https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014195888Intermediates
Intermediate 1: 3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one: To a solution of 2-(l-bromopropyl)-3-(3-fluorophenyl)-4H-chromen-4-one1 (8.80 g, 24.36 mmol ) in DMSO (85 ml), n-butanol (5 ml) was added and heated to 120° C for 3h. The reaction mixture was cooled to room temperature (RT), quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (2.10 g, 29 %) which was used without further purification in next step.
Intermediate 2: 3-(3-fluorophenyl)-2-propionyl-4H-chromen-4-one: DMSO (1.90 ml, 26.82 mmol) was added to dichloromethane (70 ml) and cooled to -78°C. Oxalyl chloride (1.14 ml, 13.41 mmol) was then added. After 10 minutes, intermediate 1 (2.00 g, 6.70 mmol) in dichloromethane (20 ml) was added dropwise and stirred for 20 min. Triethylamine (7 ml) was added and stirred for lh. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow liquid (1.20 g, 60%) which was used as such in next step.
Intermediate 3: (+)/(-)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one :
To a solution of intermediate 2 (0.600 g, 2.02 mmol) in DMF (7.65 ml) under nitrogen purging, formic acid : trietylamine 5 : 2 azeotrope (1.80 ml) was added followed by [(S,S)tethTsDpenRuCl] (3.0 mg). The reaction mixture was heated at 80°C for 1.5 hours under continuous nitrogen purging. The reaction mixture was quenched with water, extected with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (0.450 g, 74%). Mass: 299.0 (M+).
Enantiomeric excess: 78%, enriched in the late eluting isomer (retention time: 9.72 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 4: (+)/(-)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one :
The title compound was obtained as yellow solid (0.500 g, 83%) by using a procedure similar to the one described for intermediate 3, using intermediate 2 (0.600 g, 2.02 mmol), DMF (7.65 ml), formic acid : trietylamine 5 : 2 azeotrope (1.80 ml) and [(R,R)tethTsDpenRuCl] (3.0 mg). Mass: 298.9 (M+). Enantiomeric excess: 74.8%, enriched in the fast eluting isomer (retention time: 8.52 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: (R)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one:
Step 1 : (R)-2-(l-(benzyloxy)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one: To 2-(3-fluorophenyl)-l-(2-hydroxyphenyl)ethanone (2.15 g, 9.36 mmol ), in dichloromethane ( 20 ml), HATU (4.27 g, 11.23 mmol), R-(+)2-benzyloxybutyric acid (2.00 g, 10.29 mmol) were added and stirred for lOmin, then triethylamine (14.0 ml, 101.1 mmol) was added dropwise and stirred at RT for 24h. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as yellow solid (1.65 g, 45%). JH-NMR (δ ppm, CDC13, 400 MHz): 8.24 (dd, / = 7.9,1.5 Hz, 1H), 7.74 (dt, / = 7.1,1.7 Hz, 1H), 7.58 (dd, / = 8.3,0.4 Hz, 1H), 7.44-7.06 (m, 10H), 4.51 (d, / = 7.8 Hz, 1H), 4.34 (d, / = 7.8 Hz, 1H), 4.25 (dd, / = 7.8,6.2 Hz, 1H), 2.17-1.90 (m, 2H), 0.95 (t, / = 7.5 Hz, 3H). Mass: 389.0 (M+).
Step 2: (R)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one : To (R)-2-(l-(benzyloxy)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one (1.50 g, 3.86 mmol) in dichloromethane (15 ml) cooled to 0°C and aluminium chloride (1.00 g, 7.72 mmol) was added portion wise and stirred at RT for 6h. The reaction mixture was quenched with 2N HC1 solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as yellow solid (0.552 g, 48%).‘ JH-NMR (δ ppm, CDC13, 400 MHz): ‘ 8.24 (dd, / = 8.0,1.6 Hz, 1H), 7.72 (m, , 1H), 7.52 (dd, / = 8.4,0.5 Hz, 1H), 7.44 (m, 2H), 7.12-7.01(m,3H), 4.49 (t, / = 7.0 Hz, 1H), 1.94 (m, 2H), 0.93 (t, / = 7.5 Hz, 3H). Mass: (299.0(M+). Purity: 96.93%.
25[a] D -14.73 (c = 1, CHCI3). Enantiomeric excess: 85.92%, enriched in the fast eluting isomer (retention time: 8.57 min.) as determined by HPLC on a chiralpak AS-3R column.
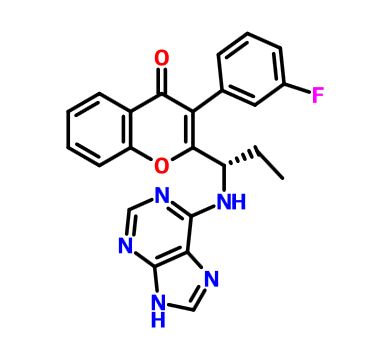
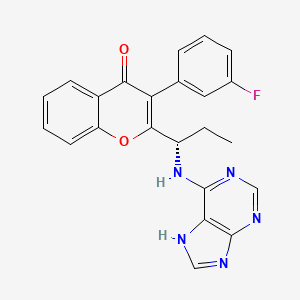
Compound A
(RS)- 2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 1 (2.50 g, 8.41 mmol) in THF (25 ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (4.81 g, 10.09 mmol) and triphenylphosphine (3.31 g, 12.62 mmol) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (2.5 ml, 12.62 mmol) was added and stirred at RT for 2h. The reaction mixture was concentrated and column chromatographed with ethyl acetate : petroleum ether to afford a yellow coloured intermediate. To the intermediate, dichloromethane (65 ml) and trifluoroacetic acid (7.9 ml) were added and the resulting mixture was stirred at RT for 12 h. The reaction mixture was then basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as pale-brown solid (1.05 g, 30 %). MP: 148-150°C. Mass: 415.6 (M+).
Compound Al
(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
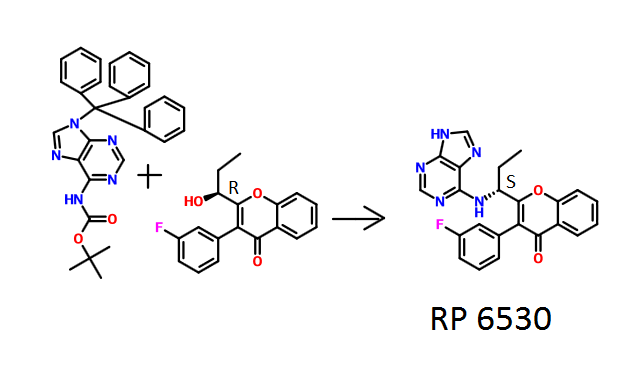
Method A: To a solution of intermediate 3 (0.250 g, 0.838 mmol) in THF (5ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (0.479 g, 1.00 mmol) and triphenylphosphine (0.329 g, 1.25 mmol) were added and the resulting mixture was stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.25 mmol) was then added and stirred at RT for 12 h. The reaction mixture was concentrated and column chromatographed with ethyl acetate: pet.ether to afford the yellow coloured intermediate. To the intermediate in dichloromethane (6 ml), trifluoroacetic acid (1.2 ml) was added stirred at RT for 12 h. The reaction mixture was basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as an off-white solid (0.015 g, 4 %). MP: 137-140°C. JH-NMR (δ ppm, DMSO- , 400 MHz): 12.94 (s, 1H), 8.12-8.10 (m, 4H), 7.84-7.80 (m, 1H), 7.61 (d, / = 8.3 Hz, 1H), 7.50-7.41 (m, 2H), 7.28-7.18 (m, 3H), 5.20-5.06 (m, 1H), 2.10-1.90 (m, 2H), 0.84 (t, / = 3.7 Hz, 3H). Enantiomeric excess: 77.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 7.90 min.).
Method B : To a solution of intermediate 5 (2.60 g, 8.68 mmol) in THF (52 ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (4.96 g, 10.42 mmol) and triphenylphosphine (2.76 g, 13.03 mmol) were added and the resulting mixture was stirred at RT for 5 min. Dusopropylazodicarboxylate (0.25 ml, 1.25 mmol) was then added and stirred at RT for 12 h. The reaction mixture was concentrated and column chromatographed with ethyl acetate: petroleum ether to afford the yellow coloured intermediate. To the intermediate in dichloromethane (55 ml), trifluoroacetic acid (14.2 ml) was added and stirred at RT for 12 h. The reaction mixture was basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as pale-yellow solid (1.00 g, 27 %). MP: 168-170°C. Mass: 416.5(M++1) Enantiomeric excess: 86.5% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 7.90 min.).
Method C : The title compound was separated by preparative SFC conditions from Compound A (1.090 g) on a CHIRALPAK AY-H column (250 x 30 mm; 5μπι) using methanol : C(¾ (35:65) as the mobile phase at a flow rate of 80 g / min. Off-white solid (0.378 g). e.e. 100%. Rt: 2.37 min. Mass: 416.1(M++1). MP: 149-152°C.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











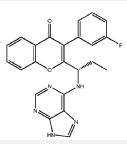 RP 6530
RP 6530