
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
 .
.

WCK ?
WATCH OUT FOR THIS POST, THIS MAY BE WCK 4234
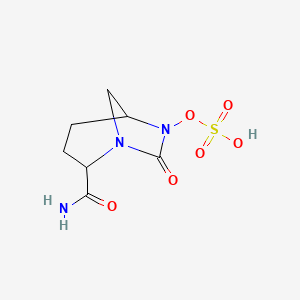
Cas 1427462-70-1, 1706523-58-1
| Molecular Formula: | C7H9N3O5S |
|---|---|
| Molecular Weight: | 247.22846 g/mol |
Sulfuric acid, mono[(1R,2S,5R)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
[(2S,5R)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl] hydrogen sulfate
CAS 1427462-59-6, 1804915-68-1, SODIUM SALT, (2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
1408/MUM/2014 and 1407/MUM/2014 INDIAN PATENT, WO2013038330
trans-7-oxo-6-(sulphooxy)-l,6-diazabicyclo[3.2.1]octane-2-carbonitrile
(2S, 5R)-7-oxo-6-(sulphooxy)-l,6-diazabicyclo [3.2.1]octane-2-carbonitrile
sulphuric acid, mono[(1R,2S,5R)-2-cyano-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl] ester
mono[(1R,2S,5R)-2-cyano-7-oxo-1,6- diazabicyclo[3.2.1]oct-6-yl] ester,
trans-7-oxo-6-(sulphoxy)-l,6-diazabicvclo[3.2.1]-octane-2- carbonitrile
Sodium salt (also known as “sodium salt of sulphuric acid, mono[(li?,25,5i?)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester” or “sulphuric acid, mono[(lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1: 1); CAS Registry Number: 1427462-59-6”); CAS 1804915-68-1

(2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
Potassium salt (also known as “potassium salt of sulphuric acid, mono[(li?,25,5i?)-2-cyano-7-oxo- l,6-diazabicyclo[3.2.1]oct-6-yl] ester” or “sulphuric acid, mono[(lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester, potassium salt (1: 1); CAS Registry Number: 1427462-60-9”); CAS 1804915-69-2
And
Other salts such as “l-butanarninium, Ν,Ν,Ν-tributyl-, (lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl sulphate (1: 1); CAS Registry Number: 1427462-72-3”.
PATENT
http://google.com/patents/WO2013038330A1?cl=en
Scheme 1

l( M = Na) a: Base,water, RT;b:Boc-anhydride,TEA,DIV1AP, DCIv1 , RT; c:LiOH, acetone; d: PivaloyI chloride, TEA; e. Ammonia(g); f:Trifluoroacetic anhydride,TEA,DC g: TFA, DC ; h: Triphosgene,TEA, D AP, DCM; i:H2, Pd/C; j:S03-DIVlF;
k: Tetrabutyl ammonium acetate, DCM; I: Dowex 50WX8 200 Na+ resin Scheme 2

a: Water, reflux, 24h; b:1-Hydroxybenzotriazole ammonium salt, DCC,D F; c: Boc-anhydride,TEA,D AP,DC ,RT; d:Trifluoroacetic anhydride,TEA, DCM;
e:TMSOI, NaH,DMSO,THF, -10°C 1 hr; f: O-Benzyl hydroxyl amine.HCI, EtOAc 60°C,2.5hr; g: Methane sulphonic acid, ethyl acetate,40°C; h:.KHC03, water, 55 °C;
i: sodium triacetoxy borohydride, STABH, H2S04; j: Triphosgene,TEA,DMAP,DCM;
Scheme-1 : further steps as depicted in scheme-1 Scheme 3

IX
: Water, reflux, 24h; b:1 -Hydroxybenzotriazole ammonium salt, DCC,D F;
: Boc-anhydride,TEA,D AP, DC ,rt; d:T SOI, NaH, D SO,THF, -1 0 °C 1 hr;
: O-Benzyl hydroxyl amine.HCI, EtOAc 60 °C, 2.5hr; f: Methane sulphonic acid, ethyl acetate, 40 °C g:.KHC03, water, 55 °C; g: sodium triacetoxy borohydride,
STABH, H2S04; h: Triphosgene,TEA,DMAP,DCIvl; i: Trifluoroacetic anhydride,
TEA, DCM; Scheme-1 : further steps as depicted in scheme-1
Step 1: Preparation of freebase and – Boc protection

The oxalate salt II (30g, 0.0697moles) was partitioned between water (300ml), and ethyl acetate (300ml) followed by addition of sodium bicarbonate (11.7gm, 0.139moles) under stirring. After lhr the organic layer was separated and the aqueous layer was extracted with ethyl acetate (150ml). The combined organic layer was washed with water (150ml) then brine (150ml), dried (over Na2S04) and the solvent evaporated under reduced pressure to obtain the free base Ila, 24gm.
To a cooled (5-10°C solution of the free base (24g, 0.0705moles) in DCM (240ml) were added triethylamine (19.68ml, 0.141moles), Boc anhydride (17.8ml, 0.0775moles) under stirring. After 30min. was added DMAP (0.86gm, 0.00705moles) and the resulting solution was allowed to warm to room temperature and stirred for a further 16hrs. The reaction mixture was diluted with saturated aqueous ammonium chloride solution (10ml), stirred well and the DCM layer was separated, washed with water (10ml) and finally with brine (10ml). The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with mixtures of ethyl acetate: hexane 25-50% and concentration of the combined fractions gave the product as a colorless oil, 25gm(yield: 80%).
MS: 439 [M+]; MF: C26H33NO5; MW: 439.
Step 2: Hydrolysis of Benzyl ester ^S | LiOH.Acetone Bn0 HN / ^-
N’^COOBn L JL
J N COOH X
To a solution of the compound lib (25gm, 0.0567moles) in acetone (500ml), at 0 °C, was added lithium hydroxide solution (3.8 lgm, 0.0908moles in mixture of 228.6ml water and 76.2 ml acetone) drop-wise under vigorous stirring. The reaction mixture was allowed to warm to RT and stirring continued further for 5hrs. The resulting mixture was cooled to 0 °C and pH adjusted to 8 to 8.5 with 2N HC1 (~10ml). The reaction mixture was diluted with brine (75ml) and toluene (250ml) under stirring, and after 10 minutes the organic layer was separated. The aqueous layer was re-extracted with toluene (2 X 120ml). The aqueous layer was acidified to pH 3-4 by using 2N HC1 and the solution extracted with ethyl acetate (3X200ml).,The combined organic layer was washed with water (200ml), and brine (200ml), dried (over Na2S04)and the solvent evaporated under reduced pressure to obtain the product as a thick oil, 21g, (quantitative yield).
MS: 349(M+); MF: C19H27NO5; MW: 349
Step 3: Conversion of Acid to Amide

IV V
To a stirred solution of compound IV (21gm, 0.06moles) in DCM (210ml) at 0°C was added TEA (25.12ml, 0.18moles) followed by slow addition of Pivaloyl chloride (11.07ml, 0.09moles). The resulting mixture was stirred further for 1.5hrs. The reaction mixture was cooled to -40°C and dry ammonia gas was bubbled through the reaction mixture for 30 min. The reaction mixture was allowed to warm to RT and the suspended white solid was filtered off. The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with a mixture of acetone: hexane system (1 :4) and concentration of the combined solvents gave the product, as thick oil, 10.2gm (yield: 49%)
MS: 348[M+] ; MF: C19H28N2O4; MW: 348.
Step 4: Conversion of Amide to Cyano

To a cooled (0°C) and stirred solution of compound VI (10.2gm, 0.0286moles) in DCM (306ml) was added Triethylamine (17.99ml, 1.289moles) and followed by the slow addition of Trifluoro acetic anhydride (12.08gm, 0.0573moles). The resulting solution was allowed to warm to RT and stirred for a further 6h. The reaction mixture was washed water (3* 100ml), Saturated ammonium chloride solution (100ml) and brine (100ml). The organic layer was dried (Na2S04) and the solvent evaporated under reduced pressure. The residue was chromatographed on a column of silica gel (60-120 mesh) using a mixture of Acetone: Hexane (1: 19). Concentration of the combined fractions gave the product, as a white solid, 9.7gm (yield – quantitative). MS: 331(M+); MF: C18H25N3O3; MW: 331
Step 5: Deprotection of Cyano

VI VII
To a chilled (-15°C) and stirred solution of compound VII (6gm,) in DCM (150ml) was added Trifluoro acetic acid (12ml) and the mixture was allowed to warm to RT. The reaction mixture was stirred for a further 4hrs. The solvent was evaporated under reduced pressure at 40± 5°C and the residue diluted with aqueous sat. sodium bicarbonate solution (60ml) and the mixture extracted with DCM (2 X 60ml). The combined extracts were washed with water (60ml), dried (over sodium sulphate) and evaporated under reduced pressure at 35± 5°C to obtain 4.2gm of compound VIII.
Step 6: Formation of bi-cyclic compound

To the cooled (0- 5°C) and stirred solution of compound VIII (4.2gm) in acetonitrile (63ml) was added triethyl amine (5.28ml) followed by a slow addition of a solution of Triphosgene (1.9gm) in Acetonitrile (16.8ml). Stirring was further continued for 30min. followed by addition of Dimethyl amino pyridine (0.178gm). The reaction mixture was allowed to warm to RT and stirred for further 16hrs. A aqueous sat. solution of sodium bicarbonate (33.6ml) was added to the reaction mixture and the resulting mixture stirred for 30min. The mixture was concentrated to l/3rd volume under reduced pressure. The residue was diluted with water (42ml) and the resulting mixture extracted with DCM (2 X 42ml). The solvent was evaporated under reduced pressure and the residue purified over a column of silica-gel (60 -120 mesh). Elution with a 1 :4 mixture of acetone: hexane and concentration of the combined fractions gave the product as white solid, 2.3g (yield: 48%).
MS: 314(M+); MF; Ci6Hi8N403; MW; 314 Step 7: Synthesis of TBA sulfate salt

To a solution of benzyl compound VIII (6 gm, 0.0233 mol) in a 1 : 1 mixture of DCM (30 ml)& DMF (30 ml), was added 1.5 gm of dry 10% Palladium charcoal and the mixture was hydrogenated under 3 kg Hydrogen pressure for 3 hour at 25-30°C.The reaction mixture was filtered through micron filter to remove catalyst and the filtrate concentrated under reduced pressure to obtain the debenzylated compound IX.
The debenzylated compound (IX) was dissolved in Ν,Ν’ -Dimethyl formamide (30 ml) under argon atmosphere and the solution cooled to 0°C. DMF: SO3 (4.26 gm, 0.0278mol) was added to the cooled solution and the stirring continued further for 30 min at 0°C. The mixture was then allowed to warm to RT and stirred for 1 hour. TLC showed complete conversion of N-Hydroxy compound to product X.
The solution containing the sulfate(X) was re-cooled to 0°C and a solution of Tetra butyl ammonium acetate (9 gm, 0.0301mol dissolved in 30ml water) was added to it. The reaction mixture was allowed to warm to 25°C and stirred for 1 hour. The volatiles were removed under reduced pressure and residue was co-evaporated with 2×50 ml Xylene to remove traces of Ν,Ν’ -Dimethyl formamide. The residue was partitioned between a 1: 1 mixture of water and dichloromethane (120ml). The aqueous layer was re-extracted with dichloromethane (30 ml). The combined organic extracts were washed with water (2x30ml), brine (30 ml). And dried over Na2S04 and the solvent evaporated under reduced pressure to obtain the crude TBA sulfate (5.2 gm). Crude compound was triturated with hexane (2×30 ml) & dried on rotavapor under 4mmHg pressure to obtain the TBA salt (XI), 5.0 g, yield-
44%.
Mass: 246 (M-H) of sulfate M.W: 488, M.F: C23H44N4O5S.
Step 8: Synthesis of Sodium salt of trans-7-oxo-6-(sulphoxy)-l,6-diazabicyclo[3.2.1]- octane-2-carbonitrile I

XI The TBA sulfate (4.4g, 0.009mol) was dissolved in 5% THF in water (2ml) and the solution was passed through column (45cm length and 2.0cm diameter) packed with Dowex 50WX8 200 Na+ resin. The column was eluted with 5% THF-water mixture (100ml). The combined fractions were evaporated under reduced pressure (4 mmHg) to obtain the product as white semi-solid, 1.5 gm, yield: 62%.
MS: 246 (M-H) of sulfate; M.W.: 269; M.F.: CyHgNaOsSNa,
XH NMR (DMSO):8 4.54 (d, 1H), 4.06 (s, 1H), 3.22 (m, 2H), 1.96 (m, 2H), 1.84 (m, 2H).
PATENT
(WO2015159167) PHARMACEUTICAL COMPOSITIONS COMPRISING ANTIBACTERIAL AGENTS
http://google.com/patents/WO2015159167A1?cl=en
PATENT
(2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
Patent
https://www.google.co.in/patents/WO2015114595A1?cl=en

EXAMPLES
Example 1
Synthesis of (25, 5R)-l,6-diaza-bicyclo r3.2.11octane-2-carbonitrile-7-oxo-6-(sulfooxy)- mono sodium salt
Step 1; Synthesis of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III):
Method 1:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (1 g, 0.00335 mol) in dichloromethane (15 ml), triethylamine hydrochloride (0.688 g, 0.00503 mol) was added in small portions at 25°C. After 30 minutes, triethylamine (0.678g, 0.0067 moles) was added, followed by addition of pivaloyl chloride (0.605 g, 0.00502 mol) at 0-5°C under stirring. After 2 hours, the reaction mass was cooled further to -20°C and aqueous ammonia (25% solution, 0.75 ml, 0.01 mol) was added slowly. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (10 ml) and the mixture was allowed to warm to room temperature. The dichloromethane layer was separated and the aqueous layer was re-extracted with dichloromethane (5 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride to obtain 0.75 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) as an off-white solid in 81 % yield.
Analysis:
Mass: 276.1 (M+l) for Molecular Weight of 275.31 and Molecular Formula of C14H17N303;
1H NMR (400MHz, CDC13): 57.43-7.35 (m, 5H), 6.56 (brs, 1H), 5.58 (brs, 1H), 5.07-4.89 (dd, 2H), 3.95-.393 (d, 1H), 3.31 (s, 1H), 3.04-3.01 (d, 1H), 2.78-2.75 (d, 1H), 2.38-2.32 (m, 1H), 2.03-1.88 (m, 2H), 1.64-1.58(m, 1H);
Purity as determined by HPLC: 98.9%.
Method 2:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo- l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in dimethylformamide (25 ml) pivaloyl chloride (3.03 g, 0.0251 mol) was added drop wise at about 0 – 5°C. After stirring for 3 hours, the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 3.75 ml, 0.0501 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (125 ml) and dichloromethane (50 ml), and allowed to warm to room temperature. The dichloromethane layer was separated and the aqueous layer extracted with fresh dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization using n-butyl chloride to obtain 0.7 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) as an off-white solid in 15 % yield.
Analysis:
Purity as determined by HPLC: 93.9%.
Method 3:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo- l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in tetrahydrofuran (50 ml), 1-methyl-2-pyrrolidinone (7.44 g, 0.0751 mol) and pivaloyl chloride (8.0 g, 0.0668 mol) was added at about 0 – 5°C. After stirring for 3 hours the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 6.2 ml, 0.0835 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (50 ml) and allowed to warm to room temperature. The tetrahydrofuran layer was separated and the aqueous layer was extracted with dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride to obtain 2.32 g of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) in 50 % yield.
Analysis:
Purity as determined by HPLC: 91.6%.
Method 4:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in tetrahydrofuran (50 ml), l-methyl-2-pyrrolidine (6.39 g, 0.0751 mol) and pivaloyl chloride (8.0 g, 0.0668 mol) was added at about 0 – 5°C. After stirring for 3 hours, the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 6.2 ml, 0.0835 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (50 ml) and allowed to warm to room temperature. The tetrahydrofuran layer was separated and the aqueous layer was extracted with dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride, to obtain 4.35 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) in 94% yield.
Analysis:
Purity as determined by HPLC: 97.6%.
Analytical data for (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide obtained from Method 2, 3 and 4 was consistent with that obtained in Method 1.
Step 2: Synthesis of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV):
Trifluoroacetic anhydride (48 ml, 0.340 mol) was added slowly to a solution of (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) (47 g, 0.170 mol) in dichloromethane, (1430 ml) containing triethylamine (107 ml, 0.765 mol), under stirring at about -5°C. After 2 hours, the reaction mixture was diluted with water (1450 ml) and the resulting mixture was stirred for further 15 minutes. The dichloromethane layer was separated, washed with aqueous saturated sodium bicarbonate solution (470 ml), brine (470 ml), dried (over anhydrous sodium sulfate) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (60-120 mesh) using acetone: hexane (0-15% acetone in hexane) solvents. The combined solvent fractions were concentrated under reduced pressure to obtain 32 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV) as a white solid in 74% yield.
Analysis:
Mass: 258 (M+l) for Molecular Weight of 257 and Molecular Formula of ![]()
1H NMR (400 MHz, DMSO): δ 7.42-7.36 (m, 5H), 5.06-4.88 (dd, 2H), 4.37-4.35 (d, 1H), 3.36-3.35 (m, 1H), 3.29-3.26 (d, 1H), 3.16-3.12 (m, 1H), 2.30-2.25 (m, 1H), 2.13-2.09(m, 1H), 1.90-1.83 (m, 2H);
Purity as determined by HPLC: 100%.
Step 3: Synthesis of (25, 5R)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (V):
A solution of (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV) (32 g, 0.124 mol) in a mixture of dimethylformamide and dichloromethane (1 : 1, 160 ml: 160 ml) containing 10% palladium on carbon (4.6 g, 50% wet) was hydro genated at 50-55 psi for 2 hours at 25 °C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of dimethylformamide and dichloromethane (1 : 1, 25 ml: 25 ml). The solvent from the combined filtrates was evaporated under reduced pressure to obtain 20.66 g of (25, 5i?)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (V) as an oil. The obtained product was used as such for the next reaction without further purification.
Step 4: Synthesis of (25, 5R)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (VI):
To a solution of (25,5i?)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (20.66 g, 0.124 mol) in dimethylformamide (160 ml), sulfur trioxide dimethylformamide complex (22.8 g, 0.149 mol) was added in one portion under stirring at about -5°C. After 60 minutes of stirring, the completion of the reaction was monitored by thin layer chromatography using mixture of chloroform and methanol (9: 1). To the resulting mixture was slowly added a solution of tetrabutylammomum acetate (48.6 g, 0.161 mol) in water (160 ml). After 1 hour of stirring, the solvent was evaporated under reduced pressure to obtain an oily residue. The oily residue was co-evaporated with xylene (2 x 200 ml), to yield a thick mass. This mass was partitioned between dichloromethane (320 ml) and water (320 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (160 ml). The combined organic extracts were washed with water (3 x 160 ml), dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure at about 35°C. The residual oily mass was triturated with ether (3 xl60 ml), each time the ether layer was decanted and finally the residue was dried under reduced pressure, to obtain 52.5 g of (25, 5i?)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutyl ammonium salt (VI) as an oil in 86% yield.
Analysis:
Mass: 246 (M-l) as free sulfonic acid; for Molecular Weight of 488 and Molecular Formula of C23H44N4O5S;
1H NMR (400 MHz, CDC13): δ 4.39 (brs, 1H), 4.34-4.32 (d, 1H), 3.41-3.33 (m, 2H), 3.27-3.22 (m, 8H), 2.28 (m, 2H), 1.89-1.84 (m, 2H), 1.67-1.59 (m, 8H), 1.47-1.37 (m, 8H), 1.00-0.96 (m, 12H);
Purity as determined by HPLC: 95.24%.
Step 5: Synthesis of (25, 5R)-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (I):
A column loaded with activated Amber lite 200 sodium resin (1200 gm) was washed with water followed by 10% tetrahydrofuran in water. A solution of (25,5i?)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammomum salt (VI) (51.5 g, 0.105 mol) in tetrahydrofuran (50 ml) was poured over the column. The column was further eluted by using 10% tetrahydrofuran in water. Tetrahydrofuran from the combined fractions was evaporated under reduced pressure and the aqueous layer extracted with ethyl acetate (5 x 250 ml). The aqueous layer was stirred with neutral charcoal (3 g) for 1 hour and then filtered through celite bed and further washed with water (100 ml). The combined filtrate was
evaporated under reduced pressure till free of moisture, to obtain 20.5 g of (25, 5i?)-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt in 72% yield.
Analysis:
Mass: 246 (M-1) as free sulfonic acid; for Molecular Weight of 269 and Molecular Formula of CvHgNsOsSNa;
1H NMR (400 MHz, DMSO): δ 4.56-4.54 (d, 1H), 4.08 (brs, 1H), 3.24-3.18 (m, 2H), 1.97-1.82 (m, 4H); and
Purity as determined by HPLC: 98.46%.
PATENT
WO 2015159265
http://google.com/patents/WO2015159265A1?cl=en
PATENT
WO 2015136387
https://www.google.co.in/patents/WO2015136387A1?cl=en
PATENT
WO 2015059642
http://www.google.com/patents/WO2015059642A1?cl=en
PATENT
http://www.google.com/patents/US20140296526
- Example 1
-

-
The oxalate salt (II) (30 gm, 0.0697 moles) was partitioned between water (300 ml), and ethyl acetate (300 ml) followed by addition of sodium bicarbonate (11.7 gm, 0.139 moles) under stirring. After 1 hour the organic layer was separated and the aqueous layer was extracted with ethyl acetate (150 ml). The combined organic layer was washed with water (150 ml) then brine (150 ml), dried (over sodium sulphate) and the solvent evaporated under reduced pressure to obtain the free base (IIa), 24 gm.
-
To a cooled (5-10° C. solution of the free base (24 gm, 0.0705 moles) in dichloromethane (240 ml) were added triethylamine (TEA) (19.68 ml, 0.141 moles), Boc anhydride ((Boc)2O) (17.8 ml, 0.0775 moles) under stiffing. After 30 minutes was added DMAP (0.86 gm, 0.00705 moles) and the resulting solution was allowed to warm to room temperature and stirred for a further 16 hours. The reaction mixture was diluted with saturated aqueous ammonium chloride solution (10 ml), stirred well and the dichloromethane layer was separated, washed with water (10 ml) and finally with brine (10 ml). The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with mixtures of ethyl acetate:hexane 25-50% and concentration of the combined fractions gave the product as colorless oil, 25 gm (yield: 80%).
-
Analysis:
-
Mass: 439 [M+]; Molecular Formula: C26H33NO5; Molecular Weight: 439.
- Preparation of Sodium salt of trans-7-oxo-6-(sulphoxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile IStep 1: Preparation of Freebase and -Boc Protection

Step 2: Hydrolysis of Benzyl Ester
-

-
To a solution of the compound (IIb) (25 gm, 0.0567 moles) in acetone (500 ml), at 0° C., was added lithium hydroxide solution (3.81 gm, 0.0908 moles in mixture of 228.6 ml water and 76.2 ml acetone) drop-wise under vigorous stiffing. The reaction mixture was allowed to warm to room temperature and stiffing continued further for 5 hours. The resulting mixture was cooled to 0° C. and pH adjusted to 8 to 8.5 with 2N HCl (about 10 ml). The reaction mixture was diluted with brine (75 ml) and toluene (250 ml) under stiffing, and after 10 minutes the organic layer was separated. The aqueous layer was re-extracted with toluene (2×120 ml). The aqueous layer was acidified to pH 3-4 by using 2N HCl and the solution extracted with ethyl acetate (3×200 ml). The combined organic layer was washed with water (200 ml), and brine (200 ml), dried (over sodium sulphate) and the solvent evaporated under reduced pressure to obtain the product (III) as a thick oil, 21 gm.
-
Analysis:
-
Mass: 349 (M+); Molecular Formula: C19H27NO5; Molecular Weight: 349.

Step 3: Conversion of Acid to Amide
-

-
To a stirred solution of compound (IV) (21 gm, 0.06 moles) in dichloromethane (210 ml) at 0° C. was added (triethylamine) TEA (25.12 ml, 0.18 moles) followed by slow addition of Pivaloyl chloride (11.07 ml, 0.09 moles). The resulting mixture was stirred further for 1.5 hours. The reaction mixture was cooled to −40° C. and dry ammonia gas was bubbled through the reaction mixture for 30 minutes. The reaction mixture was allowed to warm to room temperature and the suspended white solid was filtered off. The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with a mixture of acetone: hexane system (1:4) and concentration of the combined solvents gave the product (V), as thick oil, 10.2 gm (yield: 49%)
-
Analysis:
-
Mass: 348[M+]; Molecular Formula: C19H28N2O4; Molecular Weight: 348.

Step 4: Conversion of Amide to Cyano
-

-
To a cooled (0° C.) and stirred solution of compound (VI) (10.2 gm, 0.0286 moles) in dichloromethane (306 ml) was added triethylamine (TEA) (17.99 ml, 1.289 moles) and followed by the slow addition of trifluoroacetic anhydride (12.08 gm, 0.0573 moles). The resulting solution was allowed to warm to room temperature and stirred for a further 6 hours. The reaction mixture was washed with water (3×100 ml), Saturated ammonium chloride solution (100 ml) and brine (100 ml). The organic layer was dried (over sodium sulphate) and the solvent evaporated under reduced pressure. The residue was chromatographed on a column of silica gel (60-120 mesh) using a mixture of Acetone:Hexane (1:19). Concentration of the combined fractions gave the product, as a white solid, 9.7 gm (yield-quantitative).
-
Analysis:
-
Mass: 331(M+); Molecular Formula: C18H25N3O3; Molecular Weight: 331

Step 5: Deprotection of Cyano
-

-
To a chilled (−15° C.) and stirred solution of compound (VII) (6 gm,) in dichloromethane (150 ml) was added trifluoroacetic acid (12 ml) and the mixture was allowed to warm to room temperarture. The reaction mixture was stirred for a further 4 hours. The solvent was evaporated under reduced pressure at 40±5° C. and the residue diluted with aqueous saturated sodium bicarbonate solution (60 ml) and the mixture extracted with dichloromethane (2×60 ml). The combined extracts were washed with water (60 ml), dried (over sodium sulphate) and evaporated under reduced pressure at 35±5° C. to obtain 4.2 gm of compound (VIII).

Step 6: Formation of Bi-Cyclic Compound
-

-
To the cooled (0-5° C.) and stirred solution of compound (VIII) (4.2 gm) in acetonitrile (63 ml) was added triethyl amine (5.28 ml) followed by a slow addition of a solution of Triphosgene (1.9 gm) in Acetonitrile (16.8 ml). Stirring was further continued for 30 minutes followed by addition of Dimethylaminopyridine (DMAP) (0.178 gm). The reaction mixture was allowed to warm to room temperature and stirred for further 16 hours. A aqueous saturated solution of sodium bicarbonate (33.6 ml) was added to the reaction mixture and the resulting mixture stirred for 30 minutes. The mixture was concentrated to ⅓rd volume under reduced pressure. The residue was diluted with water (42 ml) and the resulting mixture extracted with dichloromethane (2×42 ml). The solvent was evaporated under reduced pressure and the residue purified over a column of silica-gel (60-120 mesh). Elution with a 1:4 mixture of acetone: hexane and concentration of the combined fractions gave the product as white solid, 2.3 gm (yield: 48%).
-
Analysis:
-
Mass: 314 (M+); Molecular Formula: C16H18N4O3; Molecular Weight: 314.

Step 7: Synthesis of TBA Sulfate Salt
-

-
To a solution of benzyl compound (VIII) (6 gm, 0.0233 mol) in a 1:1 mixture of dichloromethane (30 ml) and dimethylformamide (30 ml), was added 1.5 gm of dry 10% Palladium charcoal and the mixture was hydrogenated under 3 kg hydrogen pressure for 3 hour at 25-30° C. The reaction mixture was filtered through micron filter to remove catalyst and the filtrate concentrated under reduced pressure to obtain the debenzylated compound IX.
-
The debenzylated compound (IX) was dissolved in N,N′-Dimethyl formamide (30 ml) under argon atmosphere and the solution cooled to 0° C. Dimethylformamide sulfur trioxide complex (DMF: SO3) (4.26 gm, 0.0278 mol) was added to the cooled solution and the stiffing continued further for 30 minutes at 0° C. The mixture was then allowed to warm to room temperature and stirred for 1 hour. Thin layer chromatography showed complete conversion of N-Hydroxy compound to product (X).
-
The solution containing the sulfate (X) was re-cooled to 0° C. and a solution of tetra butyl ammonium acetate (TBAA) (9 gm, 0.0301 mol dissolved in 30 ml water) was added to it. The reaction mixture was allowed to warm to 25° C. and stirred for 1 hour. The volatiles were removed under reduced pressure and residue was co-evaporated with 2×50 ml xylene to remove traces of N,N′-Dimethyl formamide. The residue was partitioned between a 1:1 mixture of water and dichloromethane (120 ml). The aqueous layer was re-extracted with dichloromethane (30 ml). The combined organic extracts were washed with water (2×30 ml), brine (30 ml) and dried over sodium sulphate and the solvent evaporated under reduced pressure to obtain the crude TBA sulfate compound (XI) (5.2 gm). Crude compound was triturated with hexane (2×30 ml) and dried on rotavapor under 4 mm Hg pressure to obtain the TBA salt (XI), 5.0 gm, yield-44%.
-
Analysis:
-
Mass: 246 (M−1) of sulfate; Molecular Weight: 488, Molecular Formula: C23H44N4O5S.

Step 8: Synthesis of Sodium salt of trans-7-oxo-6-(sulphoxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile (I
-

-
The TBA sulfate compound (XI) (4.4 gm, 0.009 mol) was dissolved in 5% tetrahydrofuran (THF) in water (2 ml) and the solution was passed through column (45 cm length and 2.0 cm diameter) packed with Dowex 50WX8 200 Na+resin. The column was eluted with 5% THF-water mixture (100 ml). The combined fractions were evaporated under reduced pressure (4 mm Hg) to obtain the product (I) as white semi-solid, 1.5 gm, yield: 62%.
-
Analysis:
-
Mass: 246 (M−1) of sulfate; Molecular Weight: 269; Molecular Formula: C7H8N3O5SNa,
-
1H NMR (DMSO): δ 4.54 (d, 1H), 4.06 (s, 1H), 3.22 (m, 2H), 1.96 (m, 2H), 1.84 (m, 2H).

Example 2Preparation of Sodium salt of trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile IStep 1: Preparation of (S)-5-oxopyrrolidine-2-carboxamide (III)
-

-
To a stirred solution of L-pyroglutamic acid (II) (75 gm, 0.580 mol, commercially available) in dimethylformamide (750 ml) was added 1-hydroxy benzotriazole ammonium salt (106 gm, 0.696 mol, prepared according the literature procedure described in WO 2006100119) in one lot at 25° C. To this reaction mass, DCC was added in small portions over a period of 30 minutes at 0-5° C. The reaction mixture was allowed to warm to room temperature and stiffing continued further for 2 hours. The precipitates were removed by filtration and the filtrate concentrated under reduced pressure. The residue was treated with ethyl acetate (1000 ml) and stirred for 1 hour. The precipitate formed was filtered under suction and washed with additional ethyl acetate (2×75 ml). The combined filtrate was concentrated under reduced pressure to obtain 73 gm of (S)-5-oxopyrrolidine-2-carboxamide (III) as a white solid in 98% yield. The solid thus obtained was used without further purification in the next step.
-
Analysis:
-
Mass: 129 (M+1) for Molecular Weight: 128.13 and Molecular Formula: C5H8N2O2;
-
1H-NMR (400 MHz, DMSO): δ7.71 (s, 1H), 7.34 (s, 1H), 7.01 (s, 1H), 3.93-3.90 (m, 1H), 2.27-2.14 (m, 1H), 2.12-2.01 (m, 2H), 1.89-1.81 (m, 1H).

Step 2: Preparation of (S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate (IV)
-

-
To a cooled (0° C.), stirred solution of (S)-5-oxopyrrolidine-2-carboxamide (70 gm, 0.546 mol) in dimethylformamide (700 ml), triethylamine (TEA) (164.5 gm, 1.6 mol) was added in one lot. After stiffing for 5 minutes Boc anhydride [(Boc)2O] (225 gm, 1.031 mol) was added, followed by the addition of DMAP (6.7 gm, 0.0549 mol). Stirring was continued further for 3 hours, and the completion of the reaction was monitored by thin layer chromatography. The solvent was evaporated under reduced pressure, the residue was leached with diethyl ether (350 ml) and the same procedure repeated with additional diethyl ether (600 ml). The separated solid was filtered under suction and the residue washed with fresh diethyl ether (2×35 ml). The solid was dried at 2 mm Hg, at 45° C. for 2 hour, to obtain 102 gm of (S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate as white solid in 82% yield.
-
Analysis:
-
M.P.: 99-102° C.;
-
Mass m/z: 229 (M+H) for MW: 228 and M.F: C10H16N2O4;
-
1H NMR (400 MHz, DMSO): δ 7.60 (s, 1H), 7.15 (s, 1H), 4.42-4.39 (m, 1H), 2.48-2.32 (m, 2H), 2.20-2.15 (m, 1H), 1.77-1.72 (m, 1H), 1.38 (s, 9H).

Step 3: Preparation of (S)-tert-butyl 2-cyano-5-oxopyrrolidine-1-carboxylate (V)
-

-
Trifluoroacetic anhydride (178 gm, 0.845 mol) was added slowly to a stirred solution of (2S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate (IV) (97 gm, 0.425 mol), containing triethylamine (TEA) (193 gm, 1.907 mol) in dichloromethane (DCM) (2900 ml) at 0° C. After 2 hours of stirring, reaction mixture was diluted with water (1450 ml) and stirred further for 10 minutes. The organic layer was separated and washed with aqueous saturated solution of sodium hydrogen carbonate solution (500 ml), followed by brine (500 ml). The organic layer was dried over anhydrous sodium sulphate, and the solvent evaporated under reduced pressure. To the residue was added diethyl ether (200 ml), stirred well and the separated solid was filtered under suction to obtain the product. The filtrate was concentrated under reduced pressure and the residue was chromatographed on a column of silica gel using mixtures of ethyl acetate and hexane. The evaporation of the combined fractions gave 64.5 gm of (S)-tert-butyl 2-cyano-5-oxopyrrolidine-1-carboxylate (V) as white solid in 72% yield.
-
Analysis:
-
Melting point: 107-109° C.;
-
1H -NMR (400 MHz, DMSO): δ55.07-5.05 (m, 1H), 2.67-2.2.60 (m, 1H), 2.46-2.36 (m, 2H), 2.20-2.17 (m, 1H), 1.46 (s, 9H).

Step 4: Preparation of Sulfoxonium, [(5S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI)
-

-
Dimethyl sulfoxide (DMSO) (175 ml) was slowly added to a stirred suspension of sodium hydride (NaH) (7.3 gm, 0.182 mol, 60%) and trimethylsulfoxonium iodide (TMSOI) (40.2 gm, 0.182 mol) in tetrahydrofuran (THF) (140 ml) over a period of 1 hour at 25° C. The stirring was continued further for 1 hour and the resulting suspension cooled to −10° C. This suspension was slowly added to a stirred solution of (S)-tert-butyl-2-cyano-5-oxopyrrolidine-1-carboxylate (V) (35 gm, 0.166 mol, prepared according to the procedure described in step 3) in tetrahydrofuran (105 ml) containing triethylamine (TEA) (30 ml, 0.215 mol), over a period of 30 minutes at −10° C. Stirring was continued further for 1 hour at the same temperature. Saturated aqueous ammonium chloride solution (350 ml) was added to the reaction mass (after completion of the reaction as indicated by thin layer chromatography) and the reaction mixture was allowed to warm to 25° C. The organic layer was separated and the aqueous layer re-extracted by adding ethyl acetate (350 ml). The combined organic layer was washed with aqueous saturated solution of sodium hydrogen carbonate (350 ml) and brine (350 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. To the residual concentrate, diethyl ether (350 ml) was added and the mixture was stirred for 1 hour. The separated solid was filtered, and the residual solid was washed with additional diethyl ether (20 ml). The solid was dried under reduced pressure to provide 35 gm of Sulfoxonium, [(5S)-5[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI) as a white solid, in 70% yield.
-
Analysis:
-
Melting Point: 150-153° C.;
-
Mass: 303 (M+1) for Molecular Weight: 302 and Molecular Formula: C13H22N2O4S;
-
1H-NMR (400 MHz, CDCl3): δ 6.04 (br, 1H), 4.55 (br, 1H), 4.45 (s, 1H), 3.40-3.38 (d, 6H), 2.51-2.35 (m, 2H), 2.13-2.03 (m, 2H), 1.44 (s, 9H).

Step 5: Preparation of Carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(benzyloxy)imino]pentyl, 1,1-dimethylethyl ester (VII)
-

-
To a stirred solution of Sulfoxonium, [(5S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI) (15 gm, 0.049 mol, prepared according to the procedure described in step 4) in ethyl acetate (EtOAc) (225 ml) was added O-benzyl hydroxylamine hydrochloride (9.5 gm, 0.059 mol) in one lot, at 25° C. The reaction mixture was heated to 60° C. for 2.5 hours. After completion (checked by thin layer chromatography), the reaction mixture was allowed to cool to 25° C. and filtered to remove the precipitates. The filtrate was washed with water (75 ml) and brine (75 ml) and dried over anhydrous sodium sulphate. The solvent was evaporated under reduced pressure to obtain 17.5 gm of Carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(benzyloxy)imino]pentyl, 1,1-dimethylethyl ester (VII) as an oil in 96% yield.
-
Analysis:
-
Mass: 366 (M+1) for Molecular Weight: 365 and Molecular Formula: C18H24ClN3O3;
-
1H -NMR (400 MHz, CDCl3): δ 7.36-7.7.33 (m, 5H), 5.13 (s, 2H), 4.97 (br, 1H), 4.53 (br, 1H), 4.10 (s, 2H), 2.64-2.50 (m, 2H), 2.15-2.01 (m, 2H), 1.46 (s, 9H).

Step 6: Preparation of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX)
-

-
Methane sulphonic acid (9 ml, 0.138 mol) was slowly added to a stirred solution of carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(phenylmethoxy)imino]pentyl, 1,1-dimethylethyl ester (VII) (17 gm, 0.0465 mol, prepared according to the procedure described in step 5) in ethyl acetate (EtOAc) (130 ml), at 25° C. The resulting mixture was heated to 45° C., while monitoring the reaction with thin layer chromatography. After 45 minutes, the reaction mixture was allowed to cool to 25° C. and the resulting reaction mixture (Intermediate VIII) was slowly added to stirred aqueous suspension of potassium hydrogen carbonate (28 gm in 57 ml water). The resulting mixture was stirred and heated to 50-55° C. for 3 hours. The reaction mixture was allowed to cool to 25° C. and the organic layer was separated. The aqueous layer was re-extracted with ethyl acetate (100 ml). The combined organic layer was washed with water (75 ml) and brine (75 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure to obtain 11 gm of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX) as an oil.
-
Analysis:
-
1H-NMR (400 MHz, CDCl3): δ7.36-7.7.33 (m, 5H), 5.09 (s, 2H), 4.14-4.07 (m, 1H), 3.65-3.52 (m, 1H), 3.52-3.45 (m, 1H), 3.16-3.11 (m, 1H), 2.66-2.35 (m, 2H), 2.02-1.89 (m, 2H).

Step 7: Preparation of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X)
-

-
Sulphuric acid (11.7 ml, 0.217 mol) was slowly added to a stirred solution of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX) (10 gm, 0.0436 mol, prepared according to the procedure described in step 6) in ethyl acetate (150 ml) at −10° C. After 10 minutes of stirring, sodium triacetoxy borohydride (NaHB(OOCCH3)3) (11.7 gm, 0.0519 mol, 95% purity) was added in small portions while maintaining temperature below −5° C. After completion of the addition, stirring was further continued for 2 hour at the same temperature. The pH of the reaction mixture was adjusted to about pH 7 by using 30% aqueous potassium hydrogen carbonate solution. The mixture was allowed to warm to 25° C. and the reaction mixture was filtered under suction. The organic layer was separated and the aqueous layer extracted with fresh ethyl acetate (50 ml). The combined organic layer was washed with water (50 ml) and brine (50 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure to obtain 8.88 gm of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X) as an oil, in 88% yield. This was used as such for the next step without further purification.
-
Analysis:
-
Mass: 232 (M+1) for Molecular Weight: 231 and Molecular Formula: C13H17N3O.

Step 8: Preparation of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI)
-

-
A solution of oxalic acid dihydrate (5.28 gm, 0.0418 mol) in a mixture of ethyl acetate:acetone (1:1, 28 ml:28 ml) was slowly added to a stirred solution of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X) (8.8 gm, 0.0380 mol, prepared according to the procedure described in step 7) in ethyl acetate (35 ml) at 25° C. After 3 hour of stirring, the separated solid was filtered under suction, washed with additional 50 ml of v/v mixture of ethyl acetate: acetone solution (1:1, 25 ml: ml) and the solid dried under reduced pressure to obtain 6.7 gm of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI) in 55% yield.
-
Analysis:
-
Mass: 232 (M+1) for Molecular Weight: 321 and Molecular Formula: C13H17N3O.C2H2O4;
-
1H-NMR (400 MHz, DMSO): δ7.25 (m, 5H), 4.59 (s, 2H), 4.22 (br, 1H), 4.07-4.04 (m, 1H), 3.10-3.07 (m, 1H), 2.97-2.83 (m, 1H), 2.61-2.52 (m, 1H), 1.83-1.63 (m, 3H), 1.41-1.25 (m, 1H).

Separation of (2S,5R)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate from two isomeric (1:1) mixture of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate
-
A suspension of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI) (13 gm, 0.0404 moles) in methanol (260 ml) was heated under reflux, with stirring, for 3 hour. The resulted suspension was allowed to cool to 35° C. and the resulting suspension filtered under suction. The solid was washed with additional methanol (2×13 ml). The solid was dried under reduced pressure (4 mm Hg), to obtain (2S,5R)-5-[(benzyloxy) amino]-2-cyanopiperidine ethanedioate (XIA) as a white solid, 7.3 gm, yield 56%.
-
Analysis:
-
Mass m/z: 232.2 (M+H) for MW: 321 and M.F: C13H17N3O.C2H2O4.
-
1H-NMR (400 MHz, DMSO): δ 7.37-7.24 (m, 5H), 4.57 (s, 2H), 3.92-3.91 (m, 1H), 3.06-3.02 (m, 1H), 2.92-2.88 (m, 1H), 2.56-2.51 (m, 1H), 1.96-1.91 (m, 1H), 1.76-1.55 (m, 2H), 1.44-1.38 (m, 1H).
-
Purity as determined by HPLC: (2S,5R isomer) 88.44% (RT-9.74) and (2S,5S isomer) 5.47% (RT-8.61).
Step 9: Preparation of (2S,5R)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIII) and (2S,5S)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIV)
-

-
To a stirred suspension of (2S)-5-[(benzyloxy) amino]-2-cyanopiperidine ethanedioate (1:1) (XI) (3.7 gm, 0.0115 mol, prepared according to the procedure described in step 8) in ethyl acetate:water (1:1, 37 ml:37 ml) was added solid sodium bicarbonate (1.9 gm, 0.022 mol) at 25° C. After 30 minutes of stirring the organic layer was separated. The aqueous layer was re-extracted with ethyl acetate (20 ml). The combined organic layer was washed with water (20 ml) and brine (20 ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure to obtain 3 gm of ((2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (XII) as an oil. The oily product, (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (XII) (1 gm, 0.00432 mol, prepared as mentioned above), was dissolved in acetonitrile (ACN) (15 ml), cooled to 10° C., stirred and triethyl amine (1.8 ml, 0.0129 mol) was added in one portion. To this mixture was added slowly a solution of triphosgene (0.564 gm, 0.0019 mol) in acetonitrile (6 ml). After 15 minutes of stirring, DMAP (0.0527 gm, 0.000432 mol) was added and the reaction mixture allowed to warm to 25° C. After 16 hours of stirring, the thin layer chromatography (ethyl acetate:hexane (1:1)) showed the two separable mixture of isomers. A solution of saturated sodium bicarbonate (10 ml) was added to the reaction mass and stirring continued for another 30 minutes. The volatiles were removed under reduced pressure. The residual mass was partitioned between ethyl acetate (10 ml) and water (10 ml). The organic layer was separated and the aqueous layer re-extracted with ethyl acetate (10 ml). The combined organic layer was washed with water (10 ml) and brine (10 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The resulting mixture was dissolved in dichloromethane (15 ml) and washed with 5% potassium hydrogen sulphate solution (3×10 ml), saturated sodium hydrogen carbonate (10 ml) and water (10 ml). The organic layer was concentrated under reduced pressure, to yield 0.610 gm of crude oily product.
-
[0204]The oily mixture was purified by column chromatography using silica gel (60-120 mesh) by eluting with mixture of ethyl acetate and hexane. The upper spot was eluted out by using 25% ethyl acetate in hexane and the lower spot was eluted out by using 45% ethyl acetate in hexane. The combined pure fractions were concentrated under reduced pressure, to obtain the 0.130 gm of (2S,5R)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIII) and 0.105 gm of (2S,5S)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIV).
-
Analysis for compound of Formula (XIII):
-
Rf: 0.49;
-
Melting Point: 95-99° C.;
-
Mass: 258 (M+1) for Molecular Weight: 257 and Molecular Formula: C14H15N3O2;
-
1H-NMR (400 MHz, CDCl3): δ 7.43-7.35 (m, 5H), 5.06-5.03 (d, 1H), 4.91-4.88 (d, 1H), 4.38-4.36 (d, 1H), 3.36-3.29 (m, 2H), 3.16-3.12 (m, 1H), 2.33-2.10 (m, 2H), 1.90-1.79 (m, 2H).
-
Analysis for compound of Formula (XIV):
-
Rf: 0.12;
-
Melting Point: 115-118° C.
-
Mass: 258 (M+1) for Molecular Weight: 257 and Molecular Formula: C14H15N3O2;
-
1H-NMR (400 MHz, CDCl3): δ7.43-7.33 (m, 5H), 5.06-5.04 (d, 1H), 4.92-4.89 (d, 1H), 3.96-3.92 (dd, 1H), 3.32-3.23 (m, 2H), 2.76-2.73 (m, 1H), 2.29-2.18 (m, 2H), 2.05-1.99 (m, 1H), 1.71-1.63 (m, 1H).

Step 10: Preparation of (2S,5R)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIIIa)
-

-
A solution of (2S,5R)-6-(benzyloxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIII) (1 gm, 0.00389 mol) in a mixture of ethyl acetate and tetrahydrofuran (THF) (4:6, 4 ml:6 ml) containing 10% palladium over carbon (0.300 gm, 50% wet) was hydrogenated at 50-55 psi, for 6 hours at 25° C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of ethyl acetate and tetrahydrofuran (4:6, 4 ml:6 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain 0.649 gm of the titled compound of Formula (XIIIa) as oil, which was used as such for the next reaction without further purification.

Preparation of (2S,5S)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIVa)
-

-
A solution of (2S,5S)-6-(benzyloxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIV) (545 mg, 2.120 mol) in a mixture of ethyl acetate and tetrahydrofuran (5:5, 8 ml:8 ml) containing 10% palladium over carbon (0.109 gm, 50% wet) was hydrogenated at 50-55 psi, for 45 minutes at 25° C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of dichloromethane and dimethylformamide (5:5, 10 ml:10 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain the product as oil, which was triturated with diethyl ether (5 ml). The diethyl ether layer was decanted and the residue was dried under reduced pressure at 40° C. for 15 minutes to obtain 0.343 gm of compound of Formula (XIVa), which was used as such for the next step.

Step 11: Preparation of (2S,5R)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIII b)
-

-
To a stirred solution of (2S,5R)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIIIa) (0.649 gm, 0.00389 mol) in a mixture of dichloromethane (5 ml) and dimethylformamide (1 ml), sulfur trioxide dimethylformamide complex (1.07 gm, 0.007 mol) was added in one portion at about 10° C. After 90 minutes, the completion of the reaction was monitored by thin layer chromatography (9:1, chloroform:methanol). To the resulting reaction mass was added tetrabutylammonium hydrogen sulphate (TBAHS) in one portion (2.37 gm, 0.007 mol) under stirring. After 1 hour, water (10 ml) was added and the mixture stirred for 5 minutes. The organic layer was separated and washed with water (2×10 ml), dried (over anhydrous sodium sulphate) and the solvent evaporated under reduced pressure at 35° C. The residual oily mass was triturated with ether (2×10 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure, to obtain 0.6 gm of the titled compound of Formula (XIIIb) in 31% yield.
-
Analysis:
-
Mass: 246 (M−1), for Molecular Weight: 488 and Molecular Formula: C23H44N4O5S;
-
1H NMR (400 MHz, CDCl3): δ4.43 (brs, 1H), 4.35-4.33 (d, 1H), 3.47-3.44 (m, 2H), 3.28-3.24 (m, 8H), 2.33-2.29 (m, 2H), 1.92-1.85 (m, 2H), 1.69-1.61 (m, 8H), 1.48-1.39 (m, 8H), 1.02-0.98 (m, 12H).
-
Purity as determined by HPLC: 95.57%

Preparation of (2S,5S)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIVb)
-

-
To a stirred solution of (2S,5S)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIVa) (343 mg, 2.05 mol) in dimethylformamide (3 ml) sulfur trioxide dimethylformamide complex (390 mg, 2.549 mol) was added in one portion, at 10° C. and stirring continued further. After 60 minutes, thin layer chromatography (9:1, chloroform:methanol) showed the complete conversion. To the resulting reaction mixture was added, slowly, a solution of tetrabutylammonium acetate (TBAA) (831 mg, 2.756 mol) in water (3 ml) under stirring. After 1 hour of stirring, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2×10 ml), to yield a thick mass which was partitioned between dichloromethane (10 ml) and water (10 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (10 ml). The combined organic extracts were washed with water (3×10 ml), dried (over anhydrous sodium sulphate) and the solvent evaporated under reduced pressure at 35° C. The residual oily mass was triturated with ether (2×10 ml), each time the ether layer was decanted and finally the residue was dried under reduced pressure, to obtain 634 mg of compound of Formula (XIVb) as an oil in 61% yield.
-
Analysis:
-
Mass: 246 (M−1); for Molecular Weight: 488 and Molecular Formula: C23H44N4O5S;
-
1H NMR (400 MHz, CDCl3): δ4.38 (m, 1H), 3.98-3.93 (dd, 1H), 3.98-3.54 (m, 1H), 3.32-3.28 (m, 8H), 2.43-2.39 (m, 1H), 2.31-2.30 (m, 1H), 2.15-2.01 (m, 2H), 1.76-1.63 (m, 8H), 1.49-1.40 (m, 8H), 1.02-0.99 (m, 12H);
-
Purity as determined by HPLC: 98.22%.

Step 12: Preparation of (2S,5R)-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (I)
-

-
An activated Amberlite 200 sodium resin (20 gm) was loaded on a glass column and was washed with de-mineralized water (50 ml) followed by 10% tetrahydrofuran in water (50 ml). A solution of (2S,5R)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutyl ammonium salt (XIIIb) (575 mg, 1.176 mol) in tetrahydrofuran (THF) (1.1 ml) was loaded on column. It was eluted by using 10% tetrahydrofuran in water. The pure fractions were combined and the solvents evaporated under reduced pressure to obtain 280 mg of the compound of Formula (I) in 85% yield.
-
Analysis:
-
Mass: 246 (M−1) as free sulfonic acid, for Molecular Weight: 269 and Molecular Formula:
-
C7H8N3O5SNa;
-
1H NMR (400 MHz, DMSO): δ4.54-4.53 (d, 1H), 4.06 (brs, 1H), 3.20 (m, 2H), 1.96-1.81 (m, 4H);
-
Purity as determined by HPLC: 97.07%.

Preparation of (2S,5S)-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (Ia)
-

-
An activated Amberlite 200 sodium resin (20 gm) was loaded on a glass column and was washed with de-mineralized water (100 ml) followed by 10% tetrahydrofuran (THF) in water (100 ml). A solution of (2S,5S)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIVb) (475 mg, 0.971 mol) in tetrahydrofuran (1.5 ml) was loaded on column. It was eluted by using 10% tetrahydrofuran in water. The pure fractions were combined and the solvent evaporated under reduced pressure to obtain 242 mg of compound of Formula (Ia) as white solid, in 92% yield.
-
Analysis:
-
Mass: 246 (M−1) as free sulfonic acid, for Molecular Weight: 269 and Molecular Formula: C7H8N3O5SNa;
-
1H NMR (400 MHz, DMSO): δ 4.53-4.50 (dd, 1H), 3.98 (brs, 1H), 3.17-3.02 (dd, 2H), 1.99-1.96 (m, 2H), 1.77-1.75 (m, 2H);
-
Purity as determined by HPLC: 99.59%.

note
Avibactam is

1192500-31-4; SULFURIC ACID, MONO[(1R,2S,5R)-2-(AMINOCARBONYL)-7-OXO-1,6-DIAZABICYCLO[3.2.1]OCT-6-YL] ESTER;
COMPD IS
References
IN 2013MU03308
IN 2011MU02582
| Patent | Submitted | Granted |
|---|---|---|
| Nitrogen containing compounds and their use [US8969334] | 2014-05-04 | 2015-03-03 |
| Nitrogen containing compounds and their use [US8969567] | 2014-05-10 | 2015-03-03 |
| Nitrogen containing compounds and their use [US8754102] | 2012-09-11 | 2014-06-17 |
| WO2013014496A1 * | 4 Oct 2011 | 31 Jan 2013 | Wockhardt Limited | Pharmaceutical compositions comprising sulbactam and beta-lactamase inhibitor |
| WO2013038330A1 * | 11 Sep 2012 | 21 Mar 2013 | Wockhardt Limited | Nitrogen containing compounds and their use |
| WO2013030733A1 * | Aug 24, 2012 | Mar 7, 2013 | Wockhardt Limited | 1,6- diazabicyclo [3,2,1] octan-7-one derivatives and their use in the treatment of bacterial infections |
| WO2013038330A1 * | Sep 11, 2012 | Mar 21, 2013 | Wockhardt Limited | Nitrogen containing compounds and their use |
| WO2013149121A1 * | Mar 29, 2013 | Oct 3, 2013 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| WO2014108872A1 * | Jan 13, 2014 | Jul 17, 2014 | Wockhardt Limited | Compositions and methods for treating bacterial infections |
| CA2874279A1 * | May 30, 2013 | Dec 5, 2013 | Meiji Seika Pharma Co., Ltd. | Novel .beta.-lactamase inhibitor and process for preparing the same |
| US20130289012 * | Mar 29, 2013 | Oct 31, 2013 | Cubist Pharmaceuticals, Inc. | 1,2,4-oxadiazole and 1,2,4-thiadiazole beta-lactamase inhibitors |
////////
C1CC(N2CC1N(C2=O)OS(=O)(=O)O)C#N
or
C1C2CN(C(C1)C#N)C([C@@H]2OS(=O)(=O)O)=O
or
O=S(=O)(O)ON2C(=O)N1C[C@H]2CC[C@H]1C#N
////////

