
Home » Posts tagged 'wck'
Tag Archives: wck
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
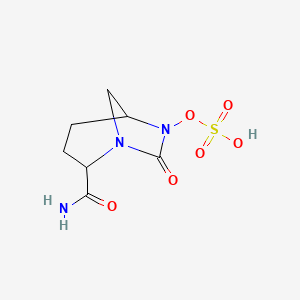
WCK ? trans-7-oxo-6-(sulphoxy)-1,6-diazabicvclo[3.2.1]-octane-2- carbonitrile from Wockhardt
 .
.

WCK ?
WATCH OUT FOR THIS POST, THIS MAY BE WCK 4234
Cas 1427462-70-1, 1706523-58-1
| Molecular Formula: | C7H9N3O5S |
|---|---|
| Molecular Weight: | 247.22846 g/mol |
Sulfuric acid, mono[(1R,2S,5R)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
[(2S,5R)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl] hydrogen sulfate
CAS 1427462-59-6, 1804915-68-1, SODIUM SALT, (2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
1408/MUM/2014 and 1407/MUM/2014 INDIAN PATENT, WO2013038330
trans-7-oxo-6-(sulphooxy)-l,6-diazabicyclo[3.2.1]octane-2-carbonitrile
(2S, 5R)-7-oxo-6-(sulphooxy)-l,6-diazabicyclo [3.2.1]octane-2-carbonitrile
sulphuric acid, mono[(1R,2S,5R)-2-cyano-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl] ester
mono[(1R,2S,5R)-2-cyano-7-oxo-1,6- diazabicyclo[3.2.1]oct-6-yl] ester,
trans-7-oxo-6-(sulphoxy)-l,6-diazabicvclo[3.2.1]-octane-2- carbonitrile
Sodium salt (also known as “sodium salt of sulphuric acid, mono[(li?,25,5i?)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester” or “sulphuric acid, mono[(lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1: 1); CAS Registry Number: 1427462-59-6”); CAS 1804915-68-1

(2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
Potassium salt (also known as “potassium salt of sulphuric acid, mono[(li?,25,5i?)-2-cyano-7-oxo- l,6-diazabicyclo[3.2.1]oct-6-yl] ester” or “sulphuric acid, mono[(lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester, potassium salt (1: 1); CAS Registry Number: 1427462-60-9”); CAS 1804915-69-2
And
Other salts such as “l-butanarninium, Ν,Ν,Ν-tributyl-, (lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl sulphate (1: 1); CAS Registry Number: 1427462-72-3”.
PATENT
http://google.com/patents/WO2013038330A1?cl=en
Scheme 1

l( M = Na) a: Base,water, RT;b:Boc-anhydride,TEA,DIV1AP, DCIv1 , RT; c:LiOH, acetone; d: PivaloyI chloride, TEA; e. Ammonia(g); f:Trifluoroacetic anhydride,TEA,DC g: TFA, DC ; h: Triphosgene,TEA, D AP, DCM; i:H2, Pd/C; j:S03-DIVlF;
k: Tetrabutyl ammonium acetate, DCM; I: Dowex 50WX8 200 Na+ resin Scheme 2

a: Water, reflux, 24h; b:1-Hydroxybenzotriazole ammonium salt, DCC,D F; c: Boc-anhydride,TEA,D AP,DC ,RT; d:Trifluoroacetic anhydride,TEA, DCM;
e:TMSOI, NaH,DMSO,THF, -10°C 1 hr; f: O-Benzyl hydroxyl amine.HCI, EtOAc 60°C,2.5hr; g: Methane sulphonic acid, ethyl acetate,40°C; h:.KHC03, water, 55 °C;
i: sodium triacetoxy borohydride, STABH, H2S04; j: Triphosgene,TEA,DMAP,DCM;
Scheme-1 : further steps as depicted in scheme-1 Scheme 3

IX
: Water, reflux, 24h; b:1 -Hydroxybenzotriazole ammonium salt, DCC,D F;
: Boc-anhydride,TEA,D AP, DC ,rt; d:T SOI, NaH, D SO,THF, -1 0 °C 1 hr;
: O-Benzyl hydroxyl amine.HCI, EtOAc 60 °C, 2.5hr; f: Methane sulphonic acid, ethyl acetate, 40 °C g:.KHC03, water, 55 °C; g: sodium triacetoxy borohydride,
STABH, H2S04; h: Triphosgene,TEA,DMAP,DCIvl; i: Trifluoroacetic anhydride,
TEA, DCM; Scheme-1 : further steps as depicted in scheme-1
Step 1: Preparation of freebase and – Boc protection

The oxalate salt II (30g, 0.0697moles) was partitioned between water (300ml), and ethyl acetate (300ml) followed by addition of sodium bicarbonate (11.7gm, 0.139moles) under stirring. After lhr the organic layer was separated and the aqueous layer was extracted with ethyl acetate (150ml). The combined organic layer was washed with water (150ml) then brine (150ml), dried (over Na2S04) and the solvent evaporated under reduced pressure to obtain the free base Ila, 24gm.
To a cooled (5-10°C solution of the free base (24g, 0.0705moles) in DCM (240ml) were added triethylamine (19.68ml, 0.141moles), Boc anhydride (17.8ml, 0.0775moles) under stirring. After 30min. was added DMAP (0.86gm, 0.00705moles) and the resulting solution was allowed to warm to room temperature and stirred for a further 16hrs. The reaction mixture was diluted with saturated aqueous ammonium chloride solution (10ml), stirred well and the DCM layer was separated, washed with water (10ml) and finally with brine (10ml). The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with mixtures of ethyl acetate: hexane 25-50% and concentration of the combined fractions gave the product as a colorless oil, 25gm(yield: 80%).
MS: 439 [M+]; MF: C26H33NO5; MW: 439.
Step 2: Hydrolysis of Benzyl ester ^S | LiOH.Acetone Bn0 HN / ^-
N’^COOBn L JL
J N COOH X
To a solution of the compound lib (25gm, 0.0567moles) in acetone (500ml), at 0 °C, was added lithium hydroxide solution (3.8 lgm, 0.0908moles in mixture of 228.6ml water and 76.2 ml acetone) drop-wise under vigorous stirring. The reaction mixture was allowed to warm to RT and stirring continued further for 5hrs. The resulting mixture was cooled to 0 °C and pH adjusted to 8 to 8.5 with 2N HC1 (~10ml). The reaction mixture was diluted with brine (75ml) and toluene (250ml) under stirring, and after 10 minutes the organic layer was separated. The aqueous layer was re-extracted with toluene (2 X 120ml). The aqueous layer was acidified to pH 3-4 by using 2N HC1 and the solution extracted with ethyl acetate (3X200ml).,The combined organic layer was washed with water (200ml), and brine (200ml), dried (over Na2S04)and the solvent evaporated under reduced pressure to obtain the product as a thick oil, 21g, (quantitative yield).
MS: 349(M+); MF: C19H27NO5; MW: 349
Step 3: Conversion of Acid to Amide

IV V
To a stirred solution of compound IV (21gm, 0.06moles) in DCM (210ml) at 0°C was added TEA (25.12ml, 0.18moles) followed by slow addition of Pivaloyl chloride (11.07ml, 0.09moles). The resulting mixture was stirred further for 1.5hrs. The reaction mixture was cooled to -40°C and dry ammonia gas was bubbled through the reaction mixture for 30 min. The reaction mixture was allowed to warm to RT and the suspended white solid was filtered off. The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with a mixture of acetone: hexane system (1 :4) and concentration of the combined solvents gave the product, as thick oil, 10.2gm (yield: 49%)
MS: 348[M+] ; MF: C19H28N2O4; MW: 348.
Step 4: Conversion of Amide to Cyano

To a cooled (0°C) and stirred solution of compound VI (10.2gm, 0.0286moles) in DCM (306ml) was added Triethylamine (17.99ml, 1.289moles) and followed by the slow addition of Trifluoro acetic anhydride (12.08gm, 0.0573moles). The resulting solution was allowed to warm to RT and stirred for a further 6h. The reaction mixture was washed water (3* 100ml), Saturated ammonium chloride solution (100ml) and brine (100ml). The organic layer was dried (Na2S04) and the solvent evaporated under reduced pressure. The residue was chromatographed on a column of silica gel (60-120 mesh) using a mixture of Acetone: Hexane (1: 19). Concentration of the combined fractions gave the product, as a white solid, 9.7gm (yield – quantitative). MS: 331(M+); MF: C18H25N3O3; MW: 331
Step 5: Deprotection of Cyano

VI VII
To a chilled (-15°C) and stirred solution of compound VII (6gm,) in DCM (150ml) was added Trifluoro acetic acid (12ml) and the mixture was allowed to warm to RT. The reaction mixture was stirred for a further 4hrs. The solvent was evaporated under reduced pressure at 40± 5°C and the residue diluted with aqueous sat. sodium bicarbonate solution (60ml) and the mixture extracted with DCM (2 X 60ml). The combined extracts were washed with water (60ml), dried (over sodium sulphate) and evaporated under reduced pressure at 35± 5°C to obtain 4.2gm of compound VIII.
Step 6: Formation of bi-cyclic compound

To the cooled (0- 5°C) and stirred solution of compound VIII (4.2gm) in acetonitrile (63ml) was added triethyl amine (5.28ml) followed by a slow addition of a solution of Triphosgene (1.9gm) in Acetonitrile (16.8ml). Stirring was further continued for 30min. followed by addition of Dimethyl amino pyridine (0.178gm). The reaction mixture was allowed to warm to RT and stirred for further 16hrs. A aqueous sat. solution of sodium bicarbonate (33.6ml) was added to the reaction mixture and the resulting mixture stirred for 30min. The mixture was concentrated to l/3rd volume under reduced pressure. The residue was diluted with water (42ml) and the resulting mixture extracted with DCM (2 X 42ml). The solvent was evaporated under reduced pressure and the residue purified over a column of silica-gel (60 -120 mesh). Elution with a 1 :4 mixture of acetone: hexane and concentration of the combined fractions gave the product as white solid, 2.3g (yield: 48%).
MS: 314(M+); MF; Ci6Hi8N403; MW; 314 Step 7: Synthesis of TBA sulfate salt

To a solution of benzyl compound VIII (6 gm, 0.0233 mol) in a 1 : 1 mixture of DCM (30 ml)& DMF (30 ml), was added 1.5 gm of dry 10% Palladium charcoal and the mixture was hydrogenated under 3 kg Hydrogen pressure for 3 hour at 25-30°C.The reaction mixture was filtered through micron filter to remove catalyst and the filtrate concentrated under reduced pressure to obtain the debenzylated compound IX.
The debenzylated compound (IX) was dissolved in Ν,Ν’ -Dimethyl formamide (30 ml) under argon atmosphere and the solution cooled to 0°C. DMF: SO3 (4.26 gm, 0.0278mol) was added to the cooled solution and the stirring continued further for 30 min at 0°C. The mixture was then allowed to warm to RT and stirred for 1 hour. TLC showed complete conversion of N-Hydroxy compound to product X.
The solution containing the sulfate(X) was re-cooled to 0°C and a solution of Tetra butyl ammonium acetate (9 gm, 0.0301mol dissolved in 30ml water) was added to it. The reaction mixture was allowed to warm to 25°C and stirred for 1 hour. The volatiles were removed under reduced pressure and residue was co-evaporated with 2×50 ml Xylene to remove traces of Ν,Ν’ -Dimethyl formamide. The residue was partitioned between a 1: 1 mixture of water and dichloromethane (120ml). The aqueous layer was re-extracted with dichloromethane (30 ml). The combined organic extracts were washed with water (2x30ml), brine (30 ml). And dried over Na2S04 and the solvent evaporated under reduced pressure to obtain the crude TBA sulfate (5.2 gm). Crude compound was triturated with hexane (2×30 ml) & dried on rotavapor under 4mmHg pressure to obtain the TBA salt (XI), 5.0 g, yield-
44%.
Mass: 246 (M-H) of sulfate M.W: 488, M.F: C23H44N4O5S.
Step 8: Synthesis of Sodium salt of trans-7-oxo-6-(sulphoxy)-l,6-diazabicyclo[3.2.1]- octane-2-carbonitrile I

XI The TBA sulfate (4.4g, 0.009mol) was dissolved in 5% THF in water (2ml) and the solution was passed through column (45cm length and 2.0cm diameter) packed with Dowex 50WX8 200 Na+ resin. The column was eluted with 5% THF-water mixture (100ml). The combined fractions were evaporated under reduced pressure (4 mmHg) to obtain the product as white semi-solid, 1.5 gm, yield: 62%.
MS: 246 (M-H) of sulfate; M.W.: 269; M.F.: CyHgNaOsSNa,
XH NMR (DMSO):8 4.54 (d, 1H), 4.06 (s, 1H), 3.22 (m, 2H), 1.96 (m, 2H), 1.84 (m, 2H).
PATENT
(WO2015159167) PHARMACEUTICAL COMPOSITIONS COMPRISING ANTIBACTERIAL AGENTS
http://google.com/patents/WO2015159167A1?cl=en
PATENT
(2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
Patent
https://www.google.co.in/patents/WO2015114595A1?cl=en

EXAMPLES
Example 1
Synthesis of (25, 5R)-l,6-diaza-bicyclo r3.2.11octane-2-carbonitrile-7-oxo-6-(sulfooxy)- mono sodium salt
Step 1; Synthesis of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III):
Method 1:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (1 g, 0.00335 mol) in dichloromethane (15 ml), triethylamine hydrochloride (0.688 g, 0.00503 mol) was added in small portions at 25°C. After 30 minutes, triethylamine (0.678g, 0.0067 moles) was added, followed by addition of pivaloyl chloride (0.605 g, 0.00502 mol) at 0-5°C under stirring. After 2 hours, the reaction mass was cooled further to -20°C and aqueous ammonia (25% solution, 0.75 ml, 0.01 mol) was added slowly. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (10 ml) and the mixture was allowed to warm to room temperature. The dichloromethane layer was separated and the aqueous layer was re-extracted with dichloromethane (5 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride to obtain 0.75 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) as an off-white solid in 81 % yield.
Analysis:
Mass: 276.1 (M+l) for Molecular Weight of 275.31 and Molecular Formula of C14H17N303;
1H NMR (400MHz, CDC13): 57.43-7.35 (m, 5H), 6.56 (brs, 1H), 5.58 (brs, 1H), 5.07-4.89 (dd, 2H), 3.95-.393 (d, 1H), 3.31 (s, 1H), 3.04-3.01 (d, 1H), 2.78-2.75 (d, 1H), 2.38-2.32 (m, 1H), 2.03-1.88 (m, 2H), 1.64-1.58(m, 1H);
Purity as determined by HPLC: 98.9%.
Method 2:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo- l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in dimethylformamide (25 ml) pivaloyl chloride (3.03 g, 0.0251 mol) was added drop wise at about 0 – 5°C. After stirring for 3 hours, the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 3.75 ml, 0.0501 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (125 ml) and dichloromethane (50 ml), and allowed to warm to room temperature. The dichloromethane layer was separated and the aqueous layer extracted with fresh dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization using n-butyl chloride to obtain 0.7 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) as an off-white solid in 15 % yield.
Analysis:
Purity as determined by HPLC: 93.9%.
Method 3:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo- l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in tetrahydrofuran (50 ml), 1-methyl-2-pyrrolidinone (7.44 g, 0.0751 mol) and pivaloyl chloride (8.0 g, 0.0668 mol) was added at about 0 – 5°C. After stirring for 3 hours the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 6.2 ml, 0.0835 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (50 ml) and allowed to warm to room temperature. The tetrahydrofuran layer was separated and the aqueous layer was extracted with dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride to obtain 2.32 g of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) in 50 % yield.
Analysis:
Purity as determined by HPLC: 91.6%.
Method 4:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in tetrahydrofuran (50 ml), l-methyl-2-pyrrolidine (6.39 g, 0.0751 mol) and pivaloyl chloride (8.0 g, 0.0668 mol) was added at about 0 – 5°C. After stirring for 3 hours, the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 6.2 ml, 0.0835 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (50 ml) and allowed to warm to room temperature. The tetrahydrofuran layer was separated and the aqueous layer was extracted with dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride, to obtain 4.35 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) in 94% yield.
Analysis:
Purity as determined by HPLC: 97.6%.
Analytical data for (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide obtained from Method 2, 3 and 4 was consistent with that obtained in Method 1.
Step 2: Synthesis of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV):
Trifluoroacetic anhydride (48 ml, 0.340 mol) was added slowly to a solution of (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) (47 g, 0.170 mol) in dichloromethane, (1430 ml) containing triethylamine (107 ml, 0.765 mol), under stirring at about -5°C. After 2 hours, the reaction mixture was diluted with water (1450 ml) and the resulting mixture was stirred for further 15 minutes. The dichloromethane layer was separated, washed with aqueous saturated sodium bicarbonate solution (470 ml), brine (470 ml), dried (over anhydrous sodium sulfate) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (60-120 mesh) using acetone: hexane (0-15% acetone in hexane) solvents. The combined solvent fractions were concentrated under reduced pressure to obtain 32 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV) as a white solid in 74% yield.
Analysis:
Mass: 258 (M+l) for Molecular Weight of 257 and Molecular Formula of ![]()
1H NMR (400 MHz, DMSO): δ 7.42-7.36 (m, 5H), 5.06-4.88 (dd, 2H), 4.37-4.35 (d, 1H), 3.36-3.35 (m, 1H), 3.29-3.26 (d, 1H), 3.16-3.12 (m, 1H), 2.30-2.25 (m, 1H), 2.13-2.09(m, 1H), 1.90-1.83 (m, 2H);
Purity as determined by HPLC: 100%.
Step 3: Synthesis of (25, 5R)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (V):
A solution of (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV) (32 g, 0.124 mol) in a mixture of dimethylformamide and dichloromethane (1 : 1, 160 ml: 160 ml) containing 10% palladium on carbon (4.6 g, 50% wet) was hydro genated at 50-55 psi for 2 hours at 25 °C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of dimethylformamide and dichloromethane (1 : 1, 25 ml: 25 ml). The solvent from the combined filtrates was evaporated under reduced pressure to obtain 20.66 g of (25, 5i?)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (V) as an oil. The obtained product was used as such for the next reaction without further purification.
Step 4: Synthesis of (25, 5R)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (VI):
To a solution of (25,5i?)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (20.66 g, 0.124 mol) in dimethylformamide (160 ml), sulfur trioxide dimethylformamide complex (22.8 g, 0.149 mol) was added in one portion under stirring at about -5°C. After 60 minutes of stirring, the completion of the reaction was monitored by thin layer chromatography using mixture of chloroform and methanol (9: 1). To the resulting mixture was slowly added a solution of tetrabutylammomum acetate (48.6 g, 0.161 mol) in water (160 ml). After 1 hour of stirring, the solvent was evaporated under reduced pressure to obtain an oily residue. The oily residue was co-evaporated with xylene (2 x 200 ml), to yield a thick mass. This mass was partitioned between dichloromethane (320 ml) and water (320 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (160 ml). The combined organic extracts were washed with water (3 x 160 ml), dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure at about 35°C. The residual oily mass was triturated with ether (3 xl60 ml), each time the ether layer was decanted and finally the residue was dried under reduced pressure, to obtain 52.5 g of (25, 5i?)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutyl ammonium salt (VI) as an oil in 86% yield.
Analysis:
Mass: 246 (M-l) as free sulfonic acid; for Molecular Weight of 488 and Molecular Formula of C23H44N4O5S;
1H NMR (400 MHz, CDC13): δ 4.39 (brs, 1H), 4.34-4.32 (d, 1H), 3.41-3.33 (m, 2H), 3.27-3.22 (m, 8H), 2.28 (m, 2H), 1.89-1.84 (m, 2H), 1.67-1.59 (m, 8H), 1.47-1.37 (m, 8H), 1.00-0.96 (m, 12H);
Purity as determined by HPLC: 95.24%.
Step 5: Synthesis of (25, 5R)-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (I):
A column loaded with activated Amber lite 200 sodium resin (1200 gm) was washed with water followed by 10% tetrahydrofuran in water. A solution of (25,5i?)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammomum salt (VI) (51.5 g, 0.105 mol) in tetrahydrofuran (50 ml) was poured over the column. The column was further eluted by using 10% tetrahydrofuran in water. Tetrahydrofuran from the combined fractions was evaporated under reduced pressure and the aqueous layer extracted with ethyl acetate (5 x 250 ml). The aqueous layer was stirred with neutral charcoal (3 g) for 1 hour and then filtered through celite bed and further washed with water (100 ml). The combined filtrate was
evaporated under reduced pressure till free of moisture, to obtain 20.5 g of (25, 5i?)-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt in 72% yield.
Analysis:
Mass: 246 (M-1) as free sulfonic acid; for Molecular Weight of 269 and Molecular Formula of CvHgNsOsSNa;
1H NMR (400 MHz, DMSO): δ 4.56-4.54 (d, 1H), 4.08 (brs, 1H), 3.24-3.18 (m, 2H), 1.97-1.82 (m, 4H); and
Purity as determined by HPLC: 98.46%.
PATENT
WO 2015159265
http://google.com/patents/WO2015159265A1?cl=en
PATENT
WO 2015136387
https://www.google.co.in/patents/WO2015136387A1?cl=en
PATENT
WO 2015059642
http://www.google.com/patents/WO2015059642A1?cl=en
PATENT
http://www.google.com/patents/US20140296526
- Example 1
-

-
The oxalate salt (II) (30 gm, 0.0697 moles) was partitioned between water (300 ml), and ethyl acetate (300 ml) followed by addition of sodium bicarbonate (11.7 gm, 0.139 moles) under stirring. After 1 hour the organic layer was separated and the aqueous layer was extracted with ethyl acetate (150 ml). The combined organic layer was washed with water (150 ml) then brine (150 ml), dried (over sodium sulphate) and the solvent evaporated under reduced pressure to obtain the free base (IIa), 24 gm.
-
To a cooled (5-10° C. solution of the free base (24 gm, 0.0705 moles) in dichloromethane (240 ml) were added triethylamine (TEA) (19.68 ml, 0.141 moles), Boc anhydride ((Boc)2O) (17.8 ml, 0.0775 moles) under stiffing. After 30 minutes was added DMAP (0.86 gm, 0.00705 moles) and the resulting solution was allowed to warm to room temperature and stirred for a further 16 hours. The reaction mixture was diluted with saturated aqueous ammonium chloride solution (10 ml), stirred well and the dichloromethane layer was separated, washed with water (10 ml) and finally with brine (10 ml). The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with mixtures of ethyl acetate:hexane 25-50% and concentration of the combined fractions gave the product as colorless oil, 25 gm (yield: 80%).
-
Analysis:
-
Mass: 439 [M+]; Molecular Formula: C26H33NO5; Molecular Weight: 439.
- Preparation of Sodium salt of trans-7-oxo-6-(sulphoxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile IStep 1: Preparation of Freebase and -Boc Protection

Step 2: Hydrolysis of Benzyl Ester
-

-
To a solution of the compound (IIb) (25 gm, 0.0567 moles) in acetone (500 ml), at 0° C., was added lithium hydroxide solution (3.81 gm, 0.0908 moles in mixture of 228.6 ml water and 76.2 ml acetone) drop-wise under vigorous stiffing. The reaction mixture was allowed to warm to room temperature and stiffing continued further for 5 hours. The resulting mixture was cooled to 0° C. and pH adjusted to 8 to 8.5 with 2N HCl (about 10 ml). The reaction mixture was diluted with brine (75 ml) and toluene (250 ml) under stiffing, and after 10 minutes the organic layer was separated. The aqueous layer was re-extracted with toluene (2×120 ml). The aqueous layer was acidified to pH 3-4 by using 2N HCl and the solution extracted with ethyl acetate (3×200 ml). The combined organic layer was washed with water (200 ml), and brine (200 ml), dried (over sodium sulphate) and the solvent evaporated under reduced pressure to obtain the product (III) as a thick oil, 21 gm.
-
Analysis:
-
Mass: 349 (M+); Molecular Formula: C19H27NO5; Molecular Weight: 349.

Step 3: Conversion of Acid to Amide
-

-
To a stirred solution of compound (IV) (21 gm, 0.06 moles) in dichloromethane (210 ml) at 0° C. was added (triethylamine) TEA (25.12 ml, 0.18 moles) followed by slow addition of Pivaloyl chloride (11.07 ml, 0.09 moles). The resulting mixture was stirred further for 1.5 hours. The reaction mixture was cooled to −40° C. and dry ammonia gas was bubbled through the reaction mixture for 30 minutes. The reaction mixture was allowed to warm to room temperature and the suspended white solid was filtered off. The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with a mixture of acetone: hexane system (1:4) and concentration of the combined solvents gave the product (V), as thick oil, 10.2 gm (yield: 49%)
-
Analysis:
-
Mass: 348[M+]; Molecular Formula: C19H28N2O4; Molecular Weight: 348.

Step 4: Conversion of Amide to Cyano
-

-
To a cooled (0° C.) and stirred solution of compound (VI) (10.2 gm, 0.0286 moles) in dichloromethane (306 ml) was added triethylamine (TEA) (17.99 ml, 1.289 moles) and followed by the slow addition of trifluoroacetic anhydride (12.08 gm, 0.0573 moles). The resulting solution was allowed to warm to room temperature and stirred for a further 6 hours. The reaction mixture was washed with water (3×100 ml), Saturated ammonium chloride solution (100 ml) and brine (100 ml). The organic layer was dried (over sodium sulphate) and the solvent evaporated under reduced pressure. The residue was chromatographed on a column of silica gel (60-120 mesh) using a mixture of Acetone:Hexane (1:19). Concentration of the combined fractions gave the product, as a white solid, 9.7 gm (yield-quantitative).
-
Analysis:
-
Mass: 331(M+); Molecular Formula: C18H25N3O3; Molecular Weight: 331

Step 5: Deprotection of Cyano
-

-
To a chilled (−15° C.) and stirred solution of compound (VII) (6 gm,) in dichloromethane (150 ml) was added trifluoroacetic acid (12 ml) and the mixture was allowed to warm to room temperarture. The reaction mixture was stirred for a further 4 hours. The solvent was evaporated under reduced pressure at 40±5° C. and the residue diluted with aqueous saturated sodium bicarbonate solution (60 ml) and the mixture extracted with dichloromethane (2×60 ml). The combined extracts were washed with water (60 ml), dried (over sodium sulphate) and evaporated under reduced pressure at 35±5° C. to obtain 4.2 gm of compound (VIII).

Step 6: Formation of Bi-Cyclic Compound
-

-
To the cooled (0-5° C.) and stirred solution of compound (VIII) (4.2 gm) in acetonitrile (63 ml) was added triethyl amine (5.28 ml) followed by a slow addition of a solution of Triphosgene (1.9 gm) in Acetonitrile (16.8 ml). Stirring was further continued for 30 minutes followed by addition of Dimethylaminopyridine (DMAP) (0.178 gm). The reaction mixture was allowed to warm to room temperature and stirred for further 16 hours. A aqueous saturated solution of sodium bicarbonate (33.6 ml) was added to the reaction mixture and the resulting mixture stirred for 30 minutes. The mixture was concentrated to ⅓rd volume under reduced pressure. The residue was diluted with water (42 ml) and the resulting mixture extracted with dichloromethane (2×42 ml). The solvent was evaporated under reduced pressure and the residue purified over a column of silica-gel (60-120 mesh). Elution with a 1:4 mixture of acetone: hexane and concentration of the combined fractions gave the product as white solid, 2.3 gm (yield: 48%).
-
Analysis:
-
Mass: 314 (M+); Molecular Formula: C16H18N4O3; Molecular Weight: 314.

Step 7: Synthesis of TBA Sulfate Salt
-

-
To a solution of benzyl compound (VIII) (6 gm, 0.0233 mol) in a 1:1 mixture of dichloromethane (30 ml) and dimethylformamide (30 ml), was added 1.5 gm of dry 10% Palladium charcoal and the mixture was hydrogenated under 3 kg hydrogen pressure for 3 hour at 25-30° C. The reaction mixture was filtered through micron filter to remove catalyst and the filtrate concentrated under reduced pressure to obtain the debenzylated compound IX.
-
The debenzylated compound (IX) was dissolved in N,N′-Dimethyl formamide (30 ml) under argon atmosphere and the solution cooled to 0° C. Dimethylformamide sulfur trioxide complex (DMF: SO3) (4.26 gm, 0.0278 mol) was added to the cooled solution and the stiffing continued further for 30 minutes at 0° C. The mixture was then allowed to warm to room temperature and stirred for 1 hour. Thin layer chromatography showed complete conversion of N-Hydroxy compound to product (X).
-
The solution containing the sulfate (X) was re-cooled to 0° C. and a solution of tetra butyl ammonium acetate (TBAA) (9 gm, 0.0301 mol dissolved in 30 ml water) was added to it. The reaction mixture was allowed to warm to 25° C. and stirred for 1 hour. The volatiles were removed under reduced pressure and residue was co-evaporated with 2×50 ml xylene to remove traces of N,N′-Dimethyl formamide. The residue was partitioned between a 1:1 mixture of water and dichloromethane (120 ml). The aqueous layer was re-extracted with dichloromethane (30 ml). The combined organic extracts were washed with water (2×30 ml), brine (30 ml) and dried over sodium sulphate and the solvent evaporated under reduced pressure to obtain the crude TBA sulfate compound (XI) (5.2 gm). Crude compound was triturated with hexane (2×30 ml) and dried on rotavapor under 4 mm Hg pressure to obtain the TBA salt (XI), 5.0 gm, yield-44%.
-
Analysis:
-
Mass: 246 (M−1) of sulfate; Molecular Weight: 488, Molecular Formula: C23H44N4O5S.

Step 8: Synthesis of Sodium salt of trans-7-oxo-6-(sulphoxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile (I
-

-
The TBA sulfate compound (XI) (4.4 gm, 0.009 mol) was dissolved in 5% tetrahydrofuran (THF) in water (2 ml) and the solution was passed through column (45 cm length and 2.0 cm diameter) packed with Dowex 50WX8 200 Na+resin. The column was eluted with 5% THF-water mixture (100 ml). The combined fractions were evaporated under reduced pressure (4 mm Hg) to obtain the product (I) as white semi-solid, 1.5 gm, yield: 62%.
-
Analysis:
-
Mass: 246 (M−1) of sulfate; Molecular Weight: 269; Molecular Formula: C7H8N3O5SNa,
-
1H NMR (DMSO): δ 4.54 (d, 1H), 4.06 (s, 1H), 3.22 (m, 2H), 1.96 (m, 2H), 1.84 (m, 2H).

Example 2Preparation of Sodium salt of trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile IStep 1: Preparation of (S)-5-oxopyrrolidine-2-carboxamide (III)
-

-
To a stirred solution of L-pyroglutamic acid (II) (75 gm, 0.580 mol, commercially available) in dimethylformamide (750 ml) was added 1-hydroxy benzotriazole ammonium salt (106 gm, 0.696 mol, prepared according the literature procedure described in WO 2006100119) in one lot at 25° C. To this reaction mass, DCC was added in small portions over a period of 30 minutes at 0-5° C. The reaction mixture was allowed to warm to room temperature and stiffing continued further for 2 hours. The precipitates were removed by filtration and the filtrate concentrated under reduced pressure. The residue was treated with ethyl acetate (1000 ml) and stirred for 1 hour. The precipitate formed was filtered under suction and washed with additional ethyl acetate (2×75 ml). The combined filtrate was concentrated under reduced pressure to obtain 73 gm of (S)-5-oxopyrrolidine-2-carboxamide (III) as a white solid in 98% yield. The solid thus obtained was used without further purification in the next step.
-
Analysis:
-
Mass: 129 (M+1) for Molecular Weight: 128.13 and Molecular Formula: C5H8N2O2;
-
1H-NMR (400 MHz, DMSO): δ7.71 (s, 1H), 7.34 (s, 1H), 7.01 (s, 1H), 3.93-3.90 (m, 1H), 2.27-2.14 (m, 1H), 2.12-2.01 (m, 2H), 1.89-1.81 (m, 1H).

Step 2: Preparation of (S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate (IV)
-

-
To a cooled (0° C.), stirred solution of (S)-5-oxopyrrolidine-2-carboxamide (70 gm, 0.546 mol) in dimethylformamide (700 ml), triethylamine (TEA) (164.5 gm, 1.6 mol) was added in one lot. After stiffing for 5 minutes Boc anhydride [(Boc)2O] (225 gm, 1.031 mol) was added, followed by the addition of DMAP (6.7 gm, 0.0549 mol). Stirring was continued further for 3 hours, and the completion of the reaction was monitored by thin layer chromatography. The solvent was evaporated under reduced pressure, the residue was leached with diethyl ether (350 ml) and the same procedure repeated with additional diethyl ether (600 ml). The separated solid was filtered under suction and the residue washed with fresh diethyl ether (2×35 ml). The solid was dried at 2 mm Hg, at 45° C. for 2 hour, to obtain 102 gm of (S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate as white solid in 82% yield.
-
Analysis:
-
M.P.: 99-102° C.;
-
Mass m/z: 229 (M+H) for MW: 228 and M.F: C10H16N2O4;
-
1H NMR (400 MHz, DMSO): δ 7.60 (s, 1H), 7.15 (s, 1H), 4.42-4.39 (m, 1H), 2.48-2.32 (m, 2H), 2.20-2.15 (m, 1H), 1.77-1.72 (m, 1H), 1.38 (s, 9H).

Step 3: Preparation of (S)-tert-butyl 2-cyano-5-oxopyrrolidine-1-carboxylate (V)
-

-
Trifluoroacetic anhydride (178 gm, 0.845 mol) was added slowly to a stirred solution of (2S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate (IV) (97 gm, 0.425 mol), containing triethylamine (TEA) (193 gm, 1.907 mol) in dichloromethane (DCM) (2900 ml) at 0° C. After 2 hours of stirring, reaction mixture was diluted with water (1450 ml) and stirred further for 10 minutes. The organic layer was separated and washed with aqueous saturated solution of sodium hydrogen carbonate solution (500 ml), followed by brine (500 ml). The organic layer was dried over anhydrous sodium sulphate, and the solvent evaporated under reduced pressure. To the residue was added diethyl ether (200 ml), stirred well and the separated solid was filtered under suction to obtain the product. The filtrate was concentrated under reduced pressure and the residue was chromatographed on a column of silica gel using mixtures of ethyl acetate and hexane. The evaporation of the combined fractions gave 64.5 gm of (S)-tert-butyl 2-cyano-5-oxopyrrolidine-1-carboxylate (V) as white solid in 72% yield.
-
Analysis:
-
Melting point: 107-109° C.;
-
1H -NMR (400 MHz, DMSO): δ55.07-5.05 (m, 1H), 2.67-2.2.60 (m, 1H), 2.46-2.36 (m, 2H), 2.20-2.17 (m, 1H), 1.46 (s, 9H).

Step 4: Preparation of Sulfoxonium, [(5S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI)
-

-
Dimethyl sulfoxide (DMSO) (175 ml) was slowly added to a stirred suspension of sodium hydride (NaH) (7.3 gm, 0.182 mol, 60%) and trimethylsulfoxonium iodide (TMSOI) (40.2 gm, 0.182 mol) in tetrahydrofuran (THF) (140 ml) over a period of 1 hour at 25° C. The stirring was continued further for 1 hour and the resulting suspension cooled to −10° C. This suspension was slowly added to a stirred solution of (S)-tert-butyl-2-cyano-5-oxopyrrolidine-1-carboxylate (V) (35 gm, 0.166 mol, prepared according to the procedure described in step 3) in tetrahydrofuran (105 ml) containing triethylamine (TEA) (30 ml, 0.215 mol), over a period of 30 minutes at −10° C. Stirring was continued further for 1 hour at the same temperature. Saturated aqueous ammonium chloride solution (350 ml) was added to the reaction mass (after completion of the reaction as indicated by thin layer chromatography) and the reaction mixture was allowed to warm to 25° C. The organic layer was separated and the aqueous layer re-extracted by adding ethyl acetate (350 ml). The combined organic layer was washed with aqueous saturated solution of sodium hydrogen carbonate (350 ml) and brine (350 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. To the residual concentrate, diethyl ether (350 ml) was added and the mixture was stirred for 1 hour. The separated solid was filtered, and the residual solid was washed with additional diethyl ether (20 ml). The solid was dried under reduced pressure to provide 35 gm of Sulfoxonium, [(5S)-5[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI) as a white solid, in 70% yield.
-
Analysis:
-
Melting Point: 150-153° C.;
-
Mass: 303 (M+1) for Molecular Weight: 302 and Molecular Formula: C13H22N2O4S;
-
1H-NMR (400 MHz, CDCl3): δ 6.04 (br, 1H), 4.55 (br, 1H), 4.45 (s, 1H), 3.40-3.38 (d, 6H), 2.51-2.35 (m, 2H), 2.13-2.03 (m, 2H), 1.44 (s, 9H).

Step 5: Preparation of Carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(benzyloxy)imino]pentyl, 1,1-dimethylethyl ester (VII)
-

-
To a stirred solution of Sulfoxonium, [(5S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI) (15 gm, 0.049 mol, prepared according to the procedure described in step 4) in ethyl acetate (EtOAc) (225 ml) was added O-benzyl hydroxylamine hydrochloride (9.5 gm, 0.059 mol) in one lot, at 25° C. The reaction mixture was heated to 60° C. for 2.5 hours. After completion (checked by thin layer chromatography), the reaction mixture was allowed to cool to 25° C. and filtered to remove the precipitates. The filtrate was washed with water (75 ml) and brine (75 ml) and dried over anhydrous sodium sulphate. The solvent was evaporated under reduced pressure to obtain 17.5 gm of Carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(benzyloxy)imino]pentyl, 1,1-dimethylethyl ester (VII) as an oil in 96% yield.
-
Analysis:
-
Mass: 366 (M+1) for Molecular Weight: 365 and Molecular Formula: C18H24ClN3O3;
-
1H -NMR (400 MHz, CDCl3): δ 7.36-7.7.33 (m, 5H), 5.13 (s, 2H), 4.97 (br, 1H), 4.53 (br, 1H), 4.10 (s, 2H), 2.64-2.50 (m, 2H), 2.15-2.01 (m, 2H), 1.46 (s, 9H).

Step 6: Preparation of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX)
-

-
Methane sulphonic acid (9 ml, 0.138 mol) was slowly added to a stirred solution of carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(phenylmethoxy)imino]pentyl, 1,1-dimethylethyl ester (VII) (17 gm, 0.0465 mol, prepared according to the procedure described in step 5) in ethyl acetate (EtOAc) (130 ml), at 25° C. The resulting mixture was heated to 45° C., while monitoring the reaction with thin layer chromatography. After 45 minutes, the reaction mixture was allowed to cool to 25° C. and the resulting reaction mixture (Intermediate VIII) was slowly added to stirred aqueous suspension of potassium hydrogen carbonate (28 gm in 57 ml water). The resulting mixture was stirred and heated to 50-55° C. for 3 hours. The reaction mixture was allowed to cool to 25° C. and the organic layer was separated. The aqueous layer was re-extracted with ethyl acetate (100 ml). The combined organic layer was washed with water (75 ml) and brine (75 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure to obtain 11 gm of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX) as an oil.
-
Analysis:
-
1H-NMR (400 MHz, CDCl3): δ7.36-7.7.33 (m, 5H), 5.09 (s, 2H), 4.14-4.07 (m, 1H), 3.65-3.52 (m, 1H), 3.52-3.45 (m, 1H), 3.16-3.11 (m, 1H), 2.66-2.35 (m, 2H), 2.02-1.89 (m, 2H).

Step 7: Preparation of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X)
-

-
Sulphuric acid (11.7 ml, 0.217 mol) was slowly added to a stirred solution of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX) (10 gm, 0.0436 mol, prepared according to the procedure described in step 6) in ethyl acetate (150 ml) at −10° C. After 10 minutes of stirring, sodium triacetoxy borohydride (NaHB(OOCCH3)3) (11.7 gm, 0.0519 mol, 95% purity) was added in small portions while maintaining temperature below −5° C. After completion of the addition, stirring was further continued for 2 hour at the same temperature. The pH of the reaction mixture was adjusted to about pH 7 by using 30% aqueous potassium hydrogen carbonate solution. The mixture was allowed to warm to 25° C. and the reaction mixture was filtered under suction. The organic layer was separated and the aqueous layer extracted with fresh ethyl acetate (50 ml). The combined organic layer was washed with water (50 ml) and brine (50 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure to obtain 8.88 gm of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X) as an oil, in 88% yield. This was used as such for the next step without further purification.
-
Analysis:
-
Mass: 232 (M+1) for Molecular Weight: 231 and Molecular Formula: C13H17N3O.

Step 8: Preparation of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI)
-

-
A solution of oxalic acid dihydrate (5.28 gm, 0.0418 mol) in a mixture of ethyl acetate:acetone (1:1, 28 ml:28 ml) was slowly added to a stirred solution of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X) (8.8 gm, 0.0380 mol, prepared according to the procedure described in step 7) in ethyl acetate (35 ml) at 25° C. After 3 hour of stirring, the separated solid was filtered under suction, washed with additional 50 ml of v/v mixture of ethyl acetate: acetone solution (1:1, 25 ml: ml) and the solid dried under reduced pressure to obtain 6.7 gm of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI) in 55% yield.
-
Analysis:
-
Mass: 232 (M+1) for Molecular Weight: 321 and Molecular Formula: C13H17N3O.C2H2O4;
-
1H-NMR (400 MHz, DMSO): δ7.25 (m, 5H), 4.59 (s, 2H), 4.22 (br, 1H), 4.07-4.04 (m, 1H), 3.10-3.07 (m, 1H), 2.97-2.83 (m, 1H), 2.61-2.52 (m, 1H), 1.83-1.63 (m, 3H), 1.41-1.25 (m, 1H).

Separation of (2S,5R)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate from two isomeric (1:1) mixture of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate
-
A suspension of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI) (13 gm, 0.0404 moles) in methanol (260 ml) was heated under reflux, with stirring, for 3 hour. The resulted suspension was allowed to cool to 35° C. and the resulting suspension filtered under suction. The solid was washed with additional methanol (2×13 ml). The solid was dried under reduced pressure (4 mm Hg), to obtain (2S,5R)-5-[(benzyloxy) amino]-2-cyanopiperidine ethanedioate (XIA) as a white solid, 7.3 gm, yield 56%.
-
Analysis:
-
Mass m/z: 232.2 (M+H) for MW: 321 and M.F: C13H17N3O.C2H2O4.
-
1H-NMR (400 MHz, DMSO): δ 7.37-7.24 (m, 5H), 4.57 (s, 2H), 3.92-3.91 (m, 1H), 3.06-3.02 (m, 1H), 2.92-2.88 (m, 1H), 2.56-2.51 (m, 1H), 1.96-1.91 (m, 1H), 1.76-1.55 (m, 2H), 1.44-1.38 (m, 1H).
-
Purity as determined by HPLC: (2S,5R isomer) 88.44% (RT-9.74) and (2S,5S isomer) 5.47% (RT-8.61).
Step 9: Preparation of (2S,5R)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIII) and (2S,5S)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIV)
-

-
To a stirred suspension of (2S)-5-[(benzyloxy) amino]-2-cyanopiperidine ethanedioate (1:1) (XI) (3.7 gm, 0.0115 mol, prepared according to the procedure described in step 8) in ethyl acetate:water (1:1, 37 ml:37 ml) was added solid sodium bicarbonate (1.9 gm, 0.022 mol) at 25° C. After 30 minutes of stirring the organic layer was separated. The aqueous layer was re-extracted with ethyl acetate (20 ml). The combined organic layer was washed with water (20 ml) and brine (20 ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure to obtain 3 gm of ((2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (XII) as an oil. The oily product, (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (XII) (1 gm, 0.00432 mol, prepared as mentioned above), was dissolved in acetonitrile (ACN) (15 ml), cooled to 10° C., stirred and triethyl amine (1.8 ml, 0.0129 mol) was added in one portion. To this mixture was added slowly a solution of triphosgene (0.564 gm, 0.0019 mol) in acetonitrile (6 ml). After 15 minutes of stirring, DMAP (0.0527 gm, 0.000432 mol) was added and the reaction mixture allowed to warm to 25° C. After 16 hours of stirring, the thin layer chromatography (ethyl acetate:hexane (1:1)) showed the two separable mixture of isomers. A solution of saturated sodium bicarbonate (10 ml) was added to the reaction mass and stirring continued for another 30 minutes. The volatiles were removed under reduced pressure. The residual mass was partitioned between ethyl acetate (10 ml) and water (10 ml). The organic layer was separated and the aqueous layer re-extracted with ethyl acetate (10 ml). The combined organic layer was washed with water (10 ml) and brine (10 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The resulting mixture was dissolved in dichloromethane (15 ml) and washed with 5% potassium hydrogen sulphate solution (3×10 ml), saturated sodium hydrogen carbonate (10 ml) and water (10 ml). The organic layer was concentrated under reduced pressure, to yield 0.610 gm of crude oily product.
-
[0204]The oily mixture was purified by column chromatography using silica gel (60-120 mesh) by eluting with mixture of ethyl acetate and hexane. The upper spot was eluted out by using 25% ethyl acetate in hexane and the lower spot was eluted out by using 45% ethyl acetate in hexane. The combined pure fractions were concentrated under reduced pressure, to obtain the 0.130 gm of (2S,5R)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIII) and 0.105 gm of (2S,5S)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIV).
-
Analysis for compound of Formula (XIII):
-
Rf: 0.49;
-
Melting Point: 95-99° C.;
-
Mass: 258 (M+1) for Molecular Weight: 257 and Molecular Formula: C14H15N3O2;
-
1H-NMR (400 MHz, CDCl3): δ 7.43-7.35 (m, 5H), 5.06-5.03 (d, 1H), 4.91-4.88 (d, 1H), 4.38-4.36 (d, 1H), 3.36-3.29 (m, 2H), 3.16-3.12 (m, 1H), 2.33-2.10 (m, 2H), 1.90-1.79 (m, 2H).
-
Analysis for compound of Formula (XIV):
-
Rf: 0.12;
-
Melting Point: 115-118° C.
-
Mass: 258 (M+1) for Molecular Weight: 257 and Molecular Formula: C14H15N3O2;
-
1H-NMR (400 MHz, CDCl3): δ7.43-7.33 (m, 5H), 5.06-5.04 (d, 1H), 4.92-4.89 (d, 1H), 3.96-3.92 (dd, 1H), 3.32-3.23 (m, 2H), 2.76-2.73 (m, 1H), 2.29-2.18 (m, 2H), 2.05-1.99 (m, 1H), 1.71-1.63 (m, 1H).

Step 10: Preparation of (2S,5R)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIIIa)
-

-
A solution of (2S,5R)-6-(benzyloxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIII) (1 gm, 0.00389 mol) in a mixture of ethyl acetate and tetrahydrofuran (THF) (4:6, 4 ml:6 ml) containing 10% palladium over carbon (0.300 gm, 50% wet) was hydrogenated at 50-55 psi, for 6 hours at 25° C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of ethyl acetate and tetrahydrofuran (4:6, 4 ml:6 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain 0.649 gm of the titled compound of Formula (XIIIa) as oil, which was used as such for the next reaction without further purification.

Preparation of (2S,5S)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIVa)
-

-
A solution of (2S,5S)-6-(benzyloxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIV) (545 mg, 2.120 mol) in a mixture of ethyl acetate and tetrahydrofuran (5:5, 8 ml:8 ml) containing 10% palladium over carbon (0.109 gm, 50% wet) was hydrogenated at 50-55 psi, for 45 minutes at 25° C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of dichloromethane and dimethylformamide (5:5, 10 ml:10 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain the product as oil, which was triturated with diethyl ether (5 ml). The diethyl ether layer was decanted and the residue was dried under reduced pressure at 40° C. for 15 minutes to obtain 0.343 gm of compound of Formula (XIVa), which was used as such for the next step.

Step 11: Preparation of (2S,5R)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIII b)
-

-
To a stirred solution of (2S,5R)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIIIa) (0.649 gm, 0.00389 mol) in a mixture of dichloromethane (5 ml) and dimethylformamide (1 ml), sulfur trioxide dimethylformamide complex (1.07 gm, 0.007 mol) was added in one portion at about 10° C. After 90 minutes, the completion of the reaction was monitored by thin layer chromatography (9:1, chloroform:methanol). To the resulting reaction mass was added tetrabutylammonium hydrogen sulphate (TBAHS) in one portion (2.37 gm, 0.007 mol) under stirring. After 1 hour, water (10 ml) was added and the mixture stirred for 5 minutes. The organic layer was separated and washed with water (2×10 ml), dried (over anhydrous sodium sulphate) and the solvent evaporated under reduced pressure at 35° C. The residual oily mass was triturated with ether (2×10 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure, to obtain 0.6 gm of the titled compound of Formula (XIIIb) in 31% yield.
-
Analysis:
-
Mass: 246 (M−1), for Molecular Weight: 488 and Molecular Formula: C23H44N4O5S;
-
1H NMR (400 MHz, CDCl3): δ4.43 (brs, 1H), 4.35-4.33 (d, 1H), 3.47-3.44 (m, 2H), 3.28-3.24 (m, 8H), 2.33-2.29 (m, 2H), 1.92-1.85 (m, 2H), 1.69-1.61 (m, 8H), 1.48-1.39 (m, 8H), 1.02-0.98 (m, 12H).
-
Purity as determined by HPLC: 95.57%

Preparation of (2S,5S)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIVb)
-

-
To a stirred solution of (2S,5S)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIVa) (343 mg, 2.05 mol) in dimethylformamide (3 ml) sulfur trioxide dimethylformamide complex (390 mg, 2.549 mol) was added in one portion, at 10° C. and stirring continued further. After 60 minutes, thin layer chromatography (9:1, chloroform:methanol) showed the complete conversion. To the resulting reaction mixture was added, slowly, a solution of tetrabutylammonium acetate (TBAA) (831 mg, 2.756 mol) in water (3 ml) under stirring. After 1 hour of stirring, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2×10 ml), to yield a thick mass which was partitioned between dichloromethane (10 ml) and water (10 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (10 ml). The combined organic extracts were washed with water (3×10 ml), dried (over anhydrous sodium sulphate) and the solvent evaporated under reduced pressure at 35° C. The residual oily mass was triturated with ether (2×10 ml), each time the ether layer was decanted and finally the residue was dried under reduced pressure, to obtain 634 mg of compound of Formula (XIVb) as an oil in 61% yield.
-
Analysis:
-
Mass: 246 (M−1); for Molecular Weight: 488 and Molecular Formula: C23H44N4O5S;
-
1H NMR (400 MHz, CDCl3): δ4.38 (m, 1H), 3.98-3.93 (dd, 1H), 3.98-3.54 (m, 1H), 3.32-3.28 (m, 8H), 2.43-2.39 (m, 1H), 2.31-2.30 (m, 1H), 2.15-2.01 (m, 2H), 1.76-1.63 (m, 8H), 1.49-1.40 (m, 8H), 1.02-0.99 (m, 12H);
-
Purity as determined by HPLC: 98.22%.

Step 12: Preparation of (2S,5R)-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (I)
-

-
An activated Amberlite 200 sodium resin (20 gm) was loaded on a glass column and was washed with de-mineralized water (50 ml) followed by 10% tetrahydrofuran in water (50 ml). A solution of (2S,5R)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutyl ammonium salt (XIIIb) (575 mg, 1.176 mol) in tetrahydrofuran (THF) (1.1 ml) was loaded on column. It was eluted by using 10% tetrahydrofuran in water. The pure fractions were combined and the solvents evaporated under reduced pressure to obtain 280 mg of the compound of Formula (I) in 85% yield.
-
Analysis:
-
Mass: 246 (M−1) as free sulfonic acid, for Molecular Weight: 269 and Molecular Formula:
-
C7H8N3O5SNa;
-
1H NMR (400 MHz, DMSO): δ4.54-4.53 (d, 1H), 4.06 (brs, 1H), 3.20 (m, 2H), 1.96-1.81 (m, 4H);
-
Purity as determined by HPLC: 97.07%.

Preparation of (2S,5S)-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (Ia)
-

-
An activated Amberlite 200 sodium resin (20 gm) was loaded on a glass column and was washed with de-mineralized water (100 ml) followed by 10% tetrahydrofuran (THF) in water (100 ml). A solution of (2S,5S)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIVb) (475 mg, 0.971 mol) in tetrahydrofuran (1.5 ml) was loaded on column. It was eluted by using 10% tetrahydrofuran in water. The pure fractions were combined and the solvent evaporated under reduced pressure to obtain 242 mg of compound of Formula (Ia) as white solid, in 92% yield.
-
Analysis:
-
Mass: 246 (M−1) as free sulfonic acid, for Molecular Weight: 269 and Molecular Formula: C7H8N3O5SNa;
-
1H NMR (400 MHz, DMSO): δ 4.53-4.50 (dd, 1H), 3.98 (brs, 1H), 3.17-3.02 (dd, 2H), 1.99-1.96 (m, 2H), 1.77-1.75 (m, 2H);
-
Purity as determined by HPLC: 99.59%.

note
Avibactam is

1192500-31-4; SULFURIC ACID, MONO[(1R,2S,5R)-2-(AMINOCARBONYL)-7-OXO-1,6-DIAZABICYCLO[3.2.1]OCT-6-YL] ESTER;
COMPD IS
References
IN 2013MU03308
IN 2011MU02582
| Patent | Submitted | Granted |
|---|---|---|
| Nitrogen containing compounds and their use [US8969334] | 2014-05-04 | 2015-03-03 |
| Nitrogen containing compounds and their use [US8969567] | 2014-05-10 | 2015-03-03 |
| Nitrogen containing compounds and their use [US8754102] | 2012-09-11 | 2014-06-17 |
| WO2013014496A1 * | 4 Oct 2011 | 31 Jan 2013 | Wockhardt Limited | Pharmaceutical compositions comprising sulbactam and beta-lactamase inhibitor |
| WO2013038330A1 * | 11 Sep 2012 | 21 Mar 2013 | Wockhardt Limited | Nitrogen containing compounds and their use |
| WO2013030733A1 * | Aug 24, 2012 | Mar 7, 2013 | Wockhardt Limited | 1,6- diazabicyclo [3,2,1] octan-7-one derivatives and their use in the treatment of bacterial infections |
| WO2013038330A1 * | Sep 11, 2012 | Mar 21, 2013 | Wockhardt Limited | Nitrogen containing compounds and their use |
| WO2013149121A1 * | Mar 29, 2013 | Oct 3, 2013 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| WO2014108872A1 * | Jan 13, 2014 | Jul 17, 2014 | Wockhardt Limited | Compositions and methods for treating bacterial infections |
| CA2874279A1 * | May 30, 2013 | Dec 5, 2013 | Meiji Seika Pharma Co., Ltd. | Novel .beta.-lactamase inhibitor and process for preparing the same |
| US20130289012 * | Mar 29, 2013 | Oct 31, 2013 | Cubist Pharmaceuticals, Inc. | 1,2,4-oxadiazole and 1,2,4-thiadiazole beta-lactamase inhibitors |
////////
C1CC(N2CC1N(C2=O)OS(=O)(=O)O)C#N
or
C1C2CN(C(C1)C#N)C([C@@H]2OS(=O)(=O)O)=O
or
O=S(=O)(O)ON2C(=O)N1C[C@H]2CC[C@H]1C#N
////////
WCK ? NEW ANTIBACTERIALS FROM WOCKHARDT

WCK ?
TRANS-SULFURIC ACID MONO-{2-[5-(2-METHYLAMINO-ETHYL)-[1,3,4]-OXADIAZOL-2-YL]-7-OXO-1,6-DIAZA-BICYCLO [3.2.1]OCT-6-YL} ESTER
Trans-sulfuric acid mono- { 2-[5-(2-methylamino-ethyl)-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester.
CAS 1824664-22-3
MW 347.35, C11 H17 N5 O6 S
|
Beta lactamase inhibitor
|
To treat
|
Bacterial infection
|
Several l,6-diazabicyclo[3.2.1]octan-7-one derivatives have been described as antibacterial agents in PCT International Patent Application No. PCT/IB2012/054296. A compound of Formula (I), chemically known as irans-sulfuric acid mono- {2- [5 -(2-methylamino-ethyl)-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl} ester has antibacterial properties and is also disclosed in PCT International Patent Application No. PCT/US2013/034562
PATENT
WO2015173663

(VII) Formula (I)
Scheme 1
Example 1
Synthesis of traras-sulfuric acid mono-{2-[5-(2-methylamino-ethyl)-[l,3,4]-oxadiazol- 2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl} ester (I)
Step 1; Preparation of tr «s-{3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane-2-carbonyl)-hydrazino]-3-oxo-propyl}-methyl-carbamic acid fert-butyl ester (IV):
Sodium salt of 6-benzyloxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylic acid (III) (5.9 g, 0.02 mol; prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) was dissolved in water (100 ml) to obtain a clear solution under stirring at 25°C. To the clear solution was added successively, (3-hydrazinocarbonyl-ethyl)-methyl-carbamic acid tert-buty\ ester (II) (4.5 g, 0.02 mol), EDC. HC1 (5.7 g, 1.5 mol), and HOBt (2.7 g, 0.02 mol) followed by water (20 ml) under stirring at 25°C. The reaction mixture was stirred at 30°C for 20 hours. As maximum precipitation was reached, thin layer chromatography (acetone: hexane, 35:65) showed completion of reaction. The suspension was filtered under suction and the wet cake was washed with additional water (100 ml) and dried under vacuum at 45°C to furnish 5.5 g of ir ns-{3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazino]-3-oxo-propyl}-methyl-carbamic acid tert-buty\ ester (IV) as a white powder in 58% yield.
Analysis:
Mass: 476.4 (M+l); for Molecular Formula: C23H33N5O6 and Molecular Weight:
475.2;
1H NMR (CDCI3): δ 7.43-7.35 (m, 5H), 5.04 (d, 1H), 4.90 (d, 1H), 4.01 (d, 1H), 3.54 (t, 2H), 3.33 (br s, 1H), 3.14-3.07 (m, 2H), 2.85 (s, 3H), 2.53 (br s, 2H), 2.33-2.30 (m, 1H), 2.07-1.94 (m, 2H), 1.64-1.61 (m, 4H), 1.40 (s, 9H), 1.25-1.17 (m, 2H).
Step 2: Preparation of tr «s-{2-[5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-methyl-carbamic acid tert-butyl ester (V):
To a solution of triphenylphosphine (3.3 g, 0.0126 mol) in dichloromethane (70 ml) at was added iodine (3.2 g, 0.0126 mol) and triethyl amine (7.0 ml, 0.0525 mol) under stirring at 25°C. Separately prepared solution of ir ns-{3-[N’-(6-benzyloxy-7-oxo-1 ,6-diaza-bicyclo[3.2.1 ]octane-2-carbonyl)-hydrazino] -3-oxo-propyl)-methyl-carbamic acid tert-butyl ester (IV) (5.5 g, 0.0105 mol) dissolved in dichloromethane (30 ml) was added to above reaction mixture and the mixture was stirred at 25°C for 30 minutes. The reaction mixture was concentrated and to this ethyl acetate (100 ml) was added. The separated triphenylphosphine oxide was filtered off. The filtrate was concentrated and the residue purified by silica gel column chromatography using mixture of ethyl acetate and hexane, to afford 5 g of the titled compound.
Analysis:
Mass: 458.3 (M+l); for Molecular Formula: C23H31N5O5 and Molecular Weight:
457.53;
1H NMR (CDCI3): δ 7.44-7.35 (m, 5H), 5.04 (d, 1H), 4.93 (d, 1H), 4.70 (t, 1H), 3.62 (br s, 2H), 3.36 (s, 1H), 3.07 (t, 2H), 2.93 (br d, 1H), 2.85 (br s, 4H), 2.32-2.27 (m, 2H), 2.12 (br d, 2H), 1.95 (br s, 1H), 1.40 (s, 9H).
Step 3: Preparation of traras-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-methyl-carbamic acid tert-butyl ester (VI):
To a solution of trans-{2-[5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-methyl-carbamic acid tert-butyl ester (V) (5 g, 0.0109 mol) in methanol (50 ml) was added 10% palladium on carbon (1.5 g) at 25°C. The reaction mixture was stirred under 1 atmospheric pressure of hydrogen at 35°C for 2 hours. The catalyst was removed by filtering the reaction mixture under suction over a celite bed. The celite bed was washed with methanol (50 ml). The combined filtrate was evaporated under vacuum below 35°C to provide 3.8 g of trans- {2- [5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-methyl-carbamic acid tert-butyl ester (VI) in 93% yield; it was used as such for the next reaction.
Step 4: Preparation of trans -tetrabutyl ammonium salt-methyl-{2-[5-(7-oxo-6-sulphooxy-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-carbamic acid tert-butyl ester (VII):
A solution of trans-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[1,3,4] oxadiazol-2-yl] -ethyl }-methyl-carbamic acid tert-butyl ester (VI) (3.8 g, 9.8 mmol), in dichloromethane (38 ml) was charged with triethylamine (2.6 ml, 19.7 mmol) under stirring to provide a clear solution. To this clear solution was added sulfur trioxide -pyridine complex (2.35 g, 14.8 mmol) under stirring at 30°C. The reaction mixture was stirred for 3 hours and to this 0.5 M aqueous potassium dihydrogen phosphate (38 ml) was added followed by ethyl acetate (76 ml). The biphasic mixture was stirred for 15 minutes at 30°C. Aqueous layer was separated and re-extracted with dichloromethane and ethyl acetate mixture (1:2 v/v, 76 ml twice). To the aqueous layer was added solid tetrabutyl ammonium hydrogen sulfate (3 g, 8.8 mmol) and stirring was continued for 1
hour at room temperature. The reaction mixture was extracted with dichloromethane (3 x 50 ml). Layers were separated and dichloromethane layer dried over sodium sulfate and then evaporated under vacuum at 35°C to provide 2.8 g of irans-tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy-l,6-diaza-bicyclo[3.2. l]oct-2-yl)-[l, 3, 4]oxadiazol -2-yl] -ethyl} -carbamic acid tert-buty\ ester (VII). This was purified by column chromatography to afford 2.0 g of pure product in 29% yield.
Analysis:
Mass: 446.5 (M-l) as free sulfonic acid; for Molecular Formula:![]()
(C4H9)4 and Molecular Weight: 688.5;
1H NMR (CDC13): δ 4.67 (d, 1H), 4.36 (br s, 1H), 3.33-3.29 (m, 8H), 3.23 (d, 1H), 3.08 (t, 2H), 2.87 (s, 3H), 2.83 (s, 1H), 2.28-2.22 (m, 3H), 2.07-2.00 (m, 8H), 1.50-1.41 (m, 17H), 1.28 (s, 3H), 1.01 (t, 12 H), 1.41-1.52 (m, 10 H).
Step 5: traras-sulfuric acid mono-{2-[5-(2-methylamino-ethyl)-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester:
irans-Tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy- 1 ,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol -2-yl] -ethyl} -carbamic acid tert-butyl ester (VII) (2.0 g, 2.9 mmol) was dissolved in dichloromethane (5 ml) and to the clear solution was slowly added trifluoroacetic acid (5 ml) at 0 to -10 °C. The reaction mixture was stirred at 0 to -10 °C for 1 hour. The solvent and excess trifluoroacetic acid was evaporated under vacuum below 40°C to approximately 1/3 of its original volume to provide pale yellow oily residue. The oily residue was stirred with diethyl ether (100 ml) for 10-15 minutes. The suspension formed was filtered under suction to provide a solid. This process was repeated twice. The solid was charged in a round bottom flask and to it was added dichloromethane (100 ml). The suspension was stirred for 15 minutes and filtered under suction to provide a solid. The obtained solid was dried under vacuum below 40°C to furnish 850 mg of trans- sulfuric acid mono-{2-[5-(2-methylamino-ethyl)-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester as white solid in 85% yield.
Analysis:
Mass: 346.3 (M-1) as a free sulfonic acid; for Molecular Formula: C11H17N5O6S and Molecular Weight: 347.35;
NMR (D20): δ 4.74 (d, IH), 4.16 (br s, IH), 3.45 (t, 2H), 3.31 (t, 2H), 3.15 (d, IH), 2.91 (d, IH), 2.98 (s, 3H), 2.27-2.22 (m, IH), 2.16-2.11 (m, 2H), 1.94-1.91 (m, IH);
Purity as determined by HPLC: 95.56%.
/////////
WCK ? New molecules from Wochkardt to treat bacterial infections

(2S, 5R)-7-OXO-N-[(3S)-PYRROLIDIN-3-YLOXY]-6-(SULFOOXY)-1,6-DIAZABICYCLO [3.2.1]OCTANE-2-CARBOXAMIDE
- (2S,5R)-7-Oxo-N-((3S)-pyrrolidin-3-yloxy)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
- C11 H18 N4 O7 S, 350.35
- Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(3S)-3-pyrrolidinyloxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
CAS 1452458-72-8
KEEP WATCHING THIS POST
SODIUM SALT CAS 1629221-44-8
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(3S)-3-pyrrolidinyloxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1:1)
Patent
http://www.google.com/patents/WO2015110886A1?cl=en

Formula (II) Formula (III) Formula (IV)
Hydrogenolysis

Formula (I)
Scheme – 1

Formula (VII) Formula (VIII)
Hydrazine hydrate

Formula I
Scheme – 2
Example 1
Synthesis of tert-butyl (3S)-2-(aminooxy)pyrrolidine-l-carboxylate (III):
Step 1; Preparation of 3-(R)-hydroxypyrrolidine hydrochloride (VIII):
To a stirred suspension of commercially available (25, 4i?)-4-hydroxy-2-pyrrolidinecarboxylic acid (L-hydroxyproline) (VII) (100 g, 0.762 mol) in anhydrous cyclohexanol (500 ml), was added 2-cyclohexen-l-one (5 ml). The resulting mixture was heated under reflux at about 154°C for about 48 hour. The obtained clear solution was allowed to cool to room temperature and then was cooled further to 10°C. To this, about 15 % solution of hydrochloric acid in ethanol (234 ml) was added and then stirred for 30 minutes. The separated solid was filtered under suction and washed with ethyl acetate (2 x 100 ml). The solid was dried under reduced pressure to obtain 47.5 g of 3-(R)-hydroxypyrrolidine hydrochloride (VIII) in 51 % yield. The solid was used without further purification in the next step.
Analysis:
Mass: 87.8 (M+l) as free base; for Molecular weight of 123.57 and Molecular Formula of C4Hi0ClNO; and
1H NMR (400MHz, DMSO): 5 9.58 – 9.32 (brd, 2H), 5.36 (brs, 1H), 4.36 – 3.39 (brs, 1H), 3.17 (brs, 2H), 3.11-2.96 (dd, 2H), 1.90 – 1.81 (m, 2H).
Step 2: Preparation of (3R)-l-(tert-butoxycarbonyl)-3-hydroxypyrrolidine (IX):
To a stirred suspension of 3-(i?)-hydroxypyrrolidine hydrochloride (VIII) (110 g, 0.9 mol) in dichloromethane (1100 ml), triethylamine (273 g, 2.7 mol) was added at 0-5°C. After 5 minute of stirring di-feri-butyldicarbonate [(Boc)20] (245 g, 1.125 mol) was added to the reaction mixture in small portions, followed by 4-dimethylaminopyridine (10.99 g, 0.09 mol). The reaction mixture was stirred for 2 hour and then poured in to water (1100 ml). The organic layer was separated and washed with saturated ammonium chloride solution (1×1100 ml) and water (1100 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The residue was purified by silica gel (60-120 mesh) column chromatography using 1-5% mixtures of acetone: hexane as an eluent. The combined fractions were evaporated, to obtain the 118 g of (3i?)-l-(ieri-butoxycarbonyl)-3-hydroxypyrrolidine (IX), as a white solid, in 71 % yield.
Analysis:
Melting point: 55 – 58°C;
Mass: 188 (M+l); for Molecular Weight of 187.24 and Molecular Formula of C9H17N03; and
1H NMR (400MHz, CDC13): 54.428 – 4.424 (s, 1H), 3.46 – 3.43 (m, 2H), 3.37 -3.28 (m, 2H), 2.36 – 2.30 (d, 1H), 2.00 – 1.86 (m, 2H), 1.44 (s, 9H).
Step 3: Preparation of (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl)oxy]pyrrolidine-l-carbox lic acid tert- butyl ester (X):
To a stirred solution of di-isopropyl azodicarboxylate (97.17 g, 0.481 mol) in tetrahydrofuran (1200 ml), a solution triphenyl phosphine (125.9 g, 0.481 mol) in tetrahydrofuran (300 ml) was added at temperature below -10°C. The resulting reaction mixture was stirred for further 45 minute at the same condition and a solution of (3i?)-l-(ieri-butoxycarbonyl)-3-hydroxypyrrolidine (IX) (60 g, 0.3204 mol) in tetrahydrofuran (300 ml) was added over a period of 15 minute. After another 45 minute of stirring, N-hydroxy phthalimide (52.4 g, 0.3204mol) was added in one portion to the reaction mass. The reaction mixture was allowed to warm to room temperature and stirred for 16 hour.
The completion of the reaction was monitored by thin layer chromatography. After completion of reaction, the solvent was evaporated under reduced pressure. The residue thus obtained was stirred with di-isopropyl ether (600 ml). The precipitate formed was filtered under suction. The filtrate was concentrated under reduced pressure and the residual mass was purified by silica gel (60-120 mesh) column chromatography using 1-5 % mixtures of acetone: hexane as an eluent. The solvent from the combined fractions was evaporated to obtain 63 g of (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl)oxy]pyrrolidine-1-carboxylic acid tert-buty\ ester (X), as a white solid, in 59% yield.
Analysis:
Melting point: 112-115°C;
Mass: 333.2 (M+l); for Molecular Weight of 332.36 and Molecular Formula of ![]()
1H NMR (400 MHz, CDC13): 57.86-7.83 (m, 2H), 7.78-7.75 (m, 2H), 4.99 – 4.94 (d, 1H), 3.80 – 3.68 (m, 2H), 3.60 – 3.53 (m, 2H), 2.28-2.25 (m, 1H), 2.02 (m, 1H), 1.48 (s, 9H).
Step 4: Preparation of tert-butyl (35)-2-(aminooxy)pyrrolidine-l-carboxylate (III):
To a stirred suspension of the (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl) oxy]pyrrolidine-l-carboxylic acid tert-buty\ ester (X) (12.68 g, 0.0381 mol) in dichloromethane (200 ml) was added 99% hydrazine hydrate (3.81 g, 0.0762 mol) drop-wise over a period of 30 minutes, at 25°C. After 2 hour of stirring, the separated solid was filtered and washed with dichloromethane (2 x 50 ml). The filtrate and washings were combined and washed with water (2 x 65 ml) and finally with brine (1 x 65 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent was evaporated under reduced pressure to obtain 7.71 g of tert-buty\ (3S)-2-(aminooxy pyrrolidine- 1-carboxylate (III) as pale yellow oil.
Analysis:
Mass: 203 (M+l); for Molecular Weight of 202.26 and Molecular Formula of C9H18N203.
Example 2
Synthesis of (25, 5R)-7-oxo-N-r(35)-pyrrolidin-3-yl-oxyl-6-(sulfooxy)-l,6-diaza bicyclor3.2. lloctane-2-carboxamide (I) :
Step 1: Preparation of fert-butyl-(35)-3-[({[25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (IV):
To a clear, stirred solution of sodium (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (11.38 g, 0.0382 mol) in water (114 ml), was added EDC.HC1 (18.24 g, 0.0955 mol) at 15°C, in small portions. After 10 minutes, a solution of feri-butyl-(35)-3-(aminooxy) pyrrolidine- 1-carboxylate (III, 7.72 g, 0.0382 mol), prepared as per the literature procedure: US5233053, Chemistry Letters, 893-896, (1986) and depicted in scheme 2), in dimethylformamide (24 ml) was added drop wise, to the above stirred solution, at about 10°C. The reaction mass was allowed to warm to 25°C and HOBt (5.15 g, 0.0382 mol) was added in small portions over a period of 15 minutes and the reaction mixture was stirred further at room temperature for 16 hour. After completion of the reaction (monitored by thin layer chromatography using solvent system acetone: hexane (35:65)) the resulting mixture was filtered and the residue was washed with water (120 ml). The residual white solid was suspended in fresh water (120 ml) and the mixture stirred at 50°C, for 3 hour. The resulting suspension was filtered and the residual solid dried under reduced pressure to obtain 16.1 g of tert-buty\ (35)-3-[({ [25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino) oxy]pyrrolidine- 1-carboxylate (IV) as off white solid in 92% yield.
Analysis:
Mass: 461.3 (M+l); for Molecular weight of 460.53 and Molecular formula of ![]()
1H NMR (400MHz, CDC13): δ 9.08-9.03 (d, 1H), 7.43-7.36 (m, 5H), 5.06-4.88 (dd, 2H), 4.63-4.57 (d, 1H), 3.97-.396 (d, 1H), 3.64-3.53 (m, 2H), 3.47-3.37 (m, 2H), 3.31 (s, 1H), 3.02-2.99 (d, 1H), 2.75-2.73 (d, 1H), 2.29(m, 2H), 2.18-2.15 (m, 1H), 2.01-1.90 (m, 3H), 1.66 (m, 1H), 1.46 (s, 9H).
Step 2: Preparation of tert-butyl-(35)-3-[({[25,5R)-6-hydroxy-7-oxo-l,6-diazabicylco
[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (V):
ieri-Butyl-(35)-3-[({ [25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (IV) (10 g, 0.02171 mol) was dissolved in a mixture of dimethylformamide and dichloromethane ( 1 : 1 , 50 ml : 50 ml) to obtain a clear solution. To this solution, was added 10% palladium on carbon (2.5 g, 50% wet) catalyst. The suspension was stirred for 4 hour, at 50 psi hydrogen atmosphere, at 25°C. After completion of the reaction (monitored by thin layer chromatography), the resulting mixture was filtered through a celite pad. The residue was washed with dichloromethane (50 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain 8.04 g of ieri-butyl(35)-3-[({ [25,5i?)-6-hydroxy-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl
amino)oxy]pyrrolidine-l-carboxylate (V) as oil. This was used as such for the next reaction without further purification.
Analysis:
Mass: 371.2 (M+l); for Molecular Weight of 370.4 and Molecular Formula of
Step 3: Preparation of tert-butyl-(35)-3-[({[25,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate, tetrabutyl ammonium salt (VI):
To a stirred solution of ieri-butyl(35)-3-[({ [25,5i?)-6-hydroxy-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (V) (8.04 g, 0.0217 mol) in dimethylformamide (50 ml), was added sulfur trioxide dimethyl formamide complex (3.98 g, 0.0260 mol) in one portion, at about 10°C. The stirring was continued further for 30 minute and then the reaction mixture was allowed to warm to room temperature. After 2 hour, a solution of tetrabutylammonium acetate (7.83 g, 0.0260 mol) in water (25.8 ml) was added to the resulting reaction mass under stirring. After additional 2 hour of stirring, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2 x 20 ml) to obtain thick mass. This mass was partitioned between dichloromethane (100 ml) and water (100 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (50 ml). The combined organic extracts were washed with water (3 x 50 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The residual oily mass was triturated with ether (3 x 50 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure to obtain 11.3 g of tert-butyl(3S)-3-[({ [2S,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy] pyrrolidine- 1-carboxylate, tetrabutylammonium salt (VI), as a white foam, in 75 % yield.
Analysis:
Mass: 449.3 (M-l, without TBA); for Molecular weight of 691.94 and Molecular formula of C32H61N5O9S; and
1H NMR (400MHz, CDC13): 59.14-9.10 (d, 1H), 4.63 (s, 1H), 4.35 (s, 1H), 3.94-3.92 (d, 1H), 3.66-3.35 (m, 5H), 3.29-3.27 (m, 8H), 2.83-2.80 (d, 1H), 2.35-2.17 (m, 3H), 1.98-1.87 (m, 2H), 1.73 (m, 1H), 1.70-1.62 (m, 8H), 1.49-1.40 (m, 17H), 1.02-0.99 (t, 12H).
Step 4: Preparation of (25,5R)-7-oxo-iV-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfooxy)-l,6-diazabicyclo [3.2.1]octane-2-carboxamide (I):
To a stirred solution of ieri-butyl(35)-3-[({ [25,5i?)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate tetrabutyl ammonium salt (VI) (11 g, 0.0158 mol) in dichloromethane (55 ml), was added trifluoroacetic acid (55 ml) drop wise at about -10 °C over a period of 1 hour. After 1 hour of stirring, the resulting mixture was poured into hexane (550 ml), stirred well for 30 minute and the separated oily layer was collected. This procedure was repeated one more time and finally the combined oily layer was added to diethyl ether (110 ml) under vigorous stirring, at about 25 °C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with diethyl ether (2 x 110 ml). The solid thus obtained was stirred with fresh dichloromethane (110 ml) for 30 minutes and filtered. The residual solid was dried at about 45 °C under reduced pressure to obtain 5.7 g of (25,5i?)-7-oxo-N-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfo-oxy)- l,6-diaza bicyclo[3.2.1] octane-2-carboxamide (I), as a white amorphous solid having XRPD as shown in Figure 1.
Analysis:
Mass: 349.2 (M-l); for Molecular Weight of 350.35 and Molecular Formula of ![]()
1H NMR (400MHz, DMSO-D6): δ 11.44 (brs, 1H), 8.80 (brs, 2H), 4.64-4.63 (m, 1H), 4.00 (s, 1H), 3.78-3.77 (d, 1H), 3.38-3.23 (m, 4H), 3.03-2.93 (dd, 2H), 2.48-2.11 (m, 1H), 2.00- 1.94 (m, 2H), 1.88- 1.86 (m, 1H), 1.71-1.65 (m, 2H).
Example 3
Preparation of Crystalline Form I of (25,5R)-7-oxo-jV-r(35)-pyrrolidin-2-yl-oxyl-6-(sulfooxy)-l,6-diaza bicyclor3.2.11 octane-2-carboxamide:
The solid (5 g) obtained in Step 4 of Example 2 was dissolved in water (30 ml) with stirring. To this solution, Isopropanol (210 ml) was slowly added at 25 °C and stirred for 12 hours. The separated solid was filtered and washed with additional isopropanol ( 10 ml) and dried under reduced pressure to obtain 3.9 g of (25,5i?)-7-oxo-N-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfo-oxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide as crystalline Form I, having XRPD as shown in Figure 2, in 78 % yield.
Analysis:
Purity as determined by HPLC: 95.56 %; and
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 10.57 (± 0.2), 12.01 (± 0.2), 13.61 (± 0.2), 15.47 (± 0.2), 17.86 (± 0.2), 18.34 (± 0.2), 19.09 (± 0.2), 19.81 (± 0.2), 22.69 (± 0.2), 24.79 (± 0.2), 27.22 (± 0.2) and 33.41 (± 0.2)
//////
WCK ? , WCK Series by Wockhardt for treating the bacterial infection

(2S,5R)-7-0X0-N-[(2S)-PYRROLLIDIN-2-YL-METHYLOXY]-6-(SULFOOXY)-1,6-DIAZABICYCLO[3.2.1 ]OCTANE-2-CARBOXAMIDE
(2S,5R)-7-Oxo-N-((2S)-pyrrolidin-2-ylmethyloxy)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(2S)-2-pyrrolidinylmethoxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
KEEP WATCHING THIS POST
MW 364.37, C12 H20 N4 O7 S
CAS 1452459-04-9 FREE FORM
CAS Na SALT 1572988-44-3
PATENTS, WO 2015079329, WO 2015079389 , WO 2015063714, US 20130225554
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistant. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in discovery of antibacterial agents.
Several compounds have been described in the prior art for use in treatment of bacterial infections (for example, see Patent Application Nos. PCT/IB2012/054296, PCT/IB2012/054290, US20130225554, PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178, PCT/US2009/041200, PCT/US2013/034562, PCT/US2013/034589, PCT/IB2013/053092 and PCT/IB2012054706). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.

PATENT
https://encrypted.google.com/patents/WO2015079329A2?cl=en



Formula (I)
Scheme -1

Formula (VII) Formula (VIII)

Formula (III) Formula (X) Formula (IX)
Scheme 2
Example 1
Synthesis of fert-butyl (25)-2-r(aminooxy)methyllpyrrolidine-l-carboxylate
Step 1: Synthesis of l-(tert-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII):
To a stirred suspension of (2S)-pyrrolidine-2-carboxylic acid (L-proline) (200 g, 1.73 mol) in 1,4-dioxan and water mixture (1: 1, 1000 ml : 1000 ml) was added a solution of sodium hydroxide (138.97 g, 3.47 mol in 740 ml water) over a period of 20 minutes at 0 °C. Bi-feri-butyl dicarbonate (415.3 ml, 1.9 mol in 400 ml 1,4-dioxan) was added to the resulting clear solution over a period of 30 minutes, at temperature of about 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 16 hours. After completion of the reaction (monitored by thin layer chromatography), the reaction mixture was concentrated to 40 % of the initial volume under reduced pressure at 40-50 °C. The pH of the residual mixture was adjusted to 2 – 2.5 using 30 % aqueous potassium hydrogen sulphate at 15 °C under continuous stirring. The separated solid was filtered under suction and washed with water (2×400 ml) and dried under reduced pressure (4 mm Hg), to obtain 370 g of l-(ieri-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII) as white solid.
Analysis:
Mass: 216 (M+l), for Molecular Weight: 215.24 and Molecular Formula:
1H NMR (400 MHz, CDC13): δ 10.60 (s, 1H), 4.35-4.24 (dd, 1H), 3.54-.3.34 (M, 2H), 2.27-1.91 (unresolved, 4H), 1.47-1.41 (d, 9H);
Purity as determined by HPLC: 99.92 %.
Step 2: Synthesis of tert-iutyl-(25)-2-(hydroxymethyl)-pyrrolidine-l-carboxylate (IX):
N-Methylmorpholine (113 ml, 1.114 mol) was added to the suspension of \-{tert-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII, 30 g, 139 mmol) in tetrahydrofuran (2000 ml) under stirring at temperature of about 0 °C. Ethyl chloroformate (106 ml, 1.114 mol) was added drop- wise to the above obtained clear solution over a period of 30 minutes. After stirring for 1 hour, the resulting suspension was filtered over celite and the residue was washed with tetrahydrofuran (2×200 ml). To the combined filtrate was added dropwise a solution of sodium borohydride (42.1 g, 1.114 mol) in 210 ml water, containing a catalytic amount of sodium hydroxide, at temperature of about -10 °C over a period of 1-2 hours under stirring. The reaction mixture was allowed to warm to room temperature and stirred further for an hour. The reaction mixture was filtered through celite bed and the filtrate concentrated under reduced pressure to yield 180 g of ieri-butyl(25)-2-(hydroxymethyl)-pyrrolidene-l-carboxylate (IX) as colorless oil.
Analysis:
Mass: 202 (M+l), for Molecular Weight: 201.2 and Molecular Formula: C10H19NO3;
1H NMR (400 MHz, CDC13): δ 3.94-.3.92 (m, 1H), 3.80 (board, 1H), 3.63-3.54 (m, 2H), 3.45-3.40 (m, 1H), 3.32-3.28 (m, 1H), 2.01-1.96 (m, 1H), 1.84-1.75 (m, 2H), 1.63 (m, 1H), 1.45 (s, 9H);
Purity as determined by HPLC: 87.7 %.
Step 3: Synthesis of fert-butyl (25)-2-[[(l,3-dihydro-l,3-dioxo-2H-isoindol-2-yl)oxy] methyl] -pyrrolidine-1 -carboxylate (X) :
Triphenylphosphine (328.4 g, 1.253 mol) in tetrahydrofuran (1260 ml) was added to solution of Diisopropyl azodicarboxylate (253.3 g, 1.253 mol) in tetrahydrofuran at temperature of -15 °C under stirring. After stirring for an hour, N-feri-butoxylcarbonyl-L-prolinol (IX) (180 g, 0.895 mol) in tetrahydrofuran (540 ml) was added to the resulting mixture over a period of 15 minutes. After stirring the mixture for 45 minutes, N-Hydroxy phthalimide (146 g, 0.895 mol) was added and the mixture was allowed to warm to room temperature and stirred further for 16 hours. The solvent was evaporated under reduced pressure and residual oil was dissolved in dichloromethane (5000 ml) and washed with an aqueous 5 % sodium hydrogen carbonate solution (2×300 ml). The organic layer was dried over anhydrous sodium sulfate and the solvent evaporated under reduced pressure to obtain viscous oil. Diisopropyl ether (720 ml) was added to the oil, the mixture was stirred well and separated solid was filtered under suction. The filtrate was concentrated under reduced pressure and the residue was further purified by chromatography over a silica gel column (60 -120 mesh) and eluted with mixtures of ethyl acetate and hexane. Upon concentration of the combined eluted fractions, 230 g of teri-butyl (25)-2-[[( l,3-dihydro- l,3-dioxo-2H-isoindol-2-yl)oxy]methyl]-pyrrolidine- l-carboxylate (X) was obtained as yellow oil.
Analysis:
Mass: 347.3 (M+l), for Molecular Weight: 346.39 and Molecular Formula: ![]()
1H NMR (400 MHz, CDCI3): δ 7.80-7.78 (m, 2H), 7.72-7.70 (m, 2H), 4.32 (brs, 1H), 4.05 (brs, 2H), 3.36-3.31 (m, 2H), 2.27-2.25 (m, 1H), 2.08(m, 1H), 1.88-1.87 (m, 2H), 1.43 (s, 9H).
Step 4: Synthesis of fert-butyl (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (HI):
To a stirred solution of the compound of Formula (X) ( 100 g, 0.288 mol) in dichloromethane (2000 ml) was added 99 % hydrazine hydrate (28.9 g, 0.577 mol) drop-wise over a period of 30 minutes at temperature of about 25 °C. The stirring was continued further for a period of 3 hours. The separated solid was filtered and the solid washed with additional dichloromethane (2 x 500 ml). The combined organic layer was collected and washed with water (2 x 500 ml). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 62.4 g of tert-butyl (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (III) as a colorless oil. This was used as such for the next reaction without further purification.
Analysis:
Mass: 215.1 (M- l), for Molecular Weight: 216.2 and Molecular Formula:
Example 2
Synthesis of (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl-methyloxyl-6-(sulfooxy)-l,6- diazabicvclor3.2.11octane-2-carboxamide (I)
Step 1: Synthesis of tert-butyl (25)-2-{[({[25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate
(IV):
Sodium(25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II, 77.4 g, 0.259 mol; prepared according to the procedure disclosed in Indian patent application No. 699/MUM/2013) was dissolved in water (774 ml) to obtain a clear solution. To the clear solution was added EDC.HC1 (120.8 g, 0.632 mol) at temperature of about 15°C and after 10 minutes a solution of tert-buty\ (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (III, 62.4 g, 0.288 moles prepared as per the literature procedure depicted in scheme 2) in dimethylformamide (125 ml) was added drop wise under continuous stirring at temperature of about 10 °C. The reaction mass was allowed to warm to temperature of about 25°C and then HOBt (38.96 g, 0.288 mol) was added in small portions over a period of 15 minutes and the resulting mixture was further stirred at room temperature for 16 hours. The reaction progress was monitored using thin layer chromatography using mixture of acetone and hexane (35: 65) as solvent system. The resulting suspension was filtered and the residue was washed with water (200 ml). The residual white solid was suspended in water (200 ml) and the mixture stirred with heating at temperatyre of about 50 °C for 3 hours. The resulting suspension was filtered, the residue dried at atmospheric temperature and then dried under vacuum to obtain 105 g of ierr-Butyl(25)-2- { [( { [25,5R)-6-(benzyloxy)-7-oxo- l,6-diazabicylco[3.2. l]oct-2-yl]carbonyl} amino)oxy]methyl}pyyrolidine-l-carboxylate (IV) as off white solid.
Analysis:
Mass: 475.4 (M+l), for Molecular Weight of 474.56 and Molecular Formula of ![]()
1H NMR (400 MHz, CDCI3): δ 10.16 (br s, 1H), 7.43-7.35 (m, 5H), 5.06-4.88 (dd, 2H), 4.12 (s, 1H), 3.94-.393 (d, 2H), 3.83 (unresolved s, 1H), 3.75-3.73 (m, 1H), 3.37-3.28 (dt, 2H), 3.02-2.86 (dd, 2H), 2.31-2.26 (m, 1H), 2.02-1.84 (m, 6H), 1.71-1.68 (m, 1H), 1.45 (s, 9H).
Step 2: Synthesis of tert-butyl(25)-2-{[({[25,5R)-6-hydroxy-7-oxo-l,6-diazabicylco
[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (V):
tert-butyl(25)-2-{ [({ [25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl] carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (IV, 85 g, 0.179 mol) was dissolved in a mixture of dimethylformamide and dichloro methane (1: 1, 425 ml : 425 ml) to obtain a clear solution. To this solution was added 10 % Pd-C (17 g, 50 % wet) catalyst. The suspension was stirred for 4 hours under 7 psi hydrogen atmosphere at temperature of about 25 °C. The resulting mixture was filtered through celite under suction. The residue was washed with dichloromethane (170 ml). The solvent from the filtrate was evaporated under reduced pressure to furnish 68.8 g of tert-buty\(2S)-2-{ [( { [25,5i?)-6-hydroxy-7-oxo- l,6-diazabicylco[3.2. l]oct-2-yl]carbonyl} amino)oxy] methyl}pyyrolidine-l-carboxylate (V) as oil. The obtained product was used as such for the next reaction without further purification.
Analysis:
Mass: 385.4 (M+l), for Molecular Weight of 384.4 and Molecular Formula of C17H28N406.
Step 3: Synthesis of tert-butyl(25)-2-{[({[25,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate, tetra butyl ammonium salt (VI):
To solution of ieri-butyl(25)-2-{ [({ [25,5i?)-6-hydroxy7-oxo-l,6-diazabicylco [3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (V, 68.8 g, 0.178 mol) in dimethylformamide, (345 ml) was added sulfur trioxide dimethylformamide complex (30 g, 0.196 mol) under stirring at temperature of about 10 °C. The reaction mass was stirred at the same temperature for 30 minutes and then allowed to warm to room temperature. After 2 hours solution of tetra butyl ammonium acetate (59.09 g, 0.196 mol) in water (178 ml) was added to the reaction mixture under stirring. After 2 hours, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2×140 ml) to obtain thick mass. This mass was partitioned between dichloromethane (690 ml) and water (690 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (345 ml). The combined organic extracts were washed with water (3×345 ml) and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure and the resulting oily mass was triturated with ether (3×140 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure to obtain 102 g of ieri-butyl(25)-2- { [({ [25,5i?)-6-(sulfooxy)-7-oxo- l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine- l-carboxylate, tetrabutyl ammonium salt (VI) as fluffy material.
Analysis:
Mass: 463.4 (M- l without TBA), for Molecular Weight of 705.96 and Molecular Formula of C33H63N5O9 S;
1H NMR (400 MHz, CDCI3): δ 10.2 (s, 1H), 4.35 (s, 1H), 4.14 (s, 1H), 3.91 -3.92 (d, 2H), 3.74 (m, 1H), 3.36-3.27 (m, 10H), 2.96-2.88 (dd, 2H), 2.31-2.26 (m, 2H), 2.19-1.98 (m, 2H), 1.95-1.70 (m, 4H), 1.68- 1.62 (p, 8H), 1.49- 1.40 (m, 17H), 1.02-0.98 (t, 12H).
Step 4: (25,5R)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diaza bicyclo [3.2.1]octane-2-carboxamide (I):
feri-butyl(25)-2-{ [( { [25,5i?)-6-(sulfooxy)-7-oxo-l ,6-diazabicylco[3.2.1]oct-2-yl] carbonyl}amino)oxy]methyl}pyyrolidine- l-carboxylate, tetrabutylammonium salt (VI) (50 g, 0.0708 mol) was dissolved in dichloromethane (250 ml) and to the clear solution was slowly added trifluoroacetic acid (250 ml) at temperature of about -10 °C over a period of 1 hour under stirring. After stirring for an hour, the resulting mixture was poured into hexane (2500 ml) and the oily layer was separated. This procedure was repeated one more time and finally the separated oily layer was added to diethyl ether (500 ml) under vigorous stirring at temperature of about 25 °C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with diethyl ether (2x500ml). The solid thus obtained was stirred with fresh dichloromethane (500 ml) for 30 minutes and filtered. The residual solid was dried at temperature of about 45 °C under reduced pressure to yield 25 g of (25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- l,6-diazabicyclo[3.2.1]octane-2-carboxamide (I) in amorphous form. The XRD of the obtained amorphous form is shown in Figure 1.
Analysis:
Mass: 363.2 (M- l), for Molecular Weight: 364.37 and Molecular Formula: C12H2oN407S;
1H NMR (400 MHz, DMSO-D6): δ 1 1.73 (s, 1H), 8.62-8.83 (d, 2H), 3.88-4.00 (m, 3H), 3.74-3.81 (m, 2H), 3.19 (t, 2H), 2.94-3.04 (dd, 2H), 1.96-2.03 (m, 2H), 1.80-1.92 (m, 3H), 1.54- 1.73 (m, 3H);
Purity as determined by HPLC: 90.30 %.
Example 3
Preparation of Crystalline Form I of (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl- methyloxyl-6-(sulfooxy)-l,6-diaza bicvclor3.2.11octane-2-carboxamide
The amorphous solid obtained in the Step 4 of Example 2 was dissolved in water (75 ml) and to this solution isopropanol (200 ml) was slowly added at temperature of about 25 °C. The solution was further stirred for 12 hours. The separated solid thus obtained was filtered and washed with additional isopropanol (25 ml) and dried under reduced pressure to obtain 19 g of (25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l ,6-diazabicyclo[3.2.1]octane-2-carboxamide as crystalline Form I. The XRD of the obtained crystalline Form I is shown in Figure 2.
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 8.08 (± 0.2), 1 1.45 (± 0.2), 16.26 (± 0.2), 17.89 (± 0.2), 18.15 (± 0.2), 19.66 (± 0.2), 21.15 (± 0.2), 23.55 (± 0.2), 24.23 (± 0.2), 24.94 (± 0.2), 25.66 (± 0.2) and 29.41 (± 0.2).
Typical X-ray analysis was performed as follows. Pass the test substance through sieve #100 BSS or gently grind it with a mortar and pestle. Place the test substance uniformly on a sample holder having cavity surface on one side, press the sample and cut into thin uniform film using a glass slide in such a way that the surface of the sample should be smooth and even. Record the X-ray diffractogram using the following instrument parameters:
Instrument : X-Ray Diffractometer
(PANalytical, Model X’Pert Pro
MPD)
Target source : CuK(a)
Antiscattering slit (Incident beam) : 1°
Programmable Divergent slit : 10 mm (fixed)
Anti- scattering slit (Diffracted beam) : 5.5 mm
Step width : 0.02°
Voltage : 40 kV
Current : 40 mA
Time per step : 30 seconds
Scan range : 3 to 40°
Example 4
Preparation of Pure (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl-methyloxyl-6-(sulfooxy)- l,6-diazabicyclor3.2.11octane-2-carboxamide
(25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- l,6-diazabicyclo [3.2.1] octane-2-carboxamide (5 g) was slowly dissolved in water (50 ml) under stirring until clear solution appears. To this clear solution 350 ml of isopropanol was added drop wise under stirring over the period of 2 hours. Formation of fine white precipitates was observed after the completion of the addition of isopropanol. The resulted fine suspension was stirred at temperature of about 25 °C for 20 hours. The formed white precipitates were filtered and vacuum dried at temperature of about 30-40 °C, under reduced pressure (2 mm Hg) to get 4.4 g of (2S,5i?)-7-oxo-N-[(2S)-pyrroMin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo [3.2.1] octane-2-carboxamide.
The above obtained (25,5i?)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide (3.4 gm) was dissolved in 34 ml of water to get clear solution. To the obtained clear solution 170 ml of isopropanol was added drop wise over a period of 1 hour. Formation of fine oily globules was observed and allowed to stand still for 15 minutes. The upper clear water and isopropanol layer was decanted from the oily mass. The clear decanted solution was allowed to stand at temperature of about 25 °C for 48 hours. Formation of crystals was observed and were collected by filtration. The collected crystals were dried at temperature of about 30-40 °C, under reduced pressure (2 mm Hg) to get 2 g of (2S,5i?)-7-oxo-N-[(2S)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide which was analyzed for content of various components using HPLC and the results are described in Table 1.
The relative % content of (25,5i?)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide with other substances (Table 1) was determined using HPLC (Agilent 1100 or equivalent). The HPLC column having 250 mm length and 4.6 mm ID packed with 5 μ particles of octa-decyl silane (ODS) was used. Mobile phase A used was a mixture of buffer (0.02 M potassium dihydrogen phosphate in HPLC grade water, pH adjusted to 2.5 with orthophosphoric acid and again readjusted to 7.0 with dilute ammonia), HPLC grade water and acetonitrile in a ratio of 40 : 60 : 0.2; v/v/v. Mobile phase B was mixture of buffer and acetonitrile in a ratio of 40 : 60; v/v. Mobile phase was run in gradient mode. Initially mobile phase A and B was run at 100 : 0 for 15 minutes, slowly ratio of mobile phase B was raised to 100 % in 10 minutes, held for 10 minutes at this concentration and brought back to initial condition in next 5 minutes and held for 10 minutes before next run. Flow rate of mobile phase was maintained at 1.0 ml/min. Column temperature was maintained at temperature of about 30°C. Detection was carried out using UV detector at wavelength 225 nm. Test solutions were prepared in mobile phase A. The method is capable of resolving diastereomers (Table 1, Sr. No. 1 and 2) with resolution of not less than 2.0.

New Antibacterial oxazolidinones in pipeline by Wockhardt

WCK ?
(5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
(5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
MF C19 H25 F2 N3 O5, MW 413.42
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-hydroxy-4-(methoxymethyl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
CAS 957796-51-9
Antibacterial oxazolidinones
Wockhardt Ltd, Innovator
THIS MAY BE WCK 4086?????….WATCHOUT THIS POST FOR UPDATION
PATENTS
WO 2015173664, US8217058, WO 2012059823, IN 2011MU03726

Oxazolidinone represent a novel chemical class of synthetic antimicrobial agents. Linezolid represents the first member of this class to be used clinically. Oxazolidinones display activity against important Gram-positive human and veterinary pathogens including Methicillin-Resistant Staphylococcus aureus (MRSA), Vancomycin Resistant Enterococci (VRE) and β-lactam Resistant Streptococcus pneumoniae (PRSP). The oxazolidinones also show activity against Gram-negative aerobic bacteria, Gram-positive and Gram-negative anaerobes. (Diekema D J et al., Lancet 2001 ; 358: 1975-82).
Various oxazolidinones and their methods of preparation are disclosed in the literature. International Publication No. WO 1995/25106 discloses substituted piperidino phenyloxazolidinones and International Publication No. WO 1996/13502 discloses phenyloxazolidinones having a multisubstituted azetidinyl or pyrrolidinyl moiety. US Patent Publication No. 2004/0063954, International Publication Nos. WO 2004/007489 and WO 2004/007488 disclose piperidinyl phenyl oxazolidinones for antimicrobial use.
Pyrrolidinyl/piperidinyl phenyl oxazohdinone antibacterial agents are also described in Kim H Y et al., Bioorg. & Med. Chem. Lett., (2003), 13:2227-2230. International Publication No. WO 1996/35691 discloses spirocyclic and bicyclic diazinyl and carbazinyl oxazolidinone derivatives. Diazepeno phenyloxazolidinone derivatives are disclosed in the International Publication No. WO 1999/24428. International Publication No. WO 2002/06278 discloses substituted aminopiperidino phenyloxazolidinone derivatives.
Various other methods of preparation of oxazolidinones are reported in US Patent No. 7087784, US Patent No. 6740754, US Patent No. 4948801 , US Patent No. 3654298, US Patent No. 5837870, Canadian Patent No. 681830, J. Med. Chem., 32, 1673 (1989), Tetrahedron, 45, 1323 (1989), J. Med. Chem., 33, 2569 (1990), Tetrahedron Letters, 37, 7937-40 (1996) and Organic Process Research and Development, 11 , 739-741(2007).
Indian Patent Application No. 2534/MUM/2007 discloses a process for the preparation of substituted piperidino phenyloxazolidinones. International Publication No. WO2012/059823 further discloses the process for the preparation of phosphoric acid mono-(L-{4-[(5)-5-(acetylaminomethyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}4-methoxymethyl piperidine-4-yl)ester.
US Patent No. 8217058 discloses (5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide as an antibacterial agent and its process for preparation.
PATENT
WO2015173664

In some embodiments, there is provided a process for preparation of a compound of Formula (I) as shown in Scheme 1

(I I) (I N)
Scheme 1

Example 1
Preparation of (55)-iV-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide (I)
To a stirred solution of lithium teri-butoxide (59.1 g, 0.74 mol) in tetrahydrofuran (500 ml) was added a solution of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-carbamic acid benzyl ester (II) (100 g, 0.25 mol) in 500 ml of tetrahydrofuran slowly at room temperature. The resulting mixture was stirred for 3 hours at room temperature (formation of lumps observed). The reaction mixture was cooled to temperature of 10°C to 15°C and acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) (95.2 g, 0.49 mol) was added in one lot, after 5 minutes methanol (2.36 g, 0.075 mol) was added in one portion. The resulting mixture was stirred further at temperature of 10°C to 15°C. After 5 hours the reaction mixture was allowed to warm to room temperature and stirring continued further for 16 hours. An aqueous solution of saturated ammonium chloride (100 ml) was added to the reaction mixture, the resulting mixture was stirred well and the solvent evaporated under reduced pressure (35°C, 150 mm Hg). The residual mixture was diluted with water (1 L stirred well and filtered under suction, the residual solid was washed with additional fresh water (100 ml). The residual mass was suspended in acetone (500 ml), stirred well and the mixture diluted with hexane (1 L), slowly. The mixture was stirred further for 1 hour and filtered under suction. The residual solid was washed with a 2:1 mixture of acetone and water (100 ml). The residual solid was dried at 45°C, for 3.5 hour at 4 mm Hg, to obtain the 78 g of (55)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l -yl)-phenyl]-2-oxo-oxazolidin-5-ylmethylj -acetamide (I) as white solid, in 77% yield.
Analysis:
Mass: 414 (M+l ); for Molecular Weight: 413 and Molecular Formula: ![]()
Melting Point: 178-179°C;
1H NMR (400 MHz, DMSO): δ 8.18-8.21 (m, 1H), 7.19-7.25 (d, 2H), 4.07-4.71 (m, 1H), 4.32 (s, 1H), 4.02-4.07 (t, 1H), 3.64-3.68 (t, 1H), 3.14 (s, 2H), 2.81-2.83 (d, 2H), 1.81 (s, 3H), 1.63-1.69 (t, 2H), 1.42-1.45 (d, 2H);
Purity as determined by HPLC: 97.65%.
Example 2
Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III)
Step-I: Preparation of l-amino-3-chloro-propan-2-ol hydrochloride (VI)
Benzaldehyde (118.67 g, 1.03 mol) was dissolved in ethanol (297 ml) under stirring and the solution was cooled to 18-19°C. To this solution aqueous ammonia solution (25%) (101.58 ml) was added slowly, followed by slow addition of S-epichlorohydrin (100 g, 1 mol). The resulting mixture was warmed to 40°C and stirred for 7 hours. The mixture was allowed to cool to room temperature and stirred further. After 16 hours, the reaction mixture was concentrated to 50% volume under reduced pressure. Toluene (228 ml) was added to the reaction mixture followed by addition of aqueous hydrochloric acid (162 ml of concentrated hydrochloric acid diluted with 152 ml of water). The mixture thus obtained for 3 hours at 45°C, the resulting mixture was allowed to cool to room temperature and the toluene layer separated. The toluene layer was further extracted with water (56 ml). The combined aqueous layer was diluted with ethanol (56 ml) and the mixture evaporated under reduced pressure. This process was repeated again. To the final concentrate was added ethanol (180 ml), stirred for 10 minutes and the mixture cooled to -28°C to -30°C and maintained at this temperature for 2 hours. The separated solid was filtered under suction and the residue washed with cold (-30°C) ethanol (50 ml). The residue was dried at 45°C, under reduced pressure (4 mm Hg) for 3 hours, to obtain 96 g of l-amino-3-chloro-propan-2-ol hydrochloride (VI) as white solid in 61% yield.
Analysis:
Mass: 110 (M+l) as free base; for Molecular Weight: 145.5 and Molecular Formula: ![]()
1H NMR (400 MHz, D20): δ 4.02-4.08 (m, 1H), 3.51-3.61 (m, 2H), 3.12-3.16 (dd, 1H), 2.93 -2.99 (dd, 1H).
Step-II: Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III).
A stirred solution of dichloromethane (220.8 ml) containing the step-I salt (96 g, 0.66 mol) was cooled to 18-20°C. Acetic anhydride (154.78 g, 1.5175 mol) was added slowly (slight exothermic). Pyridine (67.76 g, 0.8577 mol) was added slowly (exothermic) while maintaining the temperature at 18-20°C. The resulting mixture was heated to 40°C for 5 hours. The reaction mixture was allowed to cool to room temperature and stirring continued for further 16 hours. The reaction mass was cooled to 3-6°C and diluted with 170 ml of fresh water. To this was added an aqueous solution of potassium carbonate (191.2 g of K2CO3 in 382 ml water). The reaction mixture was further diluted with additional dichloromethane (170 ml) and water (425 ml). The reaction mass was stirred well and the dichloromethane layer separated. The aqueous layer was further extracted with 2×170 ml dichloromethane. The combined dichloromethane layer was washed with aqueous sodium chloride solution (13.6 g of sodium chloride in 493 ml water). The solvent was evaporated till a volume of 170 ml and the residual layer was diluted with toluene (340 ml), stirred well and the solvent was evaporated completely at 40°C under reduced pressure (4 mm Hg). To the residue ethyl acetate (170 ml) and hexane (187 ml) were added and the mixture stirred for 30 minute. The separated solid was filtered under suction and the residue washed with 50 ml of a 1 :1 mixture of ethyl acetate and hexane. The solid obtained was dried under reduced pressure (4 mm Hg) at 45°C for 3.5 hours, to obtain 96 g of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) as a white solid, in 75% yield.
Analysis:
Mass: 194 (M+l); for Molecular Weight: 193 and Molecular Formula: C7Hi2ClN03; 1H NMR (400 MHz, CDC13): 5 5.69 (s, 1H), 5.0-5.1 (m, 1H), 3.4-3.7 (m, 4H), 2.1 (s, 3H), 1.9 (s, 3H).
PATENT
http://www.google.st/patents/WO2007132314A2?cl=en



(3) (4)
Scheme -1

(6) Formula π Scheme-2

Formula II Formula in

Formula I(a) Scheme-4
Example -11 : (5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
The example- 10 (54.86 g, 0.144 mol) was suspended in methanol (1100 ml) under stirring at RT. Sodium metal (4 g, 0.174 mol) was added in small lots in 2 min to the above suspension under stirring. The reaction mixture was warmed to 40-420C and was stirred at this temperature for about 40 hrs. After completion of the reaction (TLC), the solvent was evaporated under reduced pressure to obtain a thick slurry. The thick slurry thus obtained was gradually added to water (1100 ml) under stirring. After the complete addition, the pH of the aqueous suspension was adjusted to 7 by adding sufficient quantity of glacial acetic acid. The separated solid was filtered and the residue was washed with water. The obtained solid was further purified by column chromatography over silica gel to obtain the product as a white solid, 32.7 g, 55 % yield.
M.P.: 173-1740C;
MS : M+l= 414(MH+, 100%); for M.F.: Ci9H25F2N3O5
1H-NMR (400 MHz, CDCl3): δ 7.0-7.1 (m, 2H5Ar-H), 6.0 (t, IH, NH), 4.70-4.80 (m, IH), 4.00 (t,lH), 3.70-3.75 (m, 2H), 3.5-3.7 (m, IH), 3.43 (s, 3H, OCH3), 3.37-3.42 (m, 2H), 3.30 (s, 2H, -OCH2), 3.0-3.05 (m, 2H), 2.22(bs,lH ,-OH),2.04 (s, 3H, COCH3), 1.70-1.75 (m, 4H).
Patent
INDIAN 3049/MUM/2010
Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxy methyl-piperidin-4-yl) ester

Specific intermediate compounds of the invention include:
6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane;
1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol;
[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one;
(5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one; and
(5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide.
Examples
Preparation of Intermediate-1: 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one
Chloroform (9.3 L) was charged in a 20 L reaction assembly and 4-piperidone hydrochloride (1.17 Kg, 7.62 mol) was added under stirring followed by triethylamine (2.14 Kg, 2.95 L, 21.1 mol). After 30 minutes of stirring, 3,4,5-trifluoronitrobenzene (1.5 Kg, 8.47 mol) was added to the mixture in one lot and the contents were heated to 65-70ºC for 8 h. After completion of the reaction, chloroform was removed under vacuum to obtain a syrupy mass. At this stage, water (10 L) was added to the mass and the chloroform recovery was continued under vacuum below 65oC till the chloroform was removed completely. The slurry was cooled to RT and filtered. The solid product was washed with water (3 L) followed by hexanes (2 L). The product was dried in a vacuum oven below 70oC to obtain the product as a yellow solid, 1.88 Kg ; Yield 97%.
M.P.: 130-132oC; MS: 257(M+1); M.F.: C11H10F2N2O3.
Preparation of Intermediate 3: 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
Method A:
Preparation of Intermediate–2: (Stage-I): 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane
A solution of trimethylsulfoxonium iodide (1.504kg, 6.836mol) in acetonitrile (7L) was cooled to 0 to 5oC. , under argon atmosphere. Potassium tert-butoxide (0.736kg, 6.552 mol) was added in small lots over 0.5h. The resulting solution was stirred for 2h at the same temperature. To this solution was added 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one ( 1.4kg, 5.46mol) in small lots over a period of 1h, while maintaining the temp. between 5-10oC. The resulting mixture was stirred for 1h. The solvent was evaporated to a minimum amount possible, under reduced pressure while maintaining the temperature below 10oC. The residue was poured in water( 18L) and the pH adjusted to neutral with dilute acetic acid. The resulting slurry was stirred well and the separated solid filtered under suction. The solid was washed with fresh water till the filtrate was free of acetic acid. The solid was dried at 80oC, for 6h, under reduced pressure to obtain the product as pale yellow solid, 1.264kgs, yield 85%.
M.P.: 96-97oC; MS: M+1: 271; M.F.: C12H12F2N2O3,.
Preparation of Intermediate-3: (Stage-II): 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
To a solution of sodium methoxide (236g, 4.35mol) in methanol (3L), at RT, was added 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro [2.5]octane (964g, 3.57mol) in small portions and the reaction mixture was stirred for 26h at RT. Acetic acid (265g, 4.44mol) was added slowly to neutralize the pH of the solution. The resulting mixture was poured into chilled water(18L) and stirred for 1h. The separated solid was filtered under suction. The solid was washed with additional water till the filtrate was free of acetic acid. The solid was dried for 10hat RT under reduced pressure, to obtain the product as a pale yellow solid, 973g, yield, 90%
M.P.: 84-86oC; MS: 303 (M+1); M.F.: C13H16F2N2O4
Method B:
Dimethylsulfoxide (DMSO, 100 ml) and methanol (500 ml) were charged in a 1 L glass reaction assembly. Potassium hydroxide (59.2g, 0.898 mol) was charged in the assembly followed by trimethylsulfoxonium iodide (94.5 g, 0.43 mol) and the contents were stirred for 30 minutes and then cooled to 10oC-15oC. To the cooled contents was added 1-(2,6-difluoro-4-nitrophenyl)-piperidin-4-one (100 g, 0.39 mol) in small lots. After the addition, the temperature was allowed to raise to RT and the contents were further stirred for 24 h (ring opening of the epoxide intermediate viz. 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane takes place).
[Physical data of the intermediate: M.P.: 96-970C, MS: 271(M+1); M.F.: C12H12F2N2O3, .
After completion of the reaction the contents were poured slowly in ice-water (600g crushed ice in 600 ml water). The precipitated solid product was filtered and was washed with water:methanol, 2:1 (100 ml X 2). The wet product was used in the next step.
M.P.: 84-86oC; MS: 303 (M+1);.M.F.: C13H16F2N2O4,:
Preparation of Intermediate -5: [3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester
Method A: Preparation of Intermediate 4: ( Stage-I)
Water (1.19 L) and methanol (595 ml) were charged in a 3 L glass reaction assembly, followed by 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (85 g, 0.281 mol) and the contents were stirred. Sodium dithionite (288 g, 1.407 mol) was added in one lot and the reaction mixture was heated to 80oC for 8 h. After completion of the reaction (TLC), methanol was recovered under vacuum below 65oC. After the recovery, the aqueous residue was extracted with chloroform (400 ml X 3). The combined chloroform extract (containing the intermediate 1-(4-amino-2,6-difluoro-phenyl)-4-methoxymethyl-piperidin-4-ol) was dried over anhydrous Sodium sulfate and used in the next step (carbamate formation).
Preparation of Intermediate -5: (Stage-II):
The above chloroform extract was charged in a 3 L glass reaction assembly. Sodium bicarbonate (70 g, 0.843 mol) was added to the extract and the contents were cooled to 15oC-20oC. Benzylchloroformate solution (50% in toluene, 48 g, 96 ml, 0.281 mol) was added slowly to the above mixture under stirring. After completion of the addition, the reaction mixture was stirred at RT for 2 h. After completion of the reaction (TLC), the contents were filtered on a Buchner assembly and the solid cake was washed with chloroform (85 ml X 2). The combined filtrate was evaporated under vacuum below 50oC to obtain yellowish oily mass, which was poured slowly in hexanes (850 ml) under stirring to obtain a precipitate. The precipitated product was filtered and washed with hexanes (100 ml X 2). The product was dried in a vacuum oven below 65oC to obtain 60.2 g brownish product (Yield = 38% on the basis of step-I input).
M.P.: 138-140oC; MS: 407(M+1); M.F.: C21H24F2N2O4.:.
Method B: : Preparation of Intermediate 4: ( Stage-I): To a solution of 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (973g, 3.22 mol) in ethyl acetae (10L) was added 10% Pd-C, (250g, 50% wet) and the resulting miture was hydrogenated in a pressure at 30 PSI, 45-55oC, for 3h. The catakyst was filtered and the residue was washed with additional ethyl acetate( 200ml). The combined filtrates were used as such for the next reaction (carbamate formation)
Preparation of Intermediate -5: (Stage-II):
To the above filtrate was added sodium bicarbonate(406g, 4.83 mol) and the mixture warmed to 40-45oC. To this mixture was added a 50% solution of Benzyl chloroformate in toluene(1.373L, 4.025 mol), drop-wise, over a period of 1h. Stir the resulting mixture for 1h and filter the insoluble material. The residue was washed with 300ml of ethyl acetate. The filtrates were combined and the solvent evaporated under reduced pressure, below 55oC.. Cool the residue and dilute it with hexane(10L). The resulting slurry was stirred well and the separated solid was filtered under suction. The residue was washed with additional hexane ( 2L). The solid was dried for 10h at RT, to obtain the product as dark brown solid, 1200g, yield, 96%.
M.P.: 138-140oC; MS: 407( M+1); M.F.: C21H24F2N2O.
Preparation of Intermediate -6:
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one
To a mixture of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester (100g, 0.237 mol) in dry tetrahydrofuran (THF) (2 L) at 40ºC was added drop-wise n-BuLi in hexane (1.6M, 45.5 g, 455 ml, 0.711 mol) under nitrogen atmosphere. The contents were stirred for 1 h at 40ºC and R-(-)-glycidyl butyrate (68.25 g, 0.474 mol) was added gradually. After the addition of R-(-)-glycidyl butyrate, the reaction mixture was stirred for 5-6 h at 40oC till completion of the reaction (TLC). After completion of the reaction, a solution of sodium methoxide (2 g) in methanol (66 ml) was added to the contents followed by water (8 ml) and the contents were stirred for an additional 0.5 h. Water (1 L) was added to the solution and the contents were extracted with ethyl acetate (1 L). The aqueous layer was further extracted with ethyl acetate (3 X 500 ml). The combined organic layer was evaporated under vacuum to obtain a thick residue. tert-Butyl methyl ether (1 L) was added to the residue and the contents were stirred for about 1 h to obtain a solid product, which was filtered and washed with tert-butyl methyl ether (2 X 100 ml). The product was dried under vacuum below 60ºC to obtain the product as a 46.5 g dark brown compound, 46.5g ,yield 51%.
M.P.: 117-119oC; MS: 373(M+1); M.F.: C17H22F2N2O5..
Preparation of Intermediate -7: (5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester
To a mixture of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one (45 g, 0.121 mol) in dichloromethane (0.3 L), was added triethylamine (24.5 g, 34 ml, 0.242 mol) while stirring. Methanesulfonyl chloride (18 g, 12.2 ml, 0.157 mol) was added to the above solution over a period of 1 h at 10oC -20oC and the reaction mixture was stirred for additional 2 h at RT. After completion of the reaction (TLC), the contents were evaporated under vacuum at 40oC to obtain an oily residue. Water (450 ml) was added to the residue and the traces of dichloromethane were removed under vacuum. The solid product thus obtained was filtered, washed with water (2 X 50 ml) and dried under vacuum at 70oC to obtain 50.6 g brownish compound. Yield = 93%; M.P.:106-108oC; MS: 451(M+1); M.F.: C18H24F2N2O7S.
Preparation of Intermediate 8a: (5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one
Method A:
To a solution of (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl)oxazolidin-2-one (2g, 5.3 mmol),in tetrahydrofuran (20 mL), under argon , was added diphenylphosphoryl azide (1.63mL, 5.9 mmol). The solution was cooled to 0oC in an ice-bath. 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) (0.76mL, 4.9mmol) was added drop-wise over 15min..The reaction was stirred at same temperature for 1 hr, and then warmed to room temperature and stirred under for 16 hr. The reaction mixture was diluted with ethyl acetate (20 mL), and water (20mL). After separation of water layer, the organic layer was washed with water and 0.5M citric acid monohydrate (10 mL). The organic layer was dried over sodium sulfate and the solvent evaporated under reduced pressure.The residue was triturated with ether to obtain the product as a buff colored solid, 1.32g (62%).
M.P.: 106-108oC; M.S.- 398(M+1); M.F.- C17H21F2N5O4,
Method B:
To a solution of (5R)-methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester (20 g, 0.044 mol, wet) in N,N-dimethylformamide (30 ml), was added sodium azide (8.6 g, 0.133 mol) in a single lot. The reaction mixture was gradually heated and the temperature was maintained at 70ºC for 8 h. After completion of the reaction (TLC), the contents were cooled to 20-25ºC and poured slowly into chilled water (300 ml). The solid product thus obtained was filtered and washed with water (2 x 50 ml). The wet product was air dried to obtain 16.5g dark brown compound (being an azide, it was NOT exposed to heat during drying) Yield ~ 93%.
M.P.: 106-108oC; MS : 398(M+1); M.F.: C17H21F2N5O4;:
Preparation of Intermediate 8b: (5S)-N-2-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-phthalimide
Method A:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate(10g, 0.022 mol), Potassium phthalimide (12.2g, 0.066 mol) and DMF (50ml) was heated, with stirring, at 90oC for 4h. The resulting mixture was cooled to RT and poured over ice-water mixture. The separated solid was filtered, washed with water and dried under suction to obtain the product as a white solid, 9.46g, in 85% yield.
M.P.: 154-156 oC; MS: 502 (M+1); M.F. C25H25F2N3O6.
Method B:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 minute phthalimide (1.18g, 8 mmol)) was added and after a further stirring for 10 minute, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 8 hrs ice-cold water (4 ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 1.56g, yield 58%.
M.P.: 154-156 oC; MS : 502 (M+1); M.F. C25H25F2N3O6.
Preparation of Intermediate 10: (5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40oC to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HCl solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70oC to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50oC.. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume.
To the above ethyl acetate solution was added Triethyl amine (19.1g, 0.189 mol), and acetic anhydride (16.1g, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5oC, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate(100ml) and dried at 70oC under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.011mol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(1.11g, 0.011mol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate (1gm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 ºC. for 15hrs. Then a mixture of conc. HCl (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75ºC for 5hrs. The mixture was concentrated under reduced pressure at 60-75 ºC. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 ºC for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50ºC to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Preparation of Intermediate -11: (S)-N-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryloxy-piperi din-1yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
To a solution of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)-phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.2 mmol) and tetrazole (0.6 mmol) in dichloromethane (5 ml) was added dibenzyl N,N,diisopropylphosphoramidite (0.4 mmol) and the resulting mixture was stirred for 4h. The resulting solution was cooled to 0 oC and 0.6 ml of 0.5M m-chloroperbenzoic acid solution in dichloromethane was added. After 4h, the solvent was evaporated under residue pressure and the residue chromatographed on a column of silica gel to obtain the product as a off-white solid in 75% yield,
MS: 674 (M+1); M.F. C33H38F2N3O8P;
Example A: Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxymethyl-piperidin-4-yl) ester
To a suspension of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryl- oxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.15 mmol) and 20 % palladium hydroxide (20 mg) in 20 ml of a mixture of dichloromethane /aqueous methanol was stirred at room temperature for 6h. The catalyst was filtered and the residue evaporated under reduced pressure. The residue obtained was triturated with acetone to obtain a white solid as product in 70% yield.
MP. >140 °C; MS : 494(M+1) M.F.: C19H26F2N3O8P.
PATENT
http://www.google.co.in/patents/WO2012059823A1?cl=en


c) Converting intermediate of Formula (5) into intermediate of structure (6)



f) Converting intermediate of Formula (11) into compound of Formula (A) or Pharmaceutically acceptable salts thereof



ormu a-
Scheme-1
Preparation of Intermediate 10: (5S)- N-{ 3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl- piperidin- 1 -yl)-phenyl] -2-oxo-oxazolidin-5-ylmethyl } -acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l- yl)-phenyl] -oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40°C to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HC1 solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4- hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70°C to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-5- azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50°C. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume. To the above ethyl acetate solution was added Triethyl amine (19. lg, 0.189 mol), and acetic anhydride (16. lg, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5°C, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate( 100ml) and dried at 70°C under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine- lyl)phenyl]-2-oxo-oxazolidin-5-yl methyl }-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.01 lmol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(l.l lg, 0.01 lmol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5. Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)phenyl]- 2-oxo-oxazolidin-5-yl methyl }-methanesulfonate (lgm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 °C. for 15hrs. Then a mixture of cone. HC1 (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75°C for 5hrs. The mixture was concentrated under reduced pressure at 60-75 °C. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 °C for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50°C to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.1 lg, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3- (3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-l-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5.
PATENT
http://www.google.co.in/patents/WO2008038092A2?cl=en

IV

V
‘ Scheme-1 ‘
/////////
SEE FULL ZOLID SERIES…………http://drugsynthesisint.blogspot.in/p/zolid.html
WCK Series by Wockhardt for treating the bacterial infection

Which WCK is it, WCK-4873 , WCK-4086, WCK ? for treating the bacterial infection.
(Not sure) will arrive at correct one….keep watching………..
CAS 1627163-98-7
- C14 H16 N2 O4 . Na,

- 1,6-Diazabicyclo[3.2.1]octane-2-carboxylic acid, 7-oxo-6-(phenylmethoxy)-, sodium salt (1:1), (1R,2S,5R)-
- SODIUM (2S, 5R)-6-(BENZYLOXY)-7-OXO-1,6-DIAZABICYCLO[3.2.1]OCTANE-2-CARBOXYLATE
sodium (2S,5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate…..WO2014135929

Patent
http://www.google.com/patents/WO2015136473A1?cl=en

EXAMPLES
The following examples illustrate the embodiments of the invention that are presently best known. However, it is to be understood that the following are only exemplary or illustrative of the application of the principles of the present invention. Numerous modifications and alternative compositions, methods, and systems may be devised by those skilled in the art without departing from the spirit and scope of the present invention. The appended claims are intended to cover such modifications and arrangements. Thus, while the present invention has been described above with particularity, the following examples provide further detail in connection with what are presently deemed to be the most practical and preferred embodiments of the invention.
Example 1
Synthesis of sodium (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicvclor3.2.11octane-2- carboxylate
Step 1; Preparation of -Γl-Γ(feΓt-butyldimethylsilyl -oxymethyll-5-Γdimethyl(oxido -λ-4-sulfanylidenel-4-oxo-pentyll-carbamic acid tert-butyl ester (III):
To a suspension of trimethylsulfoxonium iodide (180.36 gm, 0.819 mol) in tetrahydrofuran (900 ml), sodium hydride (32.89 g, 0.819 mol, 60% in mineral oil) was charged in one portion at 30°C temperature. The reaction mixture was stirred for 15 minutes and then dropwise addition of dimethylsulphoxide (1.125 ml) was done over a period of 3 hours at room temperature to provide a white suspension. The white suspension was added to a pre-cooled a solution of 2-(feri-butyldimethylsilyl-oxymethyl)-5-oxo-pyrrolidine-l-carboxylic acid tert-buty\ ester (II) (225 g, 0.683 mol, prepared as per J. Org Chem.; 2011, 76, 5574 and WO2009067600) in tetrahydrofuran (675 ml) and triethylamine (123.48 ml, 0.887 mol) mixture at -13°C by maintaining the reaction mixture temperature below -10°C. The resulting suspension was stirred for additional 1 hour at -10°C. The reaction mixture was carefully quenched by addition of saturated aqueous ammonium chloride (1.0 L) at -10°C to 10°C. The reaction was extracted by adding ethyl acetate (1.5 L). The layers were separated and aqueous layer was re-extracted with ethyl acetate (500 ml x 3). The combined organic layer was washed successively with saturated aqueous sodium bicarbonate (1.0 L), water (2.0 L) followed by saturated aqueous sodium chloride solution (1.0 L). Organic layer was dried over sodium sulfate and evaporated under vacuum to provide 265 g of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-buty\ ester (III) as an yellow oily mass.
Analysis:
Mass: 422.3 (M+l); for Molecular weight: 421.68 and Molecular Formula: ![]()
1H NMR (CDC13): δ 4.77 (br d, 1H), 4.38 (br s, 1H), 3.58 (br s, 3H), 3.39 (s, 3H), 3.38 (s, 3H), 2.17-2.27 (m, 2H), 1.73-1.82 (m, 2H), 1.43 (s, 9H), 0.88 (s, 9H), 0.01 (s, 3H), 0.04 (s, 3H).
Step 2: Preparation of 5-r4-benzyloxyimino-l-(fert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyll-carbamic acid tert- butyl ester (IV):
To a suspension of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-butyl ester (III) (440.0 g, 1.045 mol) in tetrahydrofuran (6.6 L), O-benzhydroxylamine hydrochloride (200.0 g, 1.254 mol) was charged. The reaction mixture was heated to 50°C for 2.5 hours. The reaction mixture was filtered through pad of celite and filtrate was concentrated to provide a residue. The residue was dissolved in ethyl acetate (5.0 L) and washed successively with saturated aqueous sodium bicarbonate (1.5 L), water (1.5 L) and saturated aqueous sodium chloride (1.5 L). Organic layer was dried over sodium sulfate. Solvent was evaporated under vacuum to yield 463.0 g of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) as an oily mass.
Analysis:
Mass: 486.1 (M+l); for Molecular weight: 485.4 and Molecular Formula: ![]()
1H NMR (CDCI3): δ 7.26-1 6 (m, 5H), 5.10 (s, 2H), 4.66 (br d, 1H), 3.58-4.27 (m, 2H), 3.56-3.58 (m, 3H), 2.40-2.57 (m, 2H), 1.68-1.89 (m, 2H), 1.44 (s, 9H), 0.89 (s, 9H), 0.02 (s, 3H), 0.04 (s, 3H).
Step 3: Preparation of 5-5-benzyloxyimino-2-(fert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V):
To a solution of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) (463.0 g 0.954 mol) in tetrahydrofuran (6.9 L), was charged potassium feri-butoxide (139.2 g, 1.241 mol) in portions over a period of 30 minutes by maintaining temperature -10°C. The resulting suspension was stirred for additional 1.5 hours at -10°C to -5°C. The reaction mixture was quenched by addition of saturated aqueous ammonium chloride (2.0 L) at -5°C to 10°C. The organic layer was separated and aqueous layer was extracted with ethyl acetate (1.0 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride solution (2.0 L). Organic layer was dried over sodium sulfate, and then evaporated under vacuum to yield 394.0 g of 5-5-benzyloxyimino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-butyl ester (V) as an yellow oily mass.
Analysis:
Mass: 449.4 (M+l) for Molecular weight: 448.68 and Molecular Formula: C24H4oN204Si;
1H NMR (CDC13): δ 7.25-1 3 (m, 5H), 5.04-5.14 (m, 2H), 4.35 (br s, 1H), 3.95 (br s, 1H), 3.63-3.74 (br d, 2H), 3.60-3.63 (m, 1H), 2.70-2.77 (m, 1H), 2.33-2.41 (m, 1H), 1.79-1.95 (m, 2H), 1.44 (s, 9H), 0.88 (s, 9H), 0.03 (s, 3H), 0.04 (s, 3H).
Step 4: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI):
To a solution of 5-5-benzyloxyimino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V) (394.0 g, 0.879 mol) in dichloromethane (5.0 L) and glacial acetic acid (788 ml), was charged sodium cyanoborohydride (70.88 g, 1.14 mol) one portion. The resulting reaction mixture was stirred at temperature of about 25 °C to 30°C for 2 hours. The mixture was quenched with adding aqueous solution of sodium bicarbonate (1.3 kg) in water (5.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (2.0 L). The combined organic layer washed successively with water (2.0 L), saturated aqueous
sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to provide a residue. The residue was purified by silica gel column chromatography to yield 208 g of (25,5i?5)-5-benzyloxyamino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-buty\ ester (VI) as pale yellow liquid.
Analysis:
Mass: 451.4 (M+l); for Molecular weight: 450.70 and Molecular Formula: C24H42N204Si;
1H NMR (CDC13): δ 7..26-7.36 (m, 5H), 4.90-5.50 (br s, 1H), 4.70 (dd, 2H), 4.09-4.25 (m, 2H), 3.56-3.72 (m, 2H), 2.55-3.14 (m, 2H), 1.21-1.94 (m, 4H), 1.45 (s, 9H), 0.89 (s, 9H), 0.05 (s, 6H).
Step 5: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine (VII):
To a solution of 5-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI) (208 g, 0.462 mol) in dichloromethane (3.0 L), boron trifluoride diethyletherate complex (114.15 ml, 0.924 mol) was charged in one portion. The resulting reaction mixture was stirred at temperature of about 25°C to 35°C temperature for 2 hours. The reaction mixture was quenched with saturated aqueous sodium bicarbonate (2.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride (1.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to yield 159 g of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) as a yellowish syrup.
Analysis:
Mass: 351.3 (M+l); for Molecular weight: 350.58 and Molecular Formula: C19H34N202Si.
Step-6: Preparation of (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-r3.2.11octane (VIII):
Part 1; Preparation of (2S,5RS)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
To a solution of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) (159.0 g, 0.454 mol) in a mixture of acetonitrile (2.38 L) and diisopropylethylamine (316.5 ml, 1.81 mol) was added triphosgene (59.27 gm, 0.199 mol) dissolved in acetonitrile (760 ml) at -15°C over 30 minutes under stirring. The resulting reaction mixture was stirred for additional 1 hour at -10°C. The reaction mixture was quenched by addition of saturated aqueous sodium bicarbonate (2.0 L) at -5°C to 10°C. Acetonitrile was evaporated from the reaction mixture under vacuum and to the left over aqueous phase, dichloromethane (2.5 L) was added. The organic layer was separated and aqueous layer extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed successively with water (2.0 L), saturated aqueous sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum and the residue was passed through a silica gel bed to yield 83.0 g of diastereomeric mixture (25, 5i?5)-6-benzyloxy-2-(feri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio as a yellow liquid.
Part-2: Separation of diastereomers to prepare (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
A mixture of diastereomers (2S,5Z?S)-6-benzyloxy-2-(teri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio (47.0 gm, 0.125 mol), was dissolved in n-hexane (141 ml) and stirred at temperature of about 10°C to 15°C for 1 hour. Precipitated solid was filtered and washed with n-hexane (47 ml) to provide 12.0 g of diastereomerically pure (25,5i?)-6-benzyloxy-2-(tert-butyl-dimethylsilyl-oxymethyl)-7-oxo- 1,6-diaza-bicyclo-[3.2.1] octane (VIII) as a white crystalline material.
Analysis:
Mass: 377.3 (M+l); for Molecular weight: 376.58 and Molecular Formula: ![]()
1H NMR (CDCI3): δ Ί -Ί.ΑΑ (m, 5H), 4.95 (dd, 2H), 3.76-3.85 (ddd, 2H), 3.37-3.40 (m, 1H), 3.28-3.31 (m, 2H), 2.89 (brd, 1H), 1.90-2.02 (m, 2H), 1.62- 1.74 (m, 2H), 1.56 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
Diastereomeric purity as determined by HPLC: 99.85%
Step-7: Preparation of (25,5R)-6-benzyloxy-2-hvdroxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane (IX):
To a solution of (25,5i?)-6-benzyloxy-2-(ieri-butyl-dimethylsilyl-oxymethyl)-7-oxo- l,6-diaza-bicyclo-[3.2.1]octane (VIII) ( 12.0 g, 31.9 rnmol) in tetrahydrofuran (180 ml) was charged tetra 7? -butyl ammonium fluoride (38.0 ml, 38 mmol, 1 M in tetrahydrofuran) at room temperature. The reaction mixture was stirred for 2 hours. It was quenched with saturated aqueous ammonium chloride ( 100 ml). The organic layer was separated and aqueous layer extracted with dichloromethane (150 ml x 3). The combined organic layer was washed with saturated aqueous sodium chloride (150 ml), dried over sodium sulfate and evaporated under vacuum to yield 24.0 g of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l ,6-diaza-bicyclo-[3.2.1]octane (IX) as a yellow liquid. The compound of Formula (IX) was purified by silica gel (60-120 mesh) column chromatography using a mixture of ethyl acetate and hexane as an eluent.
Analysis:
Mass: 263.1 (M+l); for Molecular weight: 262.31 and Molecular Formula: C14H18N203
1H NMR (CDCb): δ 7.34-7.42 (m, 5H), 4.95 (dd, 2H), 3.67-3.73 (m, 1H), 3.53-3.60 (m, 2H), 3.32-3.34 (m, 1H), 2.88-3.01 (m, 2H), 2.09 (brs, 1H), 1.57-2.03 (m, 2H), 1.53- 1.57 (m, 1H), 1.37- 1.40 (m, 1H).
Step 8: Preparation of sodium salt of (25, 5R)-6-benzyloxy-7-oxo-l,6-diaza-bicvclor3.2.11-octane-2-carboxylic acid (I):
Step I:
Compound of Formula (IX) obtained in step 8 above was used without any further purification. To the clear solution of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane (IX) (24.0 g, 31.8 mmol) (quantities added based upon theoretical basis i.e 8.3 g ) in dichloromethane (160 ml), was added Dess-Martin reagent (24.1 g, 57.24 mmol) in portions over 15 minutes. The resulting suspension was stirred for 2 hours at 25°C. The reaction was quenched by adding a solution, prepared from saturated aqueous sodium hydrogen carbonate solution (160 ml) and 72.0 g of sodium thiosulfate. Diethyl ether (160 ml) was added to the reaction mixture and it was stirred for 5-10 minutes and filtered through celite. Biphasic layer from filtrate was separated. Organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (160 ml) followed by saturated aqueous sodium chloride solution (160 ml). Organic layer was dried over sodium sulfate and evaporated to dryness at 30°C to obtain 20.0 g of intermediate aldehyde, which was used immediately for the next reaction.
Step II:
To the crude intermediate aldehyde (20.0 g, 31.6 mmol) (quantities added based upon theoretical yield i.e. 8.2 g) obtained as above, was charged i-butyl alcohol (160 ml) and cyclohexene (10.8 ml, 110.6 mmol). The reaction mixture was cooled to temperature of about 10°C to 15°C. To this mixture was added clear solution prepared from sodium hypophosphate (14.8 g, 94.8 mmol) and sodium chlorite (5.7 g, 63.2 mmol) in water (82.0 ml) over a period of 30 minutes by maintaining temperature between 10°C to 15°C. The reaction mixture was further stirred for 1 hour and was quenched with saturated aqueous ammonium chloride solution. The reaction mixture was subjected to evaporation under vacuum at 40°C to remove i-butyl alcohol. Resulting mixture was extracted with dichloromethane (3 x 150 ml). Layers were separated. Combined organic layer was washed with aqueous brine solution, dried over sodium sulfate and evaporated to dryness under vacuum to obtain 16.0 g of crude residue. To this residue was added acetone (83 ml) to provide a clear solution and to it was added dropwise a solution of sodium 2-ethyl hexanoate (4.5 g) in acetone (24 ml). The reaction mixture was stirred for 15 hours at 25°C to 30°C to provide a suspension. To the suspension was added diethyl ether (215 ml) and stirred for 30 minutes. Resulting solid was filtered over suction, and wet cake was washed with cold acetone (83 ml) followed by diethyl ether (83 ml). The solid was dried under vacuum at 40°C to provide 3.6 g of off-white colored, non-hygroscopic sodium salt of (25, 5i?)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]-octane-2-carboxylic acid (I).
Analysis:
Mass: 275.2 as M-1 (for free acid) for Molecular Weight: 298 and Molecular Formula: ![]()
NMR (DMSO-d6): δ 7.43-7.32 (m, 5H), 4.88 (q, 2H), 3.48 (s, IH), 3.21 (d, IH), 2.73 (d, IH), 2.04-2.09 (m, IH), 1.77-1.74 (m, IH), 1.65-1.72 (m, IH), 1.55-1.59 (m, IH);
Purity as determined by HPLC: 97.47%;
[a]D25: -42.34° (c 0.5, water).


Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.

/////////
New molecules from Wochkardt to treat bacterial infections

WCK ?
( Not sure) Keep watching this post………..
TRANS-SULFURIC ACID MONO-{2-[5-(3-AZETIDINYLAMINO)-METHYL-[1,3,4]- OXADIAZOL-2-YL]-7-OXO-1,6-DIAZABICYCLO[3.2.1] OCT-6-YL} ESTER TRIFLUOROACETATE
trans-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[1,3,4]- oxadiazol-2-yl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl}ester trifluoroacetate
(25,5R)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[ l,3,4]-oxadiazol-2-yl)-7-oxo- l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester
2-(1 ,3,4-OXADIAZOL-2-YL)-7-OXO-1 ,6-DIAZABICYCLO[3.2.1 ]OCTANE DER
(25,5R)-Sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[i,3,41-oxadiazol-2-yl)-7-oxo-l,6-diaza- bicvclo[3.2.11 oct-6-yll ester
PCT International Patent Application No. PCT/US2013/034562.
Indian Patent Application No. 1635/MUM/2014
Molecular Weight: 488.3 and Molecular Formula:![]()

![]()
Scheme 1. Typically, compound of Formula (I) is prepared from sodium salt of 6-benzyloxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylic acid (III).
The sodium salt of 6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylic acid
(III) is reacted with 3-(ier^butoxycarbonyl-hydrazinocarbonylmethyl-amino)-azetidine-1-carbamic acid tert-buty\ ester (II) in presence of coupling agent at a temperature ranging from -15°C to 60°C for about 1 hour to about 24 hours to provide an intermediate compound of Formula (IV). Typical, non-limiting examples of coupling agent include EDC hydrochloride, dicyclohexylcarbodiimide, diisopropylcarbodiimide (DIC), (benzotriazol-l-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), O-(benzotriazol- 1 -yl)-N,N,N’ ,Ν’ -tetramethyluroniumhexafluorophosphate (HBTU), O-(benzotriazol-l-yl)- Ν,Ν,Ν’,Ν’-tetramethyluroniumtetrafluoroborate (TBTU), 0-(7-azabenzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU), O-(6-ahlorobenzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HCTU), 0-(3,4-dihydro-4-oxo-l,2,3-benzotriazine-3-yl)-N,N,N’,N’-tetramethyl uronium tetrafluoroborate(TDBTU), 3-(diethylphosphoryloxy)- 1 ,2,3-benzotriazin-4(3H)-one (DEPBT), carbonyldiimidazole (CDI), pivalyl chloride, HOBt and the like. In some embodiments, compound of Formula (II) is reacted with a compound of Formula (III) in presence of EDC hydrochloride and HOBt at a temperature of about 25°C to about 35°C for about 15 hours to provide an intermediate compound of Formula (IV). In some embodiments, a compound of Formula (II) is reacted with a compound of Formula (III) in presence of suitable solvent such as dimethylformamide, water or a mixture thereof.
The compound of Formula (IV) is cyclized to provide a compound of Formula (V). The cyclization of a compound of Formula (IV) is effected by treating with a reagent such as p-toluene sulfonyl chloride, p-nitrobenzene sulfonyl chloride, methane sulfonyl chloride or triphenylphosphine in a suitable solvent such as toluene, chloroform, dichloromethane, or N,N-dimethyl formamide at a temperature ranging from about -10° C to about 70°C for about 15 minutes to about 4 hours to provide 1,3,4-oxadiazole intermediate compound of Formula (V). In some embodiments, a compound of Formula
(IV) is cyclized in presence of triphenylphosphine, iodine and triethylamine, at a temperature of about -10°C to about 0°C for about 30 minutes to provide a compound of Formula (V). In some embodiments, compound of Formula (IV) is cyclized to a compound of Formula (V) in presence of dichloromethane as solvent.

Sulfonation

Scheme 1
Example 1
Synthesis of traras-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]- oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl]ester trifluoroacetate (I)
Step 1; Preparation of traras-{3-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane-2-carbonyl)-hydrazinocarbonyl]-2-oxo-ethyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (IV):
A solution of 3-(ier^butoxycarbonyl-hydrazinocarbonylmethyl-amino)-azetidine-1-carbamic acid tert-butyl ester (II) (2.8 g, 0.008 mol) in dimethylformamide (7 ml) was added to a stirred solution of sodium salt of 6-benzyloxy-7-bicyclo [3.2.1] octane-2-carboxylic acid (III) (2.43 g 0.008 mol) in water (41 ml). To this EDC.HCl (2.32 g, 0.012 mol) and HOBt (1.09 g, 0.008 mol) was added and stirred for 15 hours. Dichloro methane (50 ml) was added and layers were separated. Organic layer was dried over sodium sulfate and concentrated. The residue (6.1 gm) was purified by silica gel column chromatography using mixture of acetone and hexane as eluent to afford 3.4 g of ir ns-3-({2-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazino]-2-oxo-ethyl}-teri-butoxy carbonyl-amino)-azetidine-l -carboxylic acid tert-butyl ester (IV) in 70% yield.
Analysis:
Mass: 603.3 (M+l); for Molecular Weight: 602.6; Molecular Formula: ![]()
1H NMR (400 MHz, CDC13): δ 8.45. (bs, IH), 8.20 (bs, IH) 7.38-7.45 (m, 5H), 5.04 (d, IH), 4.91 (d, IH), 4.13 (m, 2H), 3.97-4.04 (m, 5H), 3.30 (s, IH), 3.07 (s, 2H), 2.91 (d, IH), 2.31 (m, IH), 2.20 (d, IH), 1.93-2.00 (m, 2H), 1.45 (s, 18H).
Step 2: Preparation of tr «s-{2-[5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-2-oxo-ethyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (V):
Triethyl amine (3.6 ml, 0.026 mol) was added to a cooled (0 °C) solution of iodine (1.62 gm, 0.0063 mol) and triphenylphosphine (1.67 g, 0.0063 mol) in dichloromethane (64 ml). After stirring for 15 minutes a solution of 3-({2-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazino]-2-oxo-ethyl}-fert-butoxycarbonyl- amino)-azetidine-l-carboxylic acid tert-butyl ester (IV) (3.2 g, 0.0053 mol) in dichloromethane (16 ml) was added. Reaction mixture was stirred at -10°C to 0°C for another 30 minutes. Dichloromethane was concentrated and ethyl acetate (35 ml) was added; stirred and filtered to remove triphenylphosphine oxide. Filtrate was concentrated and purified by silica gel column chromatography using a mixture of methanol and chloroform as eluent to obtain 4.5 g of 3-{ [5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4] oxadiazol-2-yl-methyl]-tert-butoxycarbonyl-amino}-azetidine- 1 -carboxylic acid tert-buty\ ester (V).
Analysis:
Mass: 585.4 (M+l); for Molecular Weight: 584.6 and Molecular Formula: ![]()
1H NMR (400 MHz, CDC13): δ 7.64-7.68 (m, 6H), 7.52-7.56 (m, 3H) 7.42-7.48 (m, 7H), 7.36-7.38 (m, 2H), 5.07 (d, IH), 4.92 (d, 2H), 4.72 (s, IH), 4.68 (s, 2H), 4.15 (s, 2H), 4.01 (s, 2H), 3.36 (s, IH), 2.91 (d, IH), 2.79 (d, IH), 2.27-2.30 (m, 2H), 2.11-2.14 (m, IH), 1.97-1.99 (m, IH), 1.42 (s, 18H).
Step 3: Preparation of tr «s-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (VI):
Palladium on carbon (10%) was added to a stirred solution of 3-{ [5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl-methyl]-feri-butoxy carbonyl-amino}-azetidine-l -carboxylic acid tert-butyl ester (V) (4.5 g) in methanol (45 ml). Resulting suspension was stirred under hydrogen gas pressure of about 50 psi for 15 hours at 25°C. The reaction mixture was filtered through celite bed and washed using additional methanol (5 ml). The filtrate was concentrated to obtain 3.5 g of ir ns-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-teri-butoxy carbonyl-amino)-azetidine-l -carboxylic acid tert-butyl ester (VI) in 92% yield.
Analysis:
Mass: 495.4 (M+l); for Molecualr Weight: 494.5 and Molecular Formula: ![]()
1H NMR (400 MHz, DMSO): δ 9.86 (s, 1H), 7.51-7.62 (m, 12H), 4.70 (s, 2H), 4.58 (d, 1H), 3.99 (d, 2H), 3.65 (s, 2H), 2.92 (d, 1H), 2.67 (d, 1H), 2.31 (s, 1H), 2.00-2.11 (m, 2H), 1.84 (m, 1H), 1.31 (s, 18H).
Step-4: Preparation of traras-tetrabutyl ammonium salt-methyl-{2-[5-(7-oxo-6-sulphooxy-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-tert-butoxycarbonyl-amino )-azetidine-l-carboxylic acid fert-butyl ester (VII):
Sulfur trioxide-pyridine complex (3.17 g, 0.019 mol) and triethyl amine (4.5 ml, 0.033 mol) was added to a stirred solution of ir ns- {2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-butyl ester (VI) (2.62 g, 0.0066 mol) in dichloromethane (20 ml). The reaction mixture was stirred for 2 hours. Aqueous solution of 0.5 N potassium dihydrogen phosphate (50 ml) followed by ethyl acetate (40 ml) was added, stirred for 10 minutes and aqueous layer was separated. Aqueous layer was again extracted with the mixture of dichloromethane (10 ml) and ethyl acetate (20 ml). Combined organic layers were concentrated. The residue was dissolved in water (50 ml), washed with diethyl ether (2 x 25 ml) to remove triphenylphosphine oxide (a side product carried from the step-2) and extracted with dichloromethane (2 x25 ml). Dichloromethane was dried over sodium sulfate and concentrated to give 2.7 g of residue (87%). This residue was again dissolved in dichloromethane (50 ml) followed by addition of triethylamine (5.70 ml, 0.042 mol). Tetrabutylammonium hydrogen sulphate (1.27 g, 0.0037 mol) was added and stirred for 2 hours. Water (30 ml) was added to the reaction mixture and layers were separated. Dichloromethane layer was dried on sodium sulfate and solvent was concentrated under vacuum. The residue (2.7 g) was purified by silica gel column chromatography using methanol and chloroform as eluent to get 2.1 g of irans-tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy- 1 ,6-diaza-
bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-buty\ ester (VII) in 48% yield.
Analysis:
Mass: 575.4 (M+l) as free sulfonic acid; for Molecular Weight: 816.6 and Molecular Formula: C22H34N6O10S. Ci6H36N;
1H NMR (400 MHz, CDC13): δ 4.63-4.69 (m, 5H), 4.40 (s, 2H), 4.16 (s, 2H), 4.02 (s, 2H), 3.28-3.32 (m, 12H), 3.23 (s, 1H), 2.84 (d, 1H), 2.24-2.32 (m, 2H), 2.02-2.04 (m, 1H), 1.63-1.71 (m, 12H), 1.46-1.56 (m, 12H), 1.44 (s, 18H), 0.99-1.02 (m, 18H).
Step 5: Preparation of traras-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl]ester trifluoroacetate (I)
irans-Tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy- 1 ,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-butyl ester (VII) (2.1 g, 0.003 mol) was cooled to 0°C and to this was added trifluoro acetic acid cooled at 0°C in 15 minutes and the reaction mixture was stirred for 3 hours. The obtained reaction mixture was concentrated under high vacuum. Diethyl ether (20 ml) was added and solid precipitated was stirred and diethyl ether was decanted. This treatment was repeated twice. Solid separated was dried and dichloromethane (20 ml) was added and stirred; solid was allowed to settle and dichloromethane was decanted. Again this treatment was repeated twice and the solid was dried to get 1 g of irans-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo [3.2.1]oct-6-yl]ester trifluoroacetate (I) in 76% yield.
Analysis:
Mass: 375.2 (M+l) as free sulfonic acid; for Molecular Weight: 488.3 and Molecular Formula:![]()
CF3COOH;
1H NMR (400 MHz, DMSO): δ 4.64 (d, IH), 4.06 (s, 3H), 3.92 (s, 2H), 3.81-3.86 (m, IH), 3.73 (s, 2H), 2.94-2.97 (d, IH), 2.70 (d, IH), 2.16 -2.19 (m, IH), 1.88-2.14 (m, 2H), 1.86-1.88 (m, IH);
19F NMR (DMSO-d6): δ -74.41 (CF3COOH);
1 C NMR (DMSO-de as a TFA salt): δ 165.4, 165.1, 164.9, 159.2-158.2 (TFA-C), 57.7, 52.6 (2C), 52.3, 49.3, 46.1, 40.4, 20.1, 19.7.
PATENT
WO2015110963


Example-1
(25,5R)-Sulfuric acid mono-r2-(5-azetidin-3-ylmethyl-ri,3,41-oxadiazol-2-yl)-7-oxo-l,6-diaza- bicvclor3.2.11 oct-6-yll ester:

Step-1: Preparation of (25,5R)-2-{N’-[2-(5)-iV-tert-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazino carbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of sodium (2S, 5R)-7-oxo-6-benzyloxy-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (8.45 g, 28.3 mmol) (prepared according to the process disclosed in PCT/IB2013/059264) in water (100 ml) was added 3-(N-feri-butoxycarbonyl-azetidin-3-yl)-acetic acid hydrazide (5.9 g, 25.7 mmol), EDC hydrochloride (7.47 g, 38.6 mmol) and N-hydroxybenzotriazole (3.47 g, 25.7 mmol) at 25°C to 35°C under stirring. The reaction mixture was stirred for 18 hours. Precipitated solid was filtered under suction and washed with water (100 ml). It was dried to provide 10.01 g of (25,5R)-2-{N’-[2-(S)-N-fert-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazinocarbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane in 80% yield.
Analysis:
Mass: 486.4 (M-l), for Molecular Formula of C24H33N5O6;
Purity as determined by HPLC: 89.90%.
Step-2: Preparation of (25,5R)-2-(5-(/V-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of (25,5i?)-2-{N’-[2-(5)-N-ieri-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazinocarbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane (4 gm, 8.21 mmol) in chloroform (70 ml) was added p-toluenesulfonylchloride (2.34 gm, 12.3 mmol) followed by dnsopropylethylamine (4.4 ml, 24.6 mmol). The reaction mixture was heated under stirring at 75°C for 18 hours. The reaction mixture was concentrated under vacuum and the resulting mass was purified by using silica gel column chromatography, to provide (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane in 3.3 g quantity in 86% yield as a solid.
Analysis:
Mass: 470.4 (M+l), for Molecular Formula of C^HsiNsOs;
1H NMR: (CDCb): δ 7.36-7.44 (m, 5H), 5.08 (d, 1H), 4.93 (d, 1H), 4.68-4.71 (m, 1H), 4.10-4.15 (m, 2H), 3.68-3-72 (m, 2H), 3.37 (s, 1H), 3.13-3.15 (m, 2H), 2.90-3.11 (m, 2H), 2.77 (d, 1H), 2.25-2.31 (m, 2H), 2.10-2.19 (m, 1H), 1.87- 1.97 (m, 1H), 1.43 (s, 9H).
Step-3: Preparation of (25,5R)-2-(5-(iV-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To the solution of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane ( 3.3 g, 7.0 rnmol) in methanol (35 ml) was subjected to catalytic hydrogenolysis using 10% palladium on charcoal (350 mg) under atmospheric hydrogen gas pressure at 25°C to 35°C for 2 hours. The reaction mixture was filtered through celite bed and was washed with methanol (30 ml). The filtrate was concentrated under vacuum below 35°C to provide 2.7 g of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-hydroxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane, which was used immediately for the next reaction.
Analysis:
Mass: 378.4 (M-l), for Molecular Formula of CnH^NsOs.
Step-4: Preparation of tetrabutylammonium salt of (2S,5R)-2-(5-(V-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)- 6- hydroxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane (2.7 gm, 7.12 mmol) in dichloromethane (50 ml) was added triethylamine (5 ml, 35 mmol) followed by sulfur trioxide pyridine complex (2.26 g 14.2 mmol) under stirring at 25°C to 35°C. The reaction mixture was stirred for 2 hours. To the reaction mixture was added aqueous 0.5 N potassium dihydrogen phosphate solution (100 ml). It was stirred for about 30 minutes and tetrabutyl ammonium hydrogen sulfate (2.17 gm 6.4 mmol) was added. It was stirred for 2 hours. Layers were separated and organic layer was concentrated under vacuum to provide a crude mass, which was purified by silica gel column chromatography to furnish 2.1 g of tetrabutylammonium salt of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ 1 ,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo- 1 ,6-diaza-bicyclo[3.2.1] octane as solid in 43% yield.
Analysis:
Mass: 458.3 (M- l), as a free sulfonic acid, for Molecular Formula of C17H25N5O8S. N(C4H9)4; Purity as determined by HPLC: 94.87%.
Step-5: Preparation of (25,5R)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[l,3,4]-oxadiazol-2-yl)- 7- oxo-l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester:
To the solution of tetrabutylammonium salt of (25,5i?)-2-(5-(N-feri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane (1.0 g, 2.2 mmol) in dichloromethane (5 ml) was charged trifluoroacetic acid (5 ml) with syringe at – 10°C under stirring. The reaction mixture was stirred for 1 hour. The mixture was evaporated under vacuum by maintaining temperature below 35 °C, to provide a residue, which was suspended in diethyl ether (25 ml) twice. The suspension was filtered and the solid was suspended further in dichloromethane (50 ml) and stirred for 30 minutes. The suspension was filtered and dried to afford the 310 mg of (25,5i?)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[ l,3,4]-oxadiazol-2-yl)-7-oxo- l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester as a solid in 60% yield.
Analysis:
Mass: 358.2 (M-l), for Molecular Formula of C^HnNsOeS;
1H NMR (DMSO-d6): δ 8.50 (br s, IH), 8.62 br s, IH), 4.60 (d, IH), 4.05 (s, 3H), 3.82-3.84 (m, IH), 3.21-3.27 (m, 4H), 2.93-2.96 (m, IH), 2.75 (d, IH), 2.12-2.17 (m, IH), 1.96-2.05 (m, 2H), 1.82-1.88 (m, IH).
Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.
///////
Wockhardt, WO 2007023507, N-[[3-[3,5-difluoro-4-[4-(tetrazol-2-yl)piperidin-1-yl]phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide


Cas 928156-95-0,
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-(2H-tetrazol-2-yl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
| C18H21F2N7O3 | |
| Molecular Weight: | 421.401246 g/mol |
|---|
N-[[3-[3,5-difluoro-4-[4-(tetrazol-2-yl)piperidin-1-yl]phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide
Example- 14 and 15
(S)-N- { 3- [4-(4-(2H-tetrazol-2-yl)-piperidin- 1 -yl)-3 , 5-difluorophenyl] -2-oxo-oxazolidin-
5-ylmethyl }-acetamide and
(S)-N- { 3- [4-(4-(l H-tetrazol- 1 -yl)-piperidin- 1 -yl)-3 , 5-difluorophenyl] -2-oxo-oxazolidin-
5-ylmethyl }-acetamide

and

A mixture of (S)-N-{3-[4-methanesulphonyloxy piperidin-l-yl)-3,5-difluorophenyl]-2- oxo-oxazolidin-5-ylmethyl}-acetamide (1.12 mM), tetrazole (1.68 mM), and K2CO3 (1.68 mM) in DMF (6 ml) was heated for 22 hrs at 850C. The resulting mixture was poured into ice-water mixture, stirred for 30 min. And the separated solid was purified by column chromatography to obtain two isomeric products in 18% and 12% yields respectively. Isomer A: M.P. 234-2370C; MS(M+1)- 422 ; M.F. C18H21F2N7O3 Isomer B: M.P. 214-2170C; MS(M+1)- 422 ; M.F. C18H2JF2N7O3
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
Our New Drug Discovery team has developed a number of lead molecules, mainly in the area of anti-infectives; these are currently at various stages of development.
Of these molecules, the most advanced of the New Chemical Entities (NCE) is WCK 771, which has commenced Phase II human clinical trials.
WCK 771 is a broad-spectrum antibiotic, which has proven effective in treating diverse staphylococcal infections like MRSA and VISA.
Other lead molecules at various stages of pre-clinical trials are: WCK 2349, WCK 4873 and WCK 4086.
http://www.wockhardt.com/how-we-touch-lives/new-drug-discover.aspx


///////
