
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


DABRAFENIB
ダブラフェニブ
达拉菲尼,
1195765-45-7 BASE
1195768-06-9 cas of mesylate
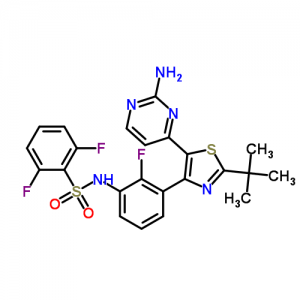
Benzenesulfonamide, N-[3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-4-thiazolyl]-2-fluorophenyl]-2,6-difluoro-
MW 519.56 BASE
MF C23 H20 F3 N5 O2 S2 BASE
- Dabarefenib
- Dabrafenib
- GSK 2118436
- Tafinlar
- UNII-QGP4HA4G1B
US FDA APPROVAL….Date of Approval: May 29, 2013
update
| Product details | |
|---|---|
| Name |
Tafinlar
|
| Agency product number |
EMEA/H/C/002604
|
| Active substance |
dabrafenib mesilate
|
| International non-proprietary name (INN) or common name |
dabrafenib
|
| Therapeutic area (MeSH) |
Melanoma
|
| Anatomical therapeutic chemical (ATC) code |
L01EC02
|
| Publication details | |
|---|---|
| Marketing-authorisation holder |
Novartis Europharm Limited
|
| Date of issue of marketing authorisation valid throughout the European Union |
26/08/2013
|
An orally bioavailable inhibitor of B-raf (BRAF) protein with potential antineoplastic activity. Dabrafenib selectively binds to and inhibits the activity of B-raf, which may inhibit the proliferation of tumor cells which contain a mutated BRAF gene. B-raf belongs to the the raf/mil family of serine/threonine protein kinases and plays a role in regulating the MAP kinase/ERKs signaling pathway, which may be constitutively activated due to BRAF gene mutations
Dabrafenib (trade name Tafinlar) is a drug for the treatment of cancers associated with a mutated version of the gene BRAF. Dabrafenib acts as an inhibitor of the associated enzyme B-Raf, which plays a role in the regulation of cell growth. Dabrafenib has clinical activity with a manageable safety profile in clinical trials of phase 1 and 2 in patients with BRAF(V600)-mutated metastatic melanoma.[1][2]
The Food and Drug Administration approved dabrafenib as a single agent treatment for patients with BRAF V600E mutation-positive advanced melanoma on May 30, 2013.[3] Clinical trial data demonstrated that resistance to dabrafinib and other BRAF inhibitors occurs within 6 to 7 months.[4] To overcome this resistance, the BRAF inhibitor dabrafenib was combined with the MEK inhibitor trametinib.[4] As a result of this research, on January 8, 2014, the FDA approved the combination of dabrafenib and trametinib for the treatment of patients with BRAF V600E/K-mutant metastatic melanoma.[5]

Inhibitor of BRAF(V600) mutants
| Active Ingredient: | DABRAFENIB MESYLATE |
| Dosage Form;Route: | CAPSULE;ORAL |
| Proprietary Name: | TAFINLAR |
| Applicant: | GLAXOSMITHKLINE |
| Strength: | EQ 75MG BASE |
| NDA Application Number: | N202806 |
| Product Number: | 002 |
| Approval Date: | May 29, 2013 |
| Reference Listed Drug | Yes |
| RX/OTC/DISCN: | RX |
Patent Data
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
Delist Requested |
|---|---|---|---|---|---|---|---|
| N202806 | 002 | 7994185 | Jan 20, 2030 | Y | Y | U – 1406 | |
| N202806 | 002 | 8415345 | Jan 20, 2030 | Y | Y | U – 1406 |
Exclusivity Data
| NDA Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N202806 | 002 | I – 678 | Jan 8, 2017 |
| N202806 | 002 | ODE | Jan 9, 2021 |
| N202806 | 002 | NCE | May 29, 2018 |
| N202806 | 002 | ODE | May 29, 2020 |
PDF……http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202806s000lbl.pdf
TERMS
I 678, TRAMETINIB, IN COMBINATION WITH DABRAFENIB, FOR THE TREATMENT OF PATIENTS WITH UNRESECTABLE OR METASTATIC MELANOMA WITH BRAF V600E OR V600K MUTATIONS AS DETECTED BY AN FDA-APPROVED TEST
ODE ORPHAN DRUG EXCLUSIVITY
NCE NEW CHEMICAL ENTITY

DABRAFENIB SYNTHESIS
WILL BE UPDATED
—
http://www.google.com/patents/WO2011047238A1?cl=en
Method 1 : Compound B (first crystal form) – A/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1 dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide

A suspension of A/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]- 2-fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) was heated in a sealed tube to 90 °C for 24 h. The reaction was diluted with DCM and added silica gel and concentrated. The crude product was chromatographed on silica gel eluting with 100% DCM to 1 :1 [DCM:(9:1 EtOAc:MeOH)]. The clean fractions were concentrated to yield the crude product. The crude product was repurified by reverse phase HPLC (a gradient of acetonitrile:water with 0.1 %TFA in both). The combined clean fractions were concentrated then partitioned between DCM and saturated NaHCO3. The DCM layer was separated and dried over Na2SO4. The title compound, /V-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1 H NMR (400 MHz, DMSO-c/6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 – 7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1 .35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Method 2: Compound B (alternative crystal form) – A/-{3-[5-(2-Amino-4-pyrimidinyl)-2- (1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide 19.6 mg of A/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 L of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-40°C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above. Method 3: Compound B (alternative crystal form, large batch) – A/-{3-[5-(2-amino-4- pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

tep A: methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate

Methyl 3-amino-2-fluorobenzoate (50 g, 1 eq) was charged to reactor followed by dichloromethane (250 mL, 5 vol). The contents were stirred and cooled to ~15°C and pyridine (26.2 mL, 1 .1 eq) was added. After addition of the pyridine, the reactor contents were adjusted to ~15°C and the addition of 2,6-diflurorobenzenesulfonyl chloride (39.7 mL, 1 .0 eq) was started via addition funnel. The temperature during addition was kept <25°C. After complete addition, the reactor contents were warmed to 20-25°C and held overnight. Ethyl acetate (150 mL) was added and dichloromethane was removed by distillation. Once distillation was complete, the reaction mixture was then diluted once more with ethyl acetate (5 vol) and concentrated. The reaction mixture was diluted with ethyl acetate (10 vol) and water (4 vol) and the contents heated to 50-55°C with stirring until all solids dissolve. The layers were settled and separated. The organic layer was diluted with water (4 vol) and the contents heated to 50-55° for 20-30 min. The layers were settled and then separated and the ethyl acetate layer was evaporated under reduced pressure to ~3 volumes. Ethyl Acetate (5 vol.) was added and again evaporated under reduced pressure to ~3 volumes.
Cyclohexane (9 vol) was then added to the reactor and the contents were heated to reflux for 30 min then cooled to 0 °C. The solids were filtered and rinsed with cyclohexane (2 x 100 mL). The solids were air dried overnight to obtain methyl 3-{[(2,6- difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (94.1 g, 91 %).
Step B: A/-{3-[(2-chloro-4-pyhmidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (490 g, 1 equiv.), prepared generally in accordance with Step A, above, was dissolved in THF (2.45 L, 5 vols) and stirred and cooled to 0-3 °C. 1 M lithium bis(trimethylsilyl)amide in THF (5.25 L, 3.7 equiv.) solution was charged to the reaction mixture followed addition of 2- chloro-4-methylpyrimidine (238 g, 1 .3 equiv.) in THF (2.45 L, 5 vols). The reaction was then stirred for 1 hr. The reaction was quenched with 4.5M HCI (3.92 L, 8 vols). The aqueous layer (bootom layer) was removed and discarded. The organic layer was concentrated under reduced pressure to ~2L. IPAC (isopropyl acetate) (2.45L) was added to the reaction mixture which was then concentrated to ~2L. IPAC (0.5L) and MTBE (2.45 L) was added and stirred overnight under N2. The solids were filtered. The solids and mother filtrate added back together and stirred for several hours. The solids were filtered and washed with MTBE (~5 vol). The solids were placed in vacuum oven at 50 °C overnight. The solids were dried in vacuum oven at 30 °C over weekend to obtain A/-{3-[(2-chloro-4-pyhmidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (479 g, 72%).
Step C: A/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged /V-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 mL). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated. The dichloromethane layers were concentrated to -120 mL. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 mL. Dimethylacetamide (270 mL) was then added to the reaction mixture and cooled to ~10°C. 2,2- Dimethylpropanethioamide (1 .3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for ~5 minutes between additions. The reaction was warmed to 20-25 °C. After 45 min, the vessel contents were heated to 75°C and held for 1 .75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 30°C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated. Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-25°C. The organic layers were then concentrated under heat and vacuum to 120 mL. The vessel contents were then heated to 50°C and heptanes (120 mL) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for ~2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 30°C to obtain /V-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide (28.8 g, 80%).
Step D: A/-{3-[5-(2-amino-4-pyhmidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide
In 1 gal pressure reactor, a mixture of A/-{3-[5-(2-chloro-4-pyrinnidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 °C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 °C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 °C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HCI to ~pH 5.4-5.5. and added water (1 vol). The aqueous layer was removed and discarded at 60-70 °C. The organic layer was washed with water (1 vol) at 60-70 °C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 °C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 °C for 10 min , then cooled to 20°C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes. The reaction mixture was maintained at ~65-70°C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 °C. The slurry was stirred at 65-70 °C for 2- 3 hours and then cooled slowly to 0-5°C. The product was filtered, washed with
EtOAc/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain A/-{3-[5-(2- amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (102.3 g, 88%).
Method 4: Compound B (mesylate salt) – A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate

To a solution of /V-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide (204 mg, 0.393 mmol) in isopropanol (2 ml_), methanesulfonic acid (0.131 ml_, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield). 1 H NMR (400 MHz, DMSO-c/6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1 .36 (s, 9 H). MS (ESI): 520.0 [M+H]+.
Method 5: Compound B (alternative mesylate salt embodiment) – A/-{3-[5-(2-amino-4- pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide methanesulfonate
A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (as may be prepared according to example 58a) (2.37g, 4.56 mmol) was combined with pre-filtered acetonitrile (5.25 vol, 12.4 ml_). A pre-filtered solution of mesic acid (1 .1 eq., 5.02 mmol, 0.48 g) in H2O (0.75 eq., 1 .78 ml_) was added at 20°C. The temperature of the resulting mixture was raised to 50- 60°C while maintaining a low agitation speed. Once the mixture temperature reached to 50-60°C, a seed slurry of A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate (1 .0 %w/w slurried in 0.2 vol of pre-filtered acetonitrile) was added, and the mixture was aged while agitating at a speed fast enough to keep solids from settling at 50-60°C for 2 hr. The mixture was then cooled to 0-5°C at 0.25°C/min and held at 0-5°C for at 6 hr. The mixture was filtered and the wet cake was washed twice with pre-filtered
acetonitrile. The first wash consisted of 14.2 ml (6 vol) pre-filtered acetonitrile and the second wash consisted of 9.5 ml (4 vol) pre-filtered acetonitrile. The wet solid was dried at 50°C under vacuum, yielding 2.39 g (85.1 % yield) of product. Typically, the salts of the present invention are pharmaceutically acceptable salts.
May 29, 2013 — GlaxoSmithKline plc announced today that the U.S. Food and Drug Administration (FDA) has approved Tafinlar (dabrafenib). Tafinlar is indicated as a single-agent oral treatment for unresectable melanoma (melanoma that cannot be removed by surgery) or metastatic melanoma (melanoma which has spread to other parts of the body) in adult patients with BRAF V600E mutation. Tafinlar is not indicated for the treatment of patients with wild-type BRAF melanoma. The mutation must be detected by an FDA-approved test, such as the companion diagnostic assay from bioMérieux S.A., THxID™-BRAF.
Among those with metastatic melanoma, approximately half have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread
Tafinlar is approved for patients with the BRAF V600E mutation, which accounts for approximately 85 percent of all BRAF V600 mutations in metastatic melanoma.
GSK will be making Tafinlar available for prescription no later than in the early third quarter of 2013.
In 2010, GSK entered a collaboration with bioMérieux to develop a companion diagnostic test to detect BRAF V600 (V600E and V600K) gene mutations found in several cancers, including melanoma. bioMérieux has received FDA pre-market approval of THxID™-BRAF. Currently, it is the only FDA-approved test that detects the V600K mutation.
The primary outcome measure was the estimation of the overall intracranial response rate (OIRR) in each cohort. The OIRR for Cohort A was 18 percent (95% CI: 9.7, 28.2). For Cohort B, the OIRR was also 18 percent (95% CI: 9.9, 30.0). The median duration of response was 4.6 months (95% CI: 2.8, Not Reached) and 4.6 months (95% CI: 1.9, 4.6) in Cohort A and Cohort B, respectively.
Melanoma is the most serious and deadly form of skin cancer. According to statistics from the National Cancer Institute, in 2013 there will be an estimated 9,480 deaths resulting from melanoma in the United States. When melanoma spreads in the body, the disease is called metastatic melanoma.Approximately half of all people with metastatic melanoma have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread.
One in two patients worldwide with metastatic melanoma is expected to survive for a year after diagnosis, while in the U.S., the five-year survival rate was 16 percent (2003-2009).The median age of a newly diagnosed metastatic melanoma patient is almost a decade younger than other cancers.
Tafinlar (dabrafenib) is now approved for the treatment of adult patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. Limitation of use: Tafinlar is not recommended for use in patients with wild-type BRAF melanoma.
Tafinlar is not approved or licensed in Europe and may not be approved in other parts of the world for the treatment of patients with BRAF V600 mutation-positive unresectable melanoma or metastatic melanoma.
Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2•CH4O3S and a molecular weight of 615.68. Dabrafenib mesylate has the following chemical structure:
 |
Dabrafenib mesylate is a white to slightly colored solid with three pKas: 6.6, 2.2, and -1.5. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
TAFINLAR (dabrafenib) capsules are supplied as 50-mg and 75-mg capsules for oral administration. Each 50-mg capsule contains 59.25 mg dabrafenib mesylate equivalent to 50 mg of dabrafenib free base. Each 75-mg capsule contains 88.88 mg dabrafenib mesylate equivalent to 75 mg of dabrafenib free base.
The inactive ingredients of TAFINLAR are colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose. Capsule shells contain hypromellose, red iron oxide (E172), and titanium dioxide (E171).
Dabrafenib mesylate
1195768-06-9 cas of mesylate
N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide;methanesulfonic acid
Chemical structure

………………….
WO2015003571
达拉菲尼甲磺酸盐的新晶型及其制备方法
Deficiencies of the prior art W, the main object of the present invention is to provide as follows has a better stability in an aqueous or aqueous systems Dara Feeney 曱

For purposes of this invention, the present invention provides Dallas Phoenix mesylate Form IV (hereinafter referred to as “Form IV”) and its preparation method. The type IV crystal is a hydrate; preferably each 摩尔达拉菲 Nepal mesylate contains about 1.5 moles of water.
Using Cu- Κα radiation, the crystalline form IV of X-ray powder diffraction pattern at diffraction angles 2Θ of 4.7 ± 0.2 °, 9.2 ± 0.2. , 12.8 ± 0.2. , 13.8 ± 0.2. 15.0 0.2 soil. And 16.3 ± 0.2 ° of the characteristic peaks.
Preferably, the crystalline form IV of the X-ray powder diffraction pattern at diffraction angles 2Θ of 4.7 ± 0.2. , 9.2 ± 0.2. , 12.8 ± 0 · 2. , 13 · 8 ± 0 · 2 o , 15.0 ± 0.2. , 16.3 ± 0 · 2. , 18.0 ± 0.2. , 18 · 6 ± 0.2. , 20 · 6 ± 0.2. , 22.9 ± 0.2 °, 23.8 ± 0.2. And 24.3 ± 0.2. Department characteristic peaks
Form IV of FIG differential scanning calorimetry (DSC) show: sample 151~ 105 ° C there is a large endothermic peak (solvent peak), the sample after dehydration melting range of 132 ~ 148 ° C, then at 200 ° C ~ 245 ° C with a heat transfer crystal peak at 249 ° C and finally melted.
The crystalline form IV has the following advantageous properties:
1) left at room temperature for one month and stable, stable for 1 month at room temperature for -97% RH;
2) known Form I in water suspension was stirred for 15 minutes into the free base monohydrate; and Form IV in water suspension was stirred for 15 minutes remain for 曱 salt Form IV, After stirring overnight converted to the free base monohydrate Form IV described more conducive to maintaining the solubility of the sample is larger than the free base state 曱 sulfonates, Form IV has a better stability in water / aqueous system or sex.
3) 0 to 22 hours compared to the elution amount, any detection points of Form IV of elution volume than the known polymorph I of the elution amount. Description Form IV has a better solubility and bioavailability.
4) 0 to 120 minutes elution amount compared to any detection points of Form IV gum Nang elution volume than the known polymorph I of the dissolution of glue Nang. Description Form IV gum Nang has better dissolution.
The Form IV was prepared using any one of the following methods:
1) The Dallas Feeney known mesylate polymorph I was dissolved in a mixed solution of tetrahydrofuran Yue alcohol, volatile crystallization, and then the precipitated crystals were separated and dried to obtain the Form IV;
The Yue alcohol and tetrahydrofuran in a volume ratio of 0.1 to 100: 1, preferably 0.5~50: 1, more preferably 0.5~5: 1;
2) The Dallas Phoenix Yue sulfonates known polymorph I was dissolved in acetone, volatile crystallization, and the precipitated crystals
Separated, dried, to give the Form IV;
3) The Dallas Phoenix 曱 known polymorph I salt is dissolved in isopropanol, after the addition of polyacrylic acid, volatile crystallization, and then the precipitated crystals were separated and dried to obtain the Form IV;
The polyacrylic acid in an amount of polymorph I of the known amount of 0.1% wt~10% wt, preferably
0.5% wt ~ 10% wt, more preferably 2% wt ~ 5% wt; an average molecular weight of the polyacrylic acid is 2000-5000.
Preparation of the above three methods, the known Dara Feeney 曱 sulfonate polymorph I at room temperature in an amount corresponding to its solubility in a solution of 0.1 to 1 times, preferably 0.5 to 1 times, more preferably 0.8 to 1 times;
The crystallization temperature of room temperature ~ 40 ° C, preferably at room temperature; the crystallization time is 1~14 days, preferably for two days; the dry, you can not vacuum or pressure, the pressure is preferably less than 0.09Mpa; temperature of 30 ° C ~ 120 ° C, preferably 4 (TC ~ 80 ° C, more preferably 40 ° C ~ 60 ° C; for 10 to 72 hours, preferably 10~48 hours, more preferably from 10- 24 hours;
4) The Dallas Phoenix Yue sulfonate polymorph Form II or V is placed to give the Form IV;
The placement of room temperature ~ 40 ° C, preferably room temperature; placement time from 15 minutes to 7 days, preferably
One day;
5) The temperature rise Dara Feeney 曱 sulfonate polymorph II to 120 ° C and then spontaneously cooled to room temperature to obtain the crystalline form
IV;
The preparation of Form I of Preparation Example 1 known
Methods Patent Document WO2009 / 137391 or CN200980126781.6 Example 58a and 58d known polymorph I. Preparation Specifically:
The N- {3- [5- (2- chloro-4-pyrimidinyl) -2- (1,1-Yue-yl-ethyl) -1,3-thiazol-4-yl] – 2-fluorophenyl 2,6-difluorophenyl sulfonamide (196 mg, 0.364mmol) and 7M ammonia in methanol (8ml, 56mmol) was added to a 25 ml autoclave, heated to 90 ° C for 24 hours, TLC showed the starting material the reaction was complete, The reaction system was cooled to room temperature, the solvent was concentrated and the residue was dry column chromatography to obtain N- {3- [5- (2- amino-4-pyrimidinyl) -2- (1,1-dimethylethyl ) -1,3-thiazol-4-yl] -2-fluorophenyl} -2,6-difluorobenzenesulfonamide 90 mg, yield: 45%.
The N- {3- [5- (2- amino-4-pyrimidinyl) -2- (1,1-Yue-yl-ethyl) -1,3-thiazol-4-yl] -2-fluorophenyl 2,6-difluorophenyl sulfonamide (204 mg, 0.393mmol) in isopropanol (2 mL) was added 曱 acid (0.131 ml, 0.393mmol) and the solution was stirred at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and washed with diethyl ether to give N- [3- [5- (2- amino-4-pyrimidinyl) -2- (t-butyl) -4-thiazol-yl] -2-fluoro phenyl] -2,6-difluorobenzenesulfonamide 曱 sulphonates crystalline solid (221 mg, 87% yield) as a white.
1HNM (400MHz, DMSO-d6)5 ppm 10.85(s, lH)7.92-8.05(m, 1H), 7.56-7.72(m, 1H), 6.91-7.50(m, 7H), 5.83-5.98(m, 1H) , 2.18-2.32(m, 3H) , 1.36(s, 9H)。
Preparation of crystal form obtained X-ray powder diffraction pattern shown in Figure 10. Report is consistent with the patent document WO2009 / 137391 or CN200980126781.6.
DSC chart is shown in Fig. Show: Known polymorphs I melt away as 247 ° C~250 ° C.
TGA spectrum shown in Figure 12. Show: decomposition temperature of 261 ° C.
Example 1
Take 10.02 mg polymorph IV (Example 7 Preparation) in 5 ml glass vial, add 0.5 ml of water, ultrasonic resulting suspension stirred at room temperature for 15 minutes, after centrifugation without drying, the present invention is to obtain crystalline form II. The yield was 10.00 mg; 99% yield.
X-ray powder diffraction pattern shown in Figure 6.
TGA pattern shown in Figure 7. Show: Form II at 50 ° C before the weight loss of about 4.6% (about 1.5 water), 50 ° C ~ 155 ° C 1.4% weight loss (about 0.5 water), the decomposition temperature of 287 ° C.
………………….
PATENT
http://www.google.com/patents/WO2009137391A2?cl=en
WO 2009137391
Example 58a: Λ/-{3-r5-(2-Amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Following a procedure analogous to the procedure described in Example 51, Step B using Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) and heating to 90 0C for 24 h, the title compound, Λ/-{3- [5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 –
7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1.35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Example 58b: Λ/-{3-r5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
19.6 mg of Λ/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 μl_ of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-400C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above. Example 58c: Λ/-{3-r5-(2-amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

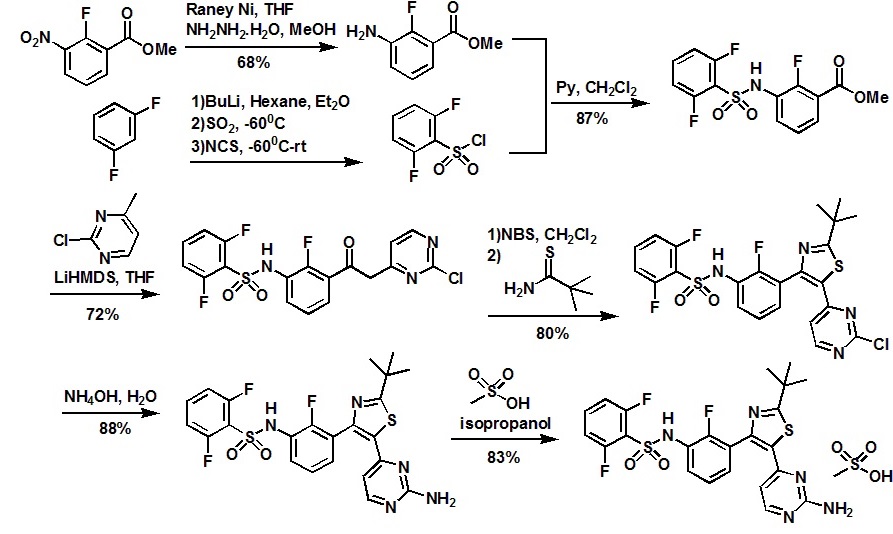
Step A: methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate

Methyl 3-amino-2-fluorobenzoate (50 g, 1 eq) was charged to reactor followed by dichloromethane (250 ml_, 5 vol). The contents were stirred and cooled to ~15°C and pyridine (26.2 ml_, 1.1 eq) was added. After addition of the pyridine, the reactor contents were adjusted to ~15°C and the addition of 2,6-diflurorobenzenesulfonyl chloride (39.7 ml_, 1.0 eq) was started via addition funnel. The temperature during addition was kept <25°C. After complete addition, the reactor contents were warmed to 20-250C and held overnight. Ethyl acetate (150 ml.) was added and dichloromethane was removed by distillation. Once distillation was complete, the reaction mixture was then diluted once more with ethyl acetate (5 vol) and concentrated. The reaction mixture was diluted with ethyl acetate (10 vol) and water (4 vol) and the contents heated to 50- 55°C with stirring until all solids dissolve. The layers were settled and separated.
The organic layer was diluted with water (4 vol) and the contents heated to 50-55° for 20-30 min. The layers were settled and then separated and the ethyl acetate layer was evaporated under reduced pressure to ~3 volumes. Ethyl Acetate (5 vol.) was added and again evaporated under reduced pressure to ~3 volumes. Cyclohexane (9 vol) was then added to the reactor and the contents were heated to reflux for 30 min then cooled to 0 0C. The solids were filtered and rinsed with cyclohexane (2 x 100 ml_). The solids were air dried overnight to obtain methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2- fluorobenzoate (94.1 g, 91 %).
Step B: Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (490 g, 1 equiv.), prepared generally in accordance with Step A, above, was dissolved in THF (2.45 L, 5 vols) and stirred and cooled to 0-3 0C. 1 M lithium bis(trimethylsilyl)amide in THF (5.25 L, 3.7 equiv.) solution was charged to the reaction mixture followed addition of 2-chloro-4- methylpyrimidine (238 g, 1.3 equiv.) in THF (2.45 L, 5 vols). The reaction was then stirred for 1 hr. The reaction was quenched with 4.5M HCI (3.92 L, 8 vols). The aqueous layer (bootom layer) was removed and discarded.
The organic layer was concentrated under reduced pressure to ~2L. IPAC (isopropyl acetate) (2.45L) was added to the reaction mixture which was then concentrated to ~2L. IPAC (0.5L) and MTBE (2.45 L) was added and stirred overnight under N2. The solids were filtered. The solids and mother filtrate added back together and stirred for several hours. The solids were filtered and washed with MTBE (~5 vol). The solids were placed in vacuum oven at 50 0C overnight. The solids were dried in vacuum oven at 30 0C over weekend to obtain Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (479 g, 72%).
Step C: Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 ml_). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated.
The dichloromethane layers were concentrated to -120 ml_. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 ml_. Dimethylacetamide (270 ml.) was then added to the reaction mixture and cooled to -1O0C. 2,2-Dimethylpropanethioamide (1.3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for -5 minutes between additions. The reaction was warmed to 20-25 0C. After 45 min, the vessel contents were heated to 75°C and held for 1.75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 300C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated.
Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-250C. The organic layers were then concentrated under heat and vacuum to 120 ml_. The vessel contents were then heated to 500C and heptanes (120 ml.) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for -2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 300C to obtain Λ/-{3-[5-(2-chloro-4-pyrimidinyl)- 2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (28.8 g, 80%).
Step D:
Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
In 1 gal pressure reactor, a mixture of Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1- dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 0C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 0C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 0C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HCI to ~pH 5.4-5.5. and added water (1vol). The aqueous layer was removed and discarded at 60-70 0C.
The organic layer was washed with water (1 vol) at 60-70 0C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 0C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 0C for 10 min , then cooled to 2O0C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes.
The reaction mixture was maintained at -65-7O0C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 0C. The slurry was stirred at 65-70 0C for 2-3 hours and then cooled slowly to 0-50C. The product was filtered, washed with EtOAc/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain Λ/-{3-[5-(2- amino-4-pyrimidinyl)-2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (102.3 g, 88%).
Example 58d:
Λ/-{3-r5-(2-amino-4-pyrimidinvn-2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate
 MESYLATE
MESYLATETo a solution of Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (204 mg, 0.393 mmol) in isopropanol (2 ml_), methanesulfonic acid (0.131 ml_, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield).
1H NMR (400 MHz, DMSO-d6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1.36 (s, 9 H). MS (ESI): 520.0 [M+H]+.WO2009137391
…………………………………………………………………
PAPER
ACS Medicinal Chemistry Letters (2013), 4(3), 358-362.
http://pubs.acs.org/doi/abs/10.1021/ml4000063

…………………………………………………………….
Patent
http://www.google.com/patents/WO2014158467A1?cl=en

Step C : N- {3-[5-(2-chloro-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)-l ,3-thiazol-4-yl]- 2-fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged N- {3-[(2-chloro-4-pyrimidinyl)acetyl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 mL). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated. The dichloromethane layers were concentrated to -120 mL. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 mL. Dimethylacetamide (270 mL) was then added to the reaction mixture and cooled to ~10°C. 2,2-Dimethylpropanethioamide (1.3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for ~5 minutes between additions. The reaction was warmed to 20-25 °C. After 45 min, the vessel contents were heated to 75°C and held for 1.75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 30°C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated. Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-25°C. The organic layers were then concentrated under heat and vacuum to 120 mL. The vessel contents were then heated to 50°C and heptanes (120 mL) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for ~2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 30°C to obtain N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (28.8 g, 80%).
Compound B is disclosed and claimed, along with pharmaceutically acceptable salts thereof, as being useful as an inhibitor of BRaf activity, particularly in the treatment of cancer, in PCT patent application PCT/US09/42682. Compound B is embodied by Examples 58a through 58e of the application. The PCT application was published on 12 November 2009 as publication WO2009/137391, and is hereby incorporated by reference.
Suitably, Compound B may be prepared according to the methods below:
Method 1 : Compound B (first crystal form) – N-{3-[5-(2-Amino-4-pyrimidinyl)-2- (1,1 -dimethylethyl)- 1 ,3-thiazol-4-yl]- -fluorophenyl} -2,6-difluorobenzenesulfonamide

A suspension of N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) was heated in a sealed tube to 90 °C for 24 h. The reaction was diluted with DCM and added silica gel and concentrated. The crude product was chromatographed on silica gel eluting with 100% DCM to 1 : 1 [DCM: (9: 1 EtOAc:MeOH)]. The clean fractions were concentrated to yield the crude product. The crude product was repurified by reverse phase HPLC (a gradient of acetonitrile: water with 0.1%TFA in both). The combined clean fractions were concentrated then partitioned between DCM and saturated NaHC03. The DCM layer was separated and dried over Na2S04. The title compound, N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)- l,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1H NMR (400 MHz, DMSO- 6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 – 7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1.35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Method 2: Compound B (alternative crystal form) – N-{3-[5-(2-Amino-4- pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide 19.6 mg of N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 L of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-40°C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above.
Method 3: Compound B (alternative crystal form, large batch) – N-{3-[5-(2-amino- 4-pyrimidinyl)-2-(l , 1 -dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6- difluorobenzenesulfonamide

Step D : N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol-4-yl]-
2-fluorophenyl}-2,6-difluorobenzenesulfonamide
In 1 gal pressure reactor, a mixture of N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide ( 120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 °C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 °C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 °C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HC1 to ~pH 5.4-5.5. and added water (lvol). The aqueous layer was removed and discarded at 60-70 °C. The organic layer was washed with water (1 vol) at 60-70 °C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 °C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 °C for 10 min , then cooled to 20°C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes. The reaction mixture was maintained at ~65-70°C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 °C. The slurry was stirred at 65-70 °C for 2-3 hours and then cooled slowly to 0- 5°C. The product was filtered, washed with EtO Ac/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (102.3 g, 88%>).
MESYLATE
Method 4: Compound B (mesylate salt) – N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide methanesulfonate

To a solution of N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (204 mg, 0.393 mmol) in isopropanol (2 mL), methanesulfonic acid (0.131 mL, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield). 1H NMR (400 MHz, DMSO- 6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1.36 (s, 9 H). MS (ESI): 520.0 [M+H]+.
Method 5: Compound B (alternative mesylate salt embodiment) – N-{3-[5-(2- amino-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)-l ,3-thiazol-4-yl]-2-fluorophenyl} -2,6- difluorobenzenesulfonamide methanesulfonate
N- {3-[5-(2-amino-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)- 1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (as may be prepared according to example 58a) (2.37g, 4.56 mmol) was combined with pre-filtered acetonitrile (5.25 vol, 12.4 mL). A pre-filtered solution of mesic acid (1.1 eq., 5.02 mmol, 0.48 g) in H20 (0.75 eq., 1.78 mL) was added at 20°C. The temperature of the resulting mixture was raised to 50-60°C while maintaining a low agitation speed. Once the mixture temperature reached to 50- 60°C, a seed slurry of N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol- 4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate (1.0 %w/w slurried in 0.2 vol of pre-filtered acetonitrile) was added, and the mixture was aged while agitating at a speed fast enough to keep solids from settling at 50-60°C for 2 hr. The mixture was then cooled to 0-5°C at 0.25°C/min and held at 0-5°C for at 6 hr. The mixture was filtered and the wet cake was washed twice with pre-filtered acetonitrile. The first wash consisted of 14.2 ml (6 vol) pre-filtered acetonitrile and the second wash consisted of 9.5 ml (4 vol) pre-filtered acetonitrile. The wet solid was dried at 50°C under vacuum, yielding 2.39 g (85.1% yield) of product
Dara Phoenix (Dabrafenib) by the British GlaxoSmithKline (GSK) has developed Sisu threonine protein kinase (BRAF) inhibitor, as monotherapy ro ー kinds of clothes capsules for carrying BRAF V600E mutation surgical unresectable melanoma or metastatic melanoma treatment of adult patients, Dara Phoenix mesylate in May 2013 was approved by the US Food and Drug Administration (FDA), and is listed on the United States, the trade name Tafinlar (Da Feina). Since the European Medicines Agency (EMA) Committee for Medicinal Products for human use (CHMP) positive evaluation of Tafinlar, making the drug is expected to become after Roche’s Weiluofeini (Vemurafinib) to enter the European market, following a second BRAF inhibitors.
The chemical name Phoenix Dallas: N- [3- [5- (2- amino-4-pyrimidinyl) -2_ (tert-butyl) ~ ~ thiazol-4-yl] _2_ fluorophenyl] – 2,6_-difluorobenzenesulfonamide.

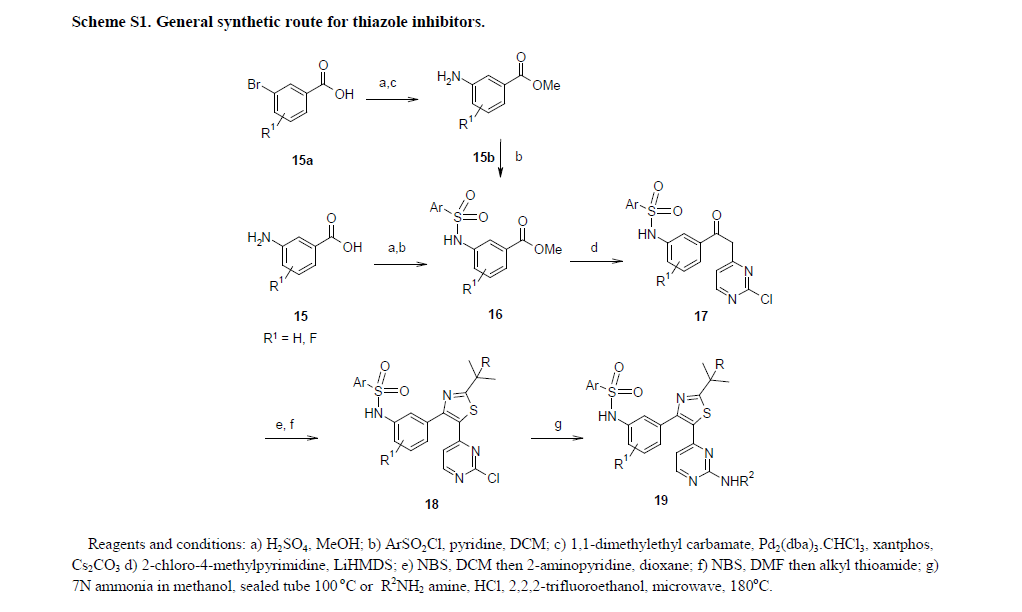
World Patent No. W02009137391, No. W02011047238 and W02012148588 number reported Dallas and Phoenix and its medicinal value synthesis method of the composition. According to the structural characteristics of Dara Phoenix and its analogues, the synthesis of such substances currently have A, B and C are three routes.

A more common route is the synthetic route, by reaction of 3-amino-2-fluorobenzoate (IX) first and 2,6_-difluorobenzene sulfonyl chloride (III) to amidation reaction occurs sulfonamide intermediate ( X); intermediate (X) with 2-chloro-4-methyl pyrimidine (XI) The condensation reaction occurs under the action of a strong base to give the intermediate (XII); intermediate (XII) to give the intermediate bromo
(XIII); intermediate (XIII) with 2,2_ dimethyl thiopropionamide (VI) to give the cyclized intermediate (XIV); and finally, the intermediate (XIV) by ammonolysis to afford the title compound Dallas Phoenix (I).

Different [0009] B is the first route by reaction of 3-amino-2-fluorobenzoate (IX) amino group protection, and thus condensation, cyclization, and bromo; then be obtained by deprotection of the amino group and the sulfonamide Intermediate (XIV); similarly, the intermediate
(XIV) obtained by ammonolysis target compound Dara Phoenix (I).

c route design features that first aminolysis reaction, and then give the desired product by deprotection and amino sulfonamide reaction. Clearly, this design is suitable for the route of these substituted amino ー aminolysis reaction, and for compounds such as Dallas Phoenix having pyrimidinylamino structure is not applicable. The reason is that if there are two aromatic amino groups will make the final sulfonamide ー reaction step to lose selectivity.

Example IV: the reaction flask was added N- [3- (5- formyl-2-t-butyl-ko -4_ thiazolyl) -2_ fluorophenyl] -2,6_ difluoro benzenesulfonamide (VIII) (5.4g, 11.5mmol), N, N- dimethylformamide dimethyl acetal (DMF-DMA) (2.74g, 23mmol) and xylene 50mL, heated to 140 ° C. About every four hours methanol was distilled out of the resulting reaction system, the reaction takes about 24 hours in total, the end of the reaction was detected by TLC. Cool, add hexane 40mL, have produced a yellow solid, filtered, and dried solids obtained after January nitrate melon (1.36,11.5mmol), sodium hydroxide (0.46g, 11.5mmol) and n-Ding enjoy 5OmL, warmed to 120 ° C, The reaction for 12 inches, TLC the reaction was complete. Cooling, with a crystal precipitated crystallized slowly for 3 inches, and filtered. The filter cake starched water, filtered and dried to yield an off-white solid Dara Phoenix (I) 3.58g, yield 60%.
References
- Gibney, G. T.; Zager, J. S. (2013). “Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies”. Expert Opinion on Drug Metabolism & Toxicology 9 (7): 1. doi:10.1517/17425255.2013.794220. PMID 23621583.
- Huang, T.; Karsy, M.; Zhuge, J.; Zhong, M.; Liu, D. (2013). “B-Raf and the inhibitors: From bench to bedside”. Journal of Hematology & Oncology 6: 30. doi:10.1186/1756-8722-6-30. PMC 3646677. PMID 23617957.
- “GSK melanoma drugs add to tally of U.S. drug approvals”. Reuters. May 30, 2013.
- “Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations” 367 (18). New England Journal of Medicine. November 1, 2012. pp. 1694–703. doi:10.1056/NEJMoa1210093. PMC 3549295. PMID 23020132.
“Dabrafenib/Trametinib Combination Approved for Advanced Melanoma”. OncLive. January 9, 2013.
Updates
Dabrafenib prediction
1H NMR PREDICT

![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_1h.png)
13C NM PREDICT
![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_13c.png)
COSY NMR PREDICT

HMBC, HSBC NMR PREDICT
INTERMEDIATES
INT 1
 Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate;Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, 1195768-23-0
Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate;Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, 1195768-23-0
 methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester;
methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester; methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3
methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, CAS No. 1195768-19-4
Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, CAS No. 1195768-19-4 CAS No. 1042055-86-6 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate
CAS No. 1042055-86-6 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoateSYN 1
![]()
 DABARAFENIB
DABARAFENIB
GLAXOSMITHKLINE LLC; HOOS, Axel; GRESHOCK, Joel Patent: WO2014/66606 A2, 2014 ; Location in patent: Page/Page column 20; 24 ;
SYN 2

![]()
WO2011/47238 A1, ;
SYN 3

![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 4
![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 5
![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 6
WO2011/47238 A1, ;
SYN 7
methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3WO2011/47238
WO2015003571
达拉菲尼甲磺酸盐的新晶型及其制备方法
Brief Description
Figure 1 Form IV of the present invention an X-ray powder diffraction pattern.
Figure 2 is a schematic diagram Form IV of DSC language.
Figure 3 Form IV of the present invention TGA profiles.
Figure 4 is a dynamic water adsorption of Form IV of the invention, FIG.
Figure 5 Form IV of the present invention 1HNMR spectrum.
Figure 6 of the present invention, Form II X-ray powder diffraction pattern.
Figure 7 of the present invention, Form II TGA profiles.
Figure 8 Form III of the present invention an X-ray powder diffraction pattern.
Figure 9 Form V of the present invention, an X-ray powder diffraction pattern.
Figure 58a and in Example 10 in accordance with Patent Document WO2009 / 137391 or in CN200980126781.6
58d method described for the preparation of polymorph I of the known X-ray powder diffraction pattern.
Figure 11 is in accordance with Patent Document WO2009 / 137391 or CN200980126781.6 in the method of Example 58a and 58d described for the preparation of polymorph I of the known DSC pattern.
12 is in accordance with Patent Document WO2009 / 137391 or CN200980126781.6 in the method of Example 58a and 58d described for the preparation of polymorph I of the known TGA profiles.
Figure 13 is a known polymorph I in Comparative Example 1 in various stages XRPD comparison chart with the sample from top to bottom in the order of: Dara Phoenix free base hydrate, a known polymorph I in water was stirred for 15 After minutes to obtain a sample, and a known polymorph I.
Figure 14 Form IV in the present invention Comparative Example 1 each stage XRPD comparison chart with the sample from top to bottom in the order of: Form IV, Form IV in water with stirring for 15 minutes to obtain a sample, Form IV After stirring overnight in water to obtain a sample, as well as the free base of the hydrate Dara Phoenix. Figure 15 is a Comparative Form IV polymorph I of the known elution compared to the situation in Figure 1 (A to Form IV, ■ known Form 1).
Figure 16 is known in the polymorph I of Comparative Example 2 in various stages XRPD comparison chart (figure from top to bottom as follows: Form I is known API by “wet granulation” process of granulation (excluding section 3-step tablet) obtained by the sample, the known polymorphs I and amount of excipients formulated physically mixed formulation obtained sample, lactose monohydrate and microcrystalline cellulose according to Formulation physical sample after mixing, Dara Feeney free base hydrate, as well as known Form 1).
17 is a crystalline form IV according to the present invention in Comparative Example 2 in various stages XRPD comparison chart (from top to bottom as follows: In Form IV according to API “wet granulation” process of granulation (not included in Step 3 tableting) after the sample obtained, Form IV and excipients Formulation amount by physically mixing the obtained sample, the sample lactose monohydrate and microcrystalline cellulose according to Formulation after physical mixing, and Form IV).
 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-{3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide | |
| Clinical data | |
| Trade names | Tafinlar |
|
|
| Legal status | |
| Identifiers | |
| CAS number | 1195765-45-7 |
| ATC code | L01XE23 |
| PubChem | CID 44462760 |
| ChemSpider | 25948204 |
| ChEBI | CHEBI:75045 |
| ChEMBL | CHEMBL2028663 |
| Chemical data | |
| Formula | C23H20F3N5O2S2 |
| Molecular mass | 519.56 g/mol |

