), 2.86 – 2.80 (m, 2H), 2.61 – 2.55 (m, 2H), 2.22 (s, 3H), 1 .94 – 1 .88 (m, 2H). (UPLC-MS 6) tR 0.72; ESI-MS 553.3 [M+H]+.
Step 14: /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-form
yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide
Concentrated hydrochloric acid (0.40 mL) was added to a solution of A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 13, 470 mg, 0.808 mmol) in THF (3 mL) and water (1 mL) at room temperature. After stirring for 3 h at room temperature saturated aqueous NaHC03 was added, the mixture extracted with DCM (3x), the organic layers dried over Na2S04 and evaporated. The residue was sonicated with EtOAc (6 mL) and pentane (6 mL) and then filtered. The white solid obtained was then dissolved in DCM (6 mL), EtOAc added (3 mL), the solution warmed, sealed and allowed to stand at room temperature for 2 h. Filtration and drying gave A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide as a white solid.
1 H NMR (400 MHz, DMSO-d6) δ 13.43 (s, 1 H), 10.06 (s, 1 H), 8.24 (s, 1 H), 7.49 (s, 1 H), 7.47 (s, 1 H), 6.96 (t, br, 1 H), 4.86 (s, 2H), 3.96 – 3.90 (m, 2H), 3.52 – 3.46 (m, 2H), 3.39 – 3.33 (m, 2H), 3.30 – 3.21 (m, 2H), 3.37 (s, 3H), 3.02 (s, 2H), 2.93 – 2.86 (m, 2H), 2.61 – 2.56 (m, 2H), 2.21 (s, 3H), 1 .95 – 1.85 (m, 2H). (UPLC-MS 6) tR0.70, ESI-MS 507.2, [M+H]+.
Step 15: A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 :1 ).
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 14, 4g, 7.896 mmol) was stirred in propionic acid (29.3 g, 29.60mL) at 70 °C until dissolution was complete (20 minutes). The solution was cooled to 55 °C and a solution of citric acid in acetone (23% w/w) was added to it. Separately, a seed suspension was prepared by adding acetone (0.2 g, 0.252mL) to A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (0.0185 g, 0.026 mmol). The seed suspension was added to the solution at 50 °C and the resulting suspension was left to stir at 50 °C for 40 minutes. A further solution of citric acid in acetone (26.6g, 2.51 % w/w, 33.63 mL) was added to the reaction over 380 minutes. The resulting suspension was stirred for a further 120 minutes and cooled to 20 °C with stirring over 4 hours. The suspension was stirred for another 12 hours
before filtering the suspension under vacuum and washing the resulting solid with a propionic acid: acetone solution (1 : 1 , 7g, 7.96ml_) at room temperature. The solid was further washed with acetone (7g, 8.85ml_) at room temperature. The resulting solid was dried in an oven at 40 °C and 5mbar to give the title compound as a light orange solid (5.2g, 7.443 mmol). (mw 698.70), mp (DSC) 168.8 °C (onset).
XRPD analysis showed the same pattern as with particles obtained by a process described in PCT/I B2014/065585 (reference example 1 ) – see Figure 5.
Example 1a
Steps 1 to 14 were carried out as described in example 1 .
Step 15a: A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 : 1 )
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 14, 5g, 9.930 mmol) was stirred in propionic acid (33.5 g, 33.84ml_) at 60 °C. Once A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide had dissolved, anhydrous citric acid powder (0.19g, 0.9889 mmol) was added. The resulting suspension was heated to 70 °C and sonicated for 5 minutes to ensure full dissolution. The resulting solution was cooled to 50 °C and a solution of citric acid in ethyl acetate (3.7 g, 1 .3% citric acid in ethyl acetate) was added over 20 minutes. Seeds of N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (0.02 g) were added to the solution and the suspension was aged for 15 minutes. Another aliquot of citric acid in ethyl acetate (128g, 1 .3% citric acid in ethyl acetate) was added to the suspension over 1 1 .85hours. The suspension was left to stir for over 4 hours. The suspension was then filtered under vacuum (500mbar) and the resulting solid was washed firstly with a propionic acid: ethyl acetate solution (1 : 1 , 7g, 7.44ml_) at room temperature and then with ethyl acetate (12g, 13.38ml_) at room temperature. The resulting solid was dried in an oven at 40 °C and 5mbar to give the title compound as a light orange solid (6.3 g, 9.074 mmol).
XRPD analysis showed the same pattern as with particles obtained by a process described in PCT/I B2014/065585 (reference example 1 ) – see Figure 5.
Reference example 1 (described in PCT/IB2014/065585) – V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 :1 )
Steps 1 to 14 were carried out as described in example 1.
Reference Step 15 – /V-(5-cvano-4-((2-methoxyethyl‘)amino‘)pyridin-2-yl‘)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl‘)methyl‘)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H‘)-carboxamide in citric acid form (1 :1 )
A solution of citric acid (96.9 mg) in acetone (5 mL) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in acetone solution (2 mL) was then added to a suspension of Λ/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in acetone (4 mL) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h before slowly cooling to room temperature. The white solid was then collected by filtration, washing 2x with acetone (2 mL), and dried for 18 h at 40 °C under vacuum to give the title salt.
Alternatively, N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (6.5 g, 12.83 mmol) was placed in a 500ml 4-flask reactor. 49 mL of glacial acetic acid was added and the resulting suspension was stirred at 23 °C until a clear mixture was obtained. In a separate flask, anhydrous 2-hydroxypropane-1 ,2,3-tricarboxylic acid (2.59 g, 13.47 mmol, 1 .05 equiv.) was dissolved in 49 mL of glacial acetic acid at 50 °C until a clear solution was obtained. This solution was then added at 23°C to the N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide solution previously prepared. This mixture was stirred for 30 min at 23 °C and then added dropwise over 1 h to 192 mL of ethyl acetate warmed to 75 °C. The temperature remained constant over the addition. At the end of the addition, the temperature of the mixture was cooled slowly to 23 °C and let 16h at this temperature under gentle stirring. The suspension was cooled to 5-10 °C and filtered. The cake was washed with 15 mL of ethyl acetate and 15 mL of acetone. The wet cake (ca 8.5g) was transferred in a 500 mL flask containing 192 mL of dry acetone. The resulting suspension was refluxed for 24h. The suspension was filtered and the cake was washed with 2 times 15 mL of dry acetone then dried at 50 °C under vacuum for several hours to give the title salt.
PATENT
WO 2016151501
The synthesis of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (abbreviated herein as CPi and also named as Example 83) and salts thereof is disclosed in PCT/IB2014/065585, the content of which are incorporated by reference, as described herein below:
Example 83: /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
Concentrated hydrochloric acid (0.40 ml) was added to a solution of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (intermediate 80, 470 mg, 0.808 mmol) in THF (3 ml) and water (1 ml) at room temperature. After stirring for 3 h at room temperature saturated aqueous NaHC03 was added, the mixture extracted with DCM (3x), the organic layers dried over Na2S04 and evaporated. The residue was sonicated with EtOAc (6 ml) and pentane (6 ml) and then filtered. The white solid obtained was then dissolved in DCM (6 ml), EtOAc added (3 ml), the solution warmed, sealed and allowed to stand at room temperature for 2 h. Filtration and drying gave the title compound as a white solid.
1H NMR (400 MHz, DMSO-c/6) δ 13.43 (s, 1 H), 10.06 (s, 1 H), 8.24 (s, 1 H), 7.49 (s, 1 H), 7.47 (s, 1 H), 6.96 (t, br, 1 H), 4.86 (s, 2H), 3.96 – 3.90 (m, 2H), 3.52 – 3.46 (m, 2H), 3.39 – 3.33 (m, 2H), 3.30 – 3.21 (m, 2H), 3.37 (s, 3H), 3.02 (s, 2H), 2.93 – 2.86 (m, 2H), 2.61
– 2.56 (m, 2H), 2.21 (s, 3H), 1 .95 – 1 .85 (m, 2H).
(UPLC-MS 6) tR 0.70, ESI-MS 507.2, [M+H]+.
The following salts were prepared from the above free form form of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide by precipitation with the appropriate counterions.
Malate with 1 :1 stoichiometry (mw 640.66), mp (DSC) 181 .1 °C (onset): Acetone (2 ml) was added to a mixture of malic acid (26.4 mg, 0.197 mmol) and /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg, 0.197 mmol) and the mixture heated on a mini-block with heating-cooling cycles from 55 to 5 °C for 7 repeat cycles (heating rate: 1 .5 °C/min, cooling rate: 0.25 °C/min). The white solid was collected by centrifugation and dried for 18 h at 40 °C to give the title salt.
Tartrate with 1 :0.5 stoichiometry (mw 581 .72), mp (DSC) 176.7 °C (onset). A solution of tartaric acid (75.7 mg) in methanol (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M tartaric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h. The white solid was then collected by filtration, washing 2x with methanol (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Tartrate with 1 :1 stoichiometry (mw 656.66), mp (DSC) 169.9 °C (onset): A solution of tartaric acid (75.7 mg) in acetone (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M tartaric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Citrate with 1 :0.5 stoichiometry (mw 602.73), mp (DSC) 168.4 °C (onset): A solution of citric acid (96.9 mg) in methanol (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in methanol solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with
stirring for 2 h. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Citrate with 1 :1 stoichiometry (mw 698.70), mp (DSC) 168.8 °C (onset): A solution of citric acid (96.9 mg) in acetone (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in acetone (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h before slowly cooling to room temperature. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Alternatively, N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (6.5 g, 12.83 mmol) was placed in a 500ml 4-flask reactor. 49 ml of glacial acetic acid was added and the resulting suspension was stirred at 23 °C until a clear mixture was obtained. In a separate flask, anhydrous 2-hydroxypropane-1 ,2,3-tricarboxylic acid (2.59 g, 13.47 mmol, 1 .05 equiv.) was dissolved in 49 ml of glacial acetic acid at 50 °C until a clear solution was obtained. This solution was then added at 23°C to the N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide solution previously prepared. This mixture was stirred for 30 min at 23 °C and then added dropwise over 1 h to 192 ml of ethyl acetate warmed to 75 °C. The temperature remained constant over the addition. At the end of the addition, the temperature of the mixture was cooled slowly to 23 °C and let 16h at this temperature under gentle stirring. The suspension was cooled to 5-10 °C and filtered. The cake was washed with 15 ml of ethyl acetate and 15 ml of acetone. The wet cake (ca 8.5g) was transferred in a 500 ml flask containing 192 ml of dry acetone. The resulting suspension was refluxed for 24h. The suspension was filtered and the cake was washed with 2 times 15 ml of dry acetone then dried at 50 °C under vacuum for several hours to give the title salt.
Intermediate 80: N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7- (dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
A solution of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile (intermediate 75, 481 mg, 2.50 mmol) in anhydrous DMF (1 .5 ml) was added drop wise over 10 minutes to a mixture of di(1 H-1 ,2,4-triazol-1 -yl)methanone (410 mg, 2.50 mmol) and DMF (1 .5 ml) cooled at 0 °C. After stirring for 45 minutes at 0 °C the reaction mixture was allowed to warm to room temperature and after a further 90 minutes at room temperature a solution of 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one (intermediate 81 , 418 mg, 1 .00 mmol) in DMF (2 ml) was added. The reaction mixture was stirred for 17.5 h at room temperature, quenched by the addition of MeOH and evaporated. The residue was applied to a 80 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 2% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, DMSO-c/6) δ 13.50 (s, 1 H), 8.27 (s, 1 H), 7.52 (s, 1 H), 7.39 (s, 1 H), 6.93 (t, 1 H), 5.45 (s, 1 H), 4.65 (s, 2H), 3.94 – 3.89 (m, 2H), 3.54 – 3.50 (m, 2H), 3.40 – 3.35 (m, 2H), 3.38 (s, 6H), 3.29 (s, 3H), 3.20 – 3.16 (m, 2H), 3.05 (s, 2H), 2.86 – 2.80 (m, 2H), 2.61 – 2.55 (m, 2H), 2.22 (s, 3H), 1 .94 – 1 .88 (m, 2H). (UPLC-MS 6) tR 0.72; ESI-MS 553.3 [M+H]+.
Intermediate 81 : 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one.
Sodium triacetoxyborohydride (3.10 g, 14.61 mmol) was added to a mixture of 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde (intermediate 41 , 2.30 g, 9.74 mmol), ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride (intermediate 82, 2.6 g, 14.61 mmol) and triethylamine (6.75 ml, 48.7 mmol) in 1 ,2-dichloroethane (20 ml) at room temperature. The reaction mixture was stirred for 21 h at room temperature and additional sodium triacetoxyborohydride (2.6 g, 9.74 mmol) was added. After a further 4 h stirring at room temperature, again additional sodium triacetoxyborohydride (1 .3 g, 4.87 mmol) was added and the reaction maintained at 4 °C for 2.5 days. The reaction mixture was then warmed to room temperature, saturated aqueous NaHC03 solution added, the mixture extracted with DCM (3x), the combined organic layers dried over Na2S04 and evaporated. The residue was applied to a 120 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 10% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, CDCI3) δ 7.08 (s, 1 H), 5.30 (s, br, 1 H), 5.20 (s, 1 H), 4.69 (s, 2H), 3.44 – 3.34 (m, 2H), 3.40 (s, 6H), 3.22 – 3.15 (m, 2H), 3.24 (s, 2H), 2.71 -2.64 (m, 2H), 2.58 – 2.50 (m, 2H), 2.31 (s, 3H), 1 .98 – 1 .82 (m, 2H). (UPLC-MS 6) tR 0.33; ESI-MS 335.3 [M+H]+.
Intermediate 82: ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride.
Concentrated hydrochloric acid (10 ml) was added to a solution of ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate (intermediate 83, 3.05 g, 1 1 .13 mmol) in THF (20 ml) and EtOH (100 ml) at room temperature. After stirring 1 h at room temperature the reaction mixture was evaporated, ethanol (20 ml) added, evaporated, further ethanol (50 ml) added and then stirred at 60 °C for 70 min. The cooled reaction
mixture was then evaporated to give the title compound as a pale-yellow glass. 1H NMR (400 MHz, DMSO-c/6) δ 8.58 (s, br, 3H), 4.19 (q, 2H), 4.26 – 4.15 (m, 2H), 3.44 (s, br, 2H), 3.21 (s, br, 2H), 2.88 (s, 3H), 1 .21 (t, 3H).
Intermediate 83: ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate.
Ethyl bromoacetate (1 .27 ml, 1 1 .48 mmol) was added to a mixture of tert-butyl (2-(methylamino)ethyl)carbamate (2.0 g, 1 1 .48 mmol), triethylamine (4.81 ml) and THF (24 ml) at 0 °C. After stirring 24 h at room temperature the reaction mixture was partitioned between saturated aqueous NaHC03 and DCM, extracted 2x with DCM, the organic layers dried over Na2S04 and evaporated to give the title compound as a clear pale-yellow oil. 1 H NMR (400 MHz, CDCI3) δ 5.20 (s, br, 1 H), 4.18 (q, 2H), 3.24 (s, 2H), 3.22 -3.16 (m, 2H), 2.65 – 2.61 (m, 2H), 2.38 (s, 3H), 1 .42 (s, 9H), 1 .24 (t, 3H).
Intermediate 41 : 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde.
To a solution of 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine
(intermediate 12, 15.0 g, 52.2 mmol) in THF (400 ml) at -78 °C under argon, was added MeLi (1 .6 M in Et20, 32.6 ml, 52.2 mmol), the solution was stirred for 5 min, then n-BuLi (1 .6 M in hexane, 35.9 ml, 57.5 mmol) was added slowly and the solution was stirred for 20 min. THF (100 ml) was added to the reaction at – 78 °C. Subsequently, n-BuLi (1 .6 M in hexane, 49.0 ml, 78 mmol) was added and the reaction mixture was stirred for 20 min, then again n-BuLi (1 .6 M in hexane, 6.53 ml, 10.45 mmol) was added and the mixture was stirred for 10 min at – 78 °C. DMF (2.10 ml, 27.2 mmol) was added and the reaction mixture was stirred at -78 °C for 45 min, then it was allowed to warm to room
temperature, poured into sat. aq. NH4CI and extracted twice with DCM. The combined organic phases were dried over Na2S04, filtered and evaporated to give the title compound as an orange oil. (UPLC-MS 3) tR 0.63 min; ESI-MS 237.2 [M+H]+.
Intermediate 12: 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
Into a 3 I 4-necked round-bottom flask was placed 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine (intermediate 4, 1 14.6 g, 550.3mmol) in acetonitrile (2 I). This was followed by the addition of NBS (103 g, 578 mol) in portions with stirring at 25 °C. The resulting solution was stirred for 30 min at 25 °C. The resulting mixture was concentrated under vacuum and the residue was diluted with 1000 ml of diethylether. The mixture was washed with 3×100 ml of ice/water. The aqueous phase was extracted with 2×100 ml of diethylether and the organic layers were combined. The resulting mixture was washed with 1 x100 ml of brine, dried over sodium sulfate and concentrated under vacuum to give the title compound as a light yellow solid. LC-MS: (ES, m/z):
286.03 [M+H]+. 1H-NMR: (300MHz, CDCI3) δ 1 .86 – 1 .94 (2H, m), 2.70 – 2.74 (2H, m), 3.9 – 3.43 (2H, m), 3.47 (6H, s), 5.23 (1 H, s), 5.58 (1 H, s), 7.29 (1 H, s).
Intermediate 4: 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 5-I pressure tank reactor (5 atm) was placed 2-(dimethoxymethyl)-1 ,8-naphthyridine (intermediate 5, 200 g, 979 mmol), ethanol (3 I), Pt02 (12 g). The reactor was evacuated and flushed three times with nitrogen, followed by flushing with hydrogen. The mixture was stirred overnight at 23 °C under an atmosphere of hydrogen. This reaction was repeated four times. The solids were filtered out and the resulting mixture was concentrated under vacuum to give the title compound as a yellow solid.
Intermediate 5: 2-(dimethoxymethyl)-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 20 I 4-necked round-bottom flask was placed 2-aminopyridine-3-carbaldehyde (1000 g, 8.19 mol), 1 ,1 -dimethoxypropan-2-one (1257 g, 10.64 mol), ethanol (10 I), and water (2 I). This was followed by the addition of a solution of sodium hydroxide (409.8 g, 10.24 mol) in water (1000 ml) drop wise with stirring at 0-15 °C. The solution was stirred for 3 h at 0-20 °C and then concentrated under vacuum. The resulting solution was extracted with 3×1200 ml of ethyl acetate and the organic layers were combined. The mixture was dried over sodium sulfate and concentrated under vacuum. The residue was washed with 3×300 ml of hexane and the solid was collected by filtration. This resulted in the title compound as a yellow solid. 1H-NMR (400 MHz, DMSO-c/6) δ 9.1 1 (dd, 1 H), 8.53 (d, 1 H), 8.50 (dd, 1 H), 7.73 (d, 1 H), 7.67 (dd, 1 H), 5.44 (s, 1 H), 3.41 (s, 6H).
Intermediate 75: 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile.
A solution of 6-amino-4-fluoronicotinonitrile (intermediate 21 , 1 .10 g, 8.02 mmol) in DMA (20 ml) was treated with 2-methoxyethylamine (2.07 ml, 24.1 mmol) and DIPEA (4.20 ml_, 24.1 mmol), heated to 50 °C and stirred for 15 h. The reaction mixture was cooled to room temperature and concentrated. The crude material was purified by normal phase chromatography (24 g silica gel cartridge, heptanes/EtOAc 100:0 to 0:100). The product containing fractions were concentrated and dried under vacuum to give the title compound as an off-white solid.
An alternative synthesis of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile is outlined below:
To fe/ -butyl N-{5-cyano-4-[(2-methoxyethyl)amino]pyridin-2-yl}carbamate (intermediate 287, 7g) was added 30-36% aqueous HCI (40 ml), the mixture stirred at room temperature for 30 minutes and monitored by chromatography until complete conversion. The solution was then basified with 20-30% NaOH solution to pH=9-10 and filtered to give a white solid. The solid was added to ethyl acetate (15 ml) and heated to 50-55 °C to form a clear solution. The solution was then cooled to 3-6 °C, stirred for 2-3 h and filtered. The wet cake was then dried to give the title compound as a white solid. 1H NMR (400 MHz, DMSO-c/6) δ 7.92 (s, 1 H), 6.39 (s, 2H), 6.15 (t, 1 H), 5.61 (s, 1 H), 3.46 (t, 2H), 3.27 (s, 3H), 3.24 (q, 2H). (UPLC-MS 3) tR 0.62; ESI-MS 193.1 [M+H]+.
1H-NMR (400 MHz, DMSO-c/6) δ 7.14 (d, 1 H), 6.51 (d, 1 H), 6.47 – 6.41 (m, 1 H), 4.98 (s, 1 H), 3.28 – 3.19 (m, 2H), 3.23 (s, 6H), 2.64 (t, 2H), 1 .73 – 1 .79 (m, 2H).
Intermediate 21 : 6-amino-4-fluoronicotinonitrile.
4-fluoro-5-iodopyridin-2-amine (intermediate 22, 240 g, 1 mol), zinc cyanide (125 g, 1 .05 mol), zinc (13 g, 0.2 mol), Pd2(dba)3 (25 g, 25 mmol) and dppf (55 g, 0.1 mol) in DMA (800 ml) were degassed and charged into the round bottom flask under nitrogen. The mixture was stirred at 100 °C for 3 h. The reaction mixture was diluted with 5% NaHC03 (2 I), extracted with EtOAc (4 x 600 ml). The combined organic layers were washed with 5% NaOH (1 I), dried over Na2S04, concentrated to 700 ml. The resulting organic phase was eluted through silica gel column with EtOAc (1 .7 I). The combined organic filtrate was washed with 2 M HCI (3 x 800 ml). The pH of the aqueous phase was adjusted to 10 with saturated NaHC03. The aqueous phase was extracted whit DCM (3 x 500 ml). The combined DCM was dried over Na2S04 and concentrated. The residue was further purified by column chromatography (eluted with pentane: EtOAc 10:1 to 3:2) followed by recrystallization from pentane/EtOAc 3/1 to give the title compound as white solid. 1H NMR (400 MHz, DMSO-c/6) δ 8.40 (d, 1 H), 7.40 (s, 2H), 6.34 (d, 1 H).
Intermediate 22: 4-fluoro-5-iodopyridin-2-amine.
A suspension of 4-fluoropyridin-2-amine (336 g, 2.5 mol) and NIS (745 g, 2.75 mol) in MeCN (9 I) was treated with TFA (1 14 g, 1 mol). The reaction mixture was then stirred at room temperature for 8 h. The reaction mixture was diluted with EtOAc (10 I), washed with sat. aq. Na2S203 (2 x 5 I), brine (4 x 5 I). The combined organic layers were dried over Na2S04, filtered and concentrated to get the crude product. The crude product was purified by recrystallization from EtOAc/pentane (1/10) to afford the title compound as a white solid. 1H NMR (400 MHz, DMSO-c/6) δ 8.14 (d, 1 H), 6.45 (s, 2H), 6.33 (d, 1 H).
Intermediate 287: fe/ -butyl (5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)carbamate.
A mixture of tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate (intermediate 288, 9.8 g, 38.6 mmol), 2-methoxyethylamine (5.8 g, 77.3 mmol) and DIPEA (6 g, 46.4 mmol) in DMSO (80 ml) was heated at 65-70 °C for 24 h and monitored by chromatography until complete conversion. The solution was then cooled to room temperature and a white solid precipitated gradually. Water (20 ml) was then added slowly within 1 h. The suspension was stirred for a further 1 h, filtered and dried to give the title compound as a white solid. 1H NMR (DMSO-d6, 400 MHz): δ 9.87 (s, 1 H), 8.18 (s, 1 H), 7.20 (s, 1 H), 6.86 (s, 9H), 3.51 (t, 2H), 3.36 (t, 2H), 3.28 (s, 3H), 1 .47 (s, 9H).
Intermediate 288: tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate.
A mixture of 2,4-dichloro-5-cyanopyridine (10g, 57.8 mmol), fe/ -butyl carbamate (8.2 g, 70.5 mmol), Pd(OAc)2 (0.26 g, 1 .1 mmol), Xantphos (1 .34 g, 2.3mmol) and K2C03 (12 g, 87 mmol) in THF (150 ml) was degassed 3x with nitrogen. The mixture was then heated at 70 °C for 4-5 h and monitored by chromatography until complete conversion. Following completion of the reaction, additional THF (100 ml) was added and heated the mixture at 70 °C for additional 1 h and then cooled to room temperature. The suspension was then filtered through a pad of celite to remove the solid. The filtrate was then concentrated and azotropically distilled with ethyl acetete before filtering to give the title compound. 1H NMR (DMSO-d6, 400 MHz): δ 10.82 (s, 1 H), 8.79 (s, 1 H), 8.09 (s, 1 H), 1 .49 (s, 9H).
/////////////FGF 401, 1708971-55-4, PHASE 1, Hepatocellular carcinoma, Solid tumours, Novartis, Novartis Oncology, Antineoplastics, Type 4 fibroblast growth factor receptor antagonists, NVP-FGF-401, Nicole Buschmann, Robin Alec Fairhurst, Pascal Furet, Thomas Knöpfel, Catherine Leblanc, Robert Mah, Pierre NIMSGERN, Sebastien RIPOCHE, Lv LIAO, Jing XIONG, Xianglin ZHAO, Bo Han, Can Wang,

Now in #MEDI 1st time disclosures Robin Fairhurst of @Novartis will also talk about an FGFR inhibitor. They are popular! #ACSSanFran
CN4CC(=O)N(Cc1cc(C=O)nc2N(CCCc12)C(=O)Nc3cc(NCCOC)c(C#N)cn3)CC4

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






















































 Born in the year 1950, Dr. Chaitanya Dutt holds an MD in Medicine. He practiced as a consulting physician before joining the company in 1982. Since then he has been associated with the Company. His rich experience spans in the areas of Pharma R&D, clinical research, manufacturing, quality assurance, etc. He is one of the key professionals in the top management team of the Company. He has been instrumental in setting up the Torrent Research Centre (TRC), the research wing of the Company. Under his prudent guidance and leadership, TRC has achieved tremendous progress in the areas of discovery research as well as development work on formulations. He does not hold any directorship in any other company.
Born in the year 1950, Dr. Chaitanya Dutt holds an MD in Medicine. He practiced as a consulting physician before joining the company in 1982. Since then he has been associated with the Company. His rich experience spans in the areas of Pharma R&D, clinical research, manufacturing, quality assurance, etc. He is one of the key professionals in the top management team of the Company. He has been instrumental in setting up the Torrent Research Centre (TRC), the research wing of the Company. Under his prudent guidance and leadership, TRC has achieved tremendous progress in the areas of discovery research as well as development work on formulations. He does not hold any directorship in any other company.




















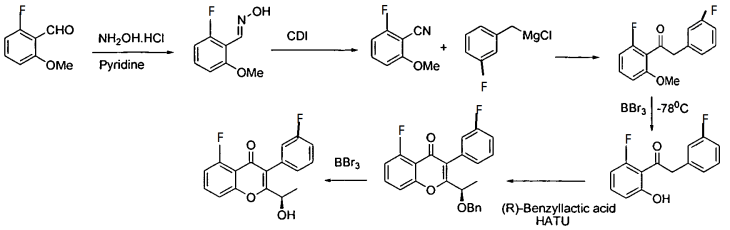
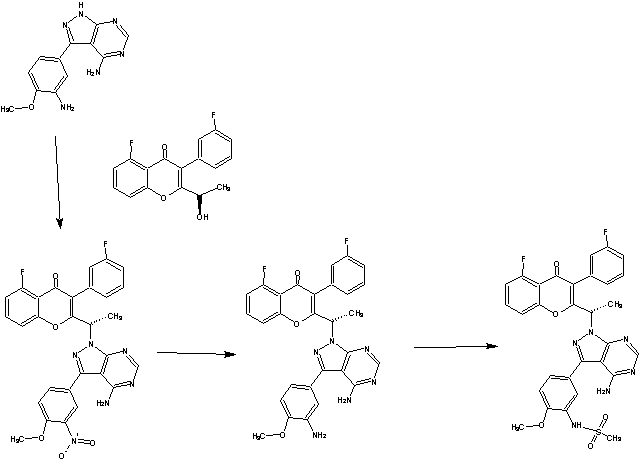







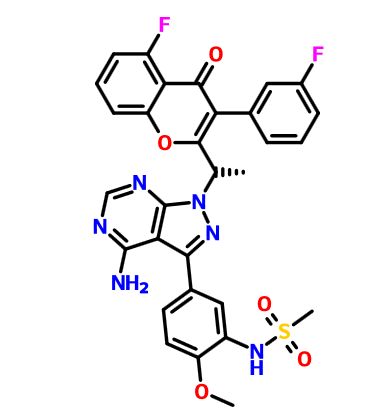
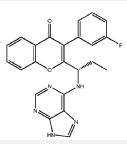 RP 6530
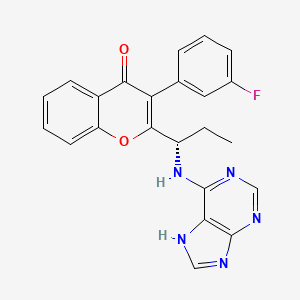
RP 6530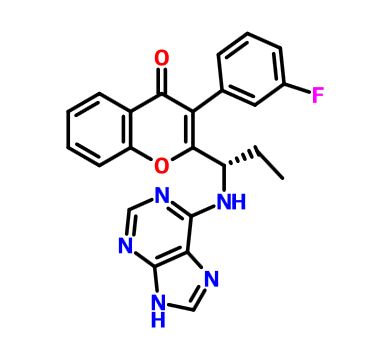

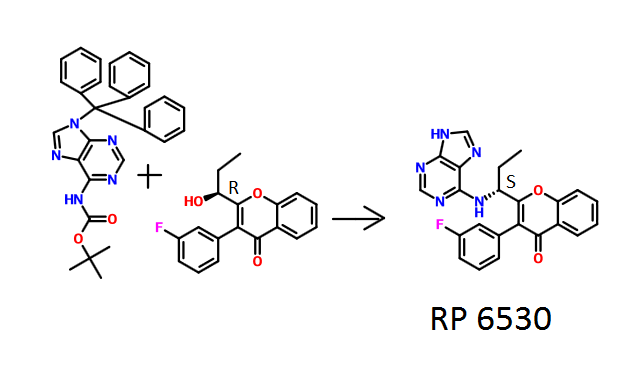






















 NADEEM SHAIKH
NADEEM SHAIKH
