
Home » Posts tagged 'Antineoplastic Agents'
Tag Archives: Antineoplastic Agents
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
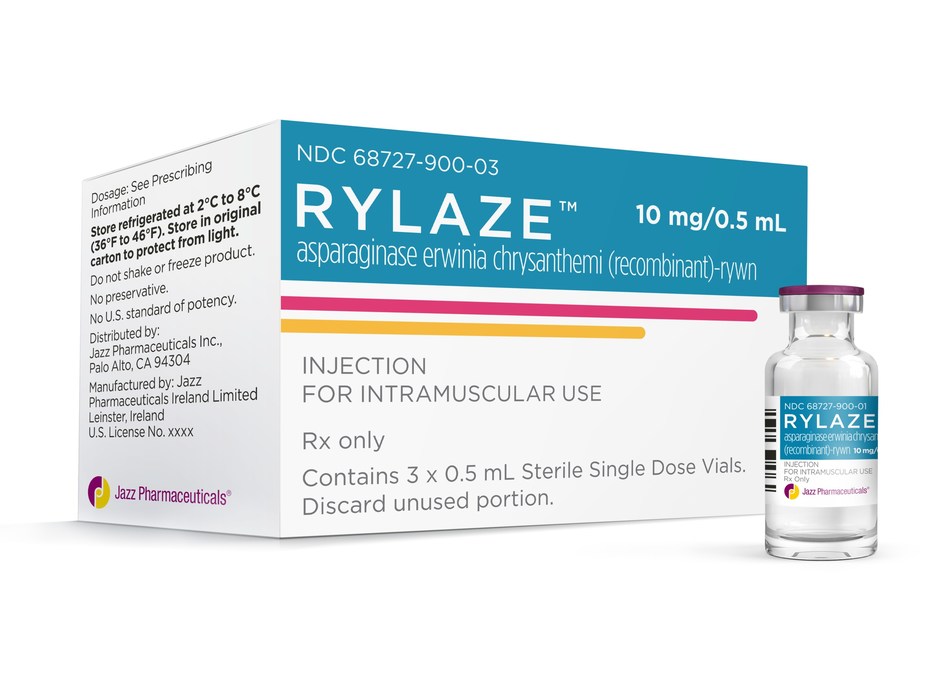
Asparaginase erwinia chrysanthemi (recombinant)-rywn
Sequence:
1ADKLPNIVIL ATGGTIAGSA ATGTQTTGYK AGALGVDTLI NAVPEVKKLA51NVKGEQFSNM ASENMTGDVV LKLSQRVNEL LARDDVDGVV ITHGTDTVEE101SAYFLHLTVK SDKPVVFVAA MRPATAISAD GPMNLLEAVR VAGDKQSRGR151GVMVVLNDRI GSARYITKTN ASTLDTFKAN EEGYLGVIIG NRIYYQNRID201KLHTTRSVFD VRGLTSLPKV DILYGYQDDP EYLYDAAIQH GVKGIVYAGM251GAGSVSVRGI AGMRKAMEKG VVVIRSTRTG NGIVPPDEEL PGLVSDSLNP301AHARILLMLA LTRTSDPKVI QEYFHTY
>Protein sequence for asparaginase (Erwinia chrysanthemi) monomer ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGEQFSNM ASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTVKSDKPVVFVAA MRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSARYITKTNASTLDTFKAN EEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKVDILYGYQDDPEYLYDAAIQH GVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNP AHARILLMLALTRTSDPKVIQEYFHTY
References:
- Therapeutic Targets Database: TTD Biologic drug sequences in fasta format [Link]
Asparaginase erwinia chrysanthemi (recombinant)-rywn
JZP458-201
JZP458
CAS Registry Number 1349719-22-7
Protein Chemical FormulaC1546H2510N432O476S9
Protein Average Weight 140000.0 Da
Rylaze, FDA APPROVED 6/30/2021, BLA 761179
L-Asparaginase (ec 3.5.1.1, L-asparagine amidohydrolase) erwinia chrysanthemi tetramer alpha4Asparaginase (Dickeya chrysanthemi subunit)
Other Names
- Asparaginase Erwinia chrysanthemi
- Crisantaspase
- Cristantaspase
- Erwinase
- Erwinaze
- L-Asparagine amidohydrolase (Erwinia chrysanthemi subunit)
Asparaginase erwinia chrysanthemi [USAN]
L-Asparaginase, erwinia chrysanthemi
Asparaginase (erwinia chrysanthemi)
Asparaginase erwinia chrysanthemi
L-Asparaginase (ec 3.5.1.1, L-asparagine amidohydrolase) erwinia chrysanthemi tetramer alpha4
Asparaginase erwinia chrysanthemi (recombinant) [USAN]
Asparaginase erwinia chrysanthemi (recombinant)
A hydrolase enzyme that converts L-asparagine and water to L-aspartate and NH3.
NCI: Asparaginase Erwinia chrysanthemi. An enzyme isolated from the bacterium Erwinia chrysanthemi (E. carotovora). Asparagine is critical to protein synthesis in leukemic cells, which cannot synthesize this amino acid due to the absence of the enzyme asparagine synthase. Asparaginase hydrolyzes L-asparagine to L-aspartic acid and ammonia, thereby depleting leukemic cells of asparagine and blocking protein synthesis and tumor cell proliferation, especially in the G1 phase of the cell cycle. This agent also induces apoptosis in tumor cells. The Erwinia-derived product is often used for those patients who have experienced a hypersensitivity reaction to the E. Coli formulation. (NCI Thesaurus)
- Treatment of Acute Lymphoblastic Leukemia (ALL)
- Antineoplastic Agents
| 10MG/0.5ML | INJECTABLE;INTRAMUSCULAR |
PATENT
WO 2011003633
https://patents.google.com/patent/WO2011003633A1/en
The present invention concerns a conjugate of a protein having substantial L-asparagine aminohydrolase activity and polyethylene glycol, particularly wherein the polyethylene glycol has a molecular weight less than or equal to about 5000 Da, particularly a conjugate wherein the protein is a L-asparaginase from Erwinia, and its use in therapy.Proteins with L-asparagine aminohydrolase activity, commonly known as L- asparaginases, have successfully been used for the treatment of Acute Lymphoblastic Leukemia(ALL) in children for many years. ALL is the most common childhood malignancy (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393).[0003] L-asparaginase has also been used to treat Hodgkin’s disease, acute myelocytic leukemia, acute myelomonocytic leukemia, chronic lymphocytic leukemia, lymphosarcoma, reticulosarcoma, and melanosarcoma (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).The anti-tumor activity of L-asparaginase is believed to be due to the inability or reduced ability of certain malignant cells to synthesize L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669). These malignant cells rely on an extracellular supply of L-asparagine. However, the L-asparaginase enzyme catalyzes the hydrolysis of L-asparagine to aspartic acid and ammonia, thereby depleting circulating pools of L-asparagine and killing tumor cells which cannot perform protein synthesis without L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0004] L-asparaginase from E. coli was the first enzyme drug used in ALL therapy and has been marketed as Elspar® in the USA or as Kidrolase® and L-asparaginase Medac® in Europe. L- asparaginases have also been isolated from other microorganisms, e.g., an L-asparaginase protein from Erwinia chrysanthemi, named crisantaspase, that has been marketed as Erwinase® (Wriston Jr., J.C. (1985) “L-asparaginase” Meth. Enzymol. 113, 608-618; Goward, CR. et al. (1992) “Rapid large scale preparation of recombinant Erwinia chrysanthemi L-asparaginase”, Bioseparation 2, 335-341). L-asparaginases from other species of Erwinia have also been identified, including, for example, Erwinia chrysanthemi 3937 (Genbank Accession#AAS67028), Erwinia chrysanthemi NCPPB 1125 (Genbank Accession #CAA31239), Erwinia carotovora (Genbank Accession #AAP92666), and Erwinia carotovora subsp. Astroseptica (Genbank Accession #AAS67027). These Erwinia chrysanthemi L-asparaginases have about 91-98% amino acid sequence identity with each other, while the Erwinia carotovora L- asparaginases have approximately 75-77% amino acid sequence identity with the Erwinia chrysanthemi L-asparaginases (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0005] L-asparaginases of bacterial origin have a high immunogenic and antigenic potential and frequently provoke adverse reactions ranging from mild allergic reaction to anaphylactic shock in sensitized patients (Wang, B. et al. (2003) “Evaluation of immunologic cross reaction of anti- asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),Leukemia 17, 1583-1588). E. coli L-asparaginase is particularly immunogenic, with reports of the presence of anti-asparaginase antibodies to E. coli L-asparaginase following i.v. or i.m. administration reaching as high as 78% in adults and 70% in children (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0006] L-asparaginases from Escherichia coli and Erwinia chrysanthemi differ in their pharmacokinetic properties and have distinct immunogenic profiles, respectively (Klug Albertsen, B. et al. (2001) “Comparison of intramuscular therapy with Erwinia asparaginase and asparaginase Medac: pharmacokinetics, pharmacodynamics, formation of antibodies and influence on the coagulation system” Brit. J. Haematol. 115, 983-990). Furthermore, it has been shown that antibodies that developed after a treatment with L-asparaginase from E. coli do not cross react with L-Asparaginase from Erwinia (Wang, B. et al., Leukemia 17 (2003) 1583-1588). Thus, L-asparaginase from Erwinia (crisantaspase) has been used as a second line treatment of ALL in patients that react to E. coli L-asparaginase (Duval, M. et al. (2002) “Comparison of Escherichia co/z-asparaginase with £Vwzm‘α-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment ofCancer, Children’s Leukemia Group phase 3 trial” Blood 15, 2734-2739; Avramis and Panosyan,Clin. Pharmacokinet. (2005) 44:367-393).[0007] In another attempt to reduce immunogenicity associated with administration of microbial L-asparaginases, an E. coli L-asparaginase has been developed that is modified with methoxy- polyethyleneglycol (mPEG). This method is commonly known as “PEGylation” and has been shown to alter the immunological properties of proteins (Abuchowski, A. et al. (1977) “Alteration of Immunological Properties of Bovine Serum Albumin by Covalent Attachment of Polyethylene Glycol,” J.Biol.Chem. 252 (11), 3578-3581). This so-called mPEG-L- asparaginase, or pegaspargase, marketed as Oncaspar® (Enzon Inc., USA), was first approved in the U.S. for second line treatment of ALL in 1994, and has been approved for first- line therapy of ALL in children and adults since 2006. Oncaspar® has a prolonged in vivo half-life and a reduced immunogenicity/antigenicity.[0008] Oncaspar® is E. coli L-asparaginase that has been modified at multiple lysine residues using 5 kDa mPEG-succinimidyl succinate (SS-PEG) (U.S. Patent No. 4,179,337). SS-PEG is aPEG reagent of the first generation that contains an instable ester linkage that is sensitive to hydro lysis by enzymes or at slightly alkaline pH values (U.S. Patent No. 4,670,417; Makromol. Chem. 1986, 187, 1131-1144). These properties decrease both in vitro and in vivo stability and can impair drug safety.[0009] Furthermore, it has been demonstrated that antibodies developed against L-asparaginase from E. coli will cross react with Oncaspar® (Wang, B. et al. (2003) “Evaluation of immunologic cross-reaction of anti-asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),” Leukemia 17, 1583-1588). Even though these antibodies were not neutralizing, this finding clearly demonstrated the high potential for cross-hypersensitivity or cross-inactivation in vivo. Indeed, in one report 30-41% of children who received pegaspargase had an allergic reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0010] In addition to outward allergic reactions, the problem of “silent hypersensitivity” was recently reported, whereby patients develop anti-asparaginase antibodies without showing any clinical evidence of a hypersensitivity reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588). This reaction can result in the formation of neutralizing antibodies to E. coli L-asparaginase and pegaspargase; however, these patients are not switched to Erwinia L-asparaginase because there are not outward signs of hypersensitivity, and therefore they receive a shorter duration of effective treatment (Holcenberg, J., J. Pediatr. Hematol. Oncol. 26 (2004) 273-274).[0011] Erwinia chrysanthemi L-asparaginase treatment is often used in the event of hypersensitivity to E. co/z-derived L-asparaginases. However, it has been observed that as many as 30-50% of patients receiving Erwinia L-asparaginase are antibody-positive (Avramis andPanosyan, Clin. Pharmacokinet. (2005) 44:367-393). Moreover, because Erwinia chrysanthemi L-asparaginase has a significantly shorter elimination half-life than the E. coli L-asparaginases, it must be administered more frequently (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393). In a study by Avramis et al., Erwinia asparaginase was associated with inferior pharmacokinetic profiles (Avramis et al., J. Pediatr. Hematol. Oncol. 29 (2007) 239-247). E. coli L-asparaginase and pegaspargase therefore have been the preferred first-line therapies for ALL over Erwinia L-asparaginase.[0012] Numerous biopharmaceuticals have successfully been PEGylated and marketed for many years. In order to couple PEG to a protein, the PEG has to be activated at its OH terminus. The activation group is chosen based on the available reactive group on the protein that will bePEGylated. In the case of proteins, the most important amino acids are lysine, cysteine, glutamic acid, aspartic acid, C-terminal carboxylic acid and the N-terminal amino group. In view of the wide range of reactive groups in a protein nearly the entire peptide chemistry has been applied to activate the PEG moiety. Examples for this activated PEG-reagents are activated carbonates, e.g., p-nitrophenyl carbonate, succinimidyl carbonate; active esters, e.g., succinimidyl ester; and for site specific coupling aldehydes and maleimides have been developed (Harris, M., Adv. Drug – A -DeI. Rev. 54 (2002), 459-476). The availability of various chemical methods for PEG modification shows that each new development of a PEGylated protein will be a case by case study. In addition to the chemistry the molecular weight of the PEG that is attached to the protein has a strong impact on the pharmaceutical properties of the PEGylated protein. In most cases it is expected that, the higher the molecular weight of the PEG, the better the improvement of the pharmaceutical properties (Sherman, M. R., Adv. Drug Del. Rev. 60 (2008), 59-68; Holtsberg, F. W., Journal of Controlled Release 80 (2002), 259-271). For example, Holtsberg et al. found that, when PEG was conjugated to arginine deaminase, another amino acid degrading enzyme isolated from a microbial source, pharmacokinetic and pharmacodynamic function of the enzyme increased as the size of the PEG attachment increased from a molecular weight of 5000Da to 20,000 Da (Holtsberg, F.W., Journal of Controlled Release 80 (2002), 259-271).[0013] However, in many cases, PEGylated biopharmaceuticals show significantly reduced activity compared to the unmodified biopharmaceutical (Fishburn, CS. (2008) Review “The Pharmacology of PEGylation: Balancing PD with PK to Generate Novel Therapeutics” J. Pharm. Sd., 1-17). In the case of L-asparaginase from Erwinia carotovora, it has been observed that PEGylation reduced its in vitro activity to approximately 57% (Kuchumova, A.V. et al. (2007) “Modification of Recombinant asparaginase from Erwinia carotovora with Polyethylene Glycol 5000” Biochemistry (Moscow) Supplement Series B: Biomedical Chemistry, 1, 230-232). The L-asparaginase from Erwinia carotovora has only about 75% homology to the Erwinia chrysanthemi L-asparaginase (crisantaspase). For Oncaspar® it is also known that its in vitro activity is approximately 50% compared to the unmodified E. coli L-asparaginase.[0014] The currently available L-asparaginase preparations do not provide alternative or complementary therapies— particularly therapies to treat ALL— that are characterized by high catalytic activity and significantly improved pharmacological and pharmacokinetic properties, as well as reduced immunogenicity. L-asparaginase protein has at least about 80% homology or identity with the protein comprising the sequence of SEQ ID NO:1, more specifically at least about 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% homology or identity with the protein comprising the sequence of SEQ ID NO:1. SEQ ID NO:1 is as follows:ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGE QFSNMASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTV KSDKPVVFVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSA RYITKTNASTLDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKV DILYGYQDDPEYLYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY (SEQ ID NO:1) [0048] The term “comprising the sequence of SEQ ID NO:1” means that the amino-acid sequence of the protein may not be strictly limited to SEQ ID NO:1 but may contain additional amino-acids.ExamplesExample 1 : Preparation of Recombinant Crisantaspase [0100] The recombinant bacterial strain used to manufacture the naked recombinant Erwinia chrysanthemi L-asparaginase protein (also referred to herein as “r-crisantaspase”) was an E. coli BL21 strain with a deleted ansB gene (the gene encoding the endogenous E. coli type II L- asparaginase) to avoid potential contamination of the recombinant Erwinia chrysanthemi L- asparaginase with this enzyme. The deletion of the ansB gene relies on homologous recombination methods and phage transduction performed according to the three following steps:1) a bacterial strain (NMI lOO) expressing a defective lambda phage which supplies functions that protect and recombine electroporated linear DNA substrate in the bacterial cell was transformed with a linear plasmid (kanamycin cassette) containing the kanamycin gene flanked by an FLP recognition target sequence (FRT). Recombination occurs to replace the ansB gene by the kanamycin cassette in the bacterial genome, resulting in a ΛansB strain; 2) phage transduction was used to integrate the integrated kanamycin cassette region from the ΛansB NMI lOO strain to the ansB locus in BL21 strain. This results in an E. coli BL21 strain with a deleted ansB gene and resistant to kanamycin; 3) this strain was transformed with a FLP -helper plasmid to remove the kanamycin gene by homologous recombination at the FRT sequence. The genome of the final strain (BL21 ΛansB strain) was sequenced, confirming full deletion of the endogenous ansB gene.[0101] The E. co/z‘-optimized DNA sequence encoding for the mature Erwinia chrysanthemi L- asparaginase fused with the ENX signal peptide from Bacillus subtilis was inserted into an expression vector. This vector allows expression of recombinant Erwinia chrysanthemi L- asparaginase under the control of hybrid T5/lac promoter induced by the addition of Isopropyl β- D-1-thiogalactopyranoside (IPTG) and confers resistance to kanamycin.[0102] BL21 ΛansB strain was transformed with this expression vector. The transformed cells were used for production of the r-crisantaspase by feed batch glucose fermentation in Reisenberg medium. The induction of the cell was done 16h at 23°C with IPTG as inducer. After cell harvest and lysis by homogenization in 1OmM sodium phosphate buffer pH6 5mM EDTA (Buffer A), the protein solution was clarified by centrifugation twice at 1500Og, followed by 0.45μm and 0.22μm filtration steps. The recombinant Erwinia chrysanthemi L-asparaginase was next purified using a sequence of chromatography and concentration steps. Briefly, the theoretical isoelectric point of the Erwinia chrysanthemi L-asparaginase (7.23) permits the recombinant enzyme to adsorb to cation exchange resins at pH6. Thus, the recombinant enzyme was captured on a Capto S column (cation exchange chromatography) and eluted with salt gradient in Buffer A. Fractions containing the recombinant enzyme were pooled. The pooled solution was next purified on Capto MMC column (cation exchange chromatography) in Buffer A with salt gradient. . The eluted fractions containing Erwinia chrysanthemi L-asparaginase were pooled and concentrated before protein separation on Superdex 200pg size exclusion chromatography as polishing step. Fractions containing recombinant enzymes were pooled, concentrated, and diafiltered against 10OmM sodium phosphate buffer pH8. The purity of the final Erwinia chrysanthemi L-asparaginase preparation was evaluated by SDS-PAGE (Figure 1) and RP-HPLC and was at least 90%. The integrity of the recombinant enzyme was verified byN-terminal sequencing and LC-MS. Enzyme activity was measured at 37°C using Nessler’s reagent. The specific activity of the purified recombinant Erwinia chrysanthemi L-asparaginase was around 600 U/mg. One unit of enzyme activity is defined as the amount of enzyme that liberates lμmol of ammonia from L-asparagine per minute at 37°C. Example 2: Preparation of 10 kDa mPEG-L- Asparaginase Conjugates[0103] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration between 2.5 and 4 mg/mL, in the presence of 150 mg/mL or 36 mg/mL 10 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 10 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 10 kDa mPEG-L-asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) residues being conjugated corresponding to PEGylation of 78% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (39% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 50% of accessible amino groups (e.g., lysine residues and/or the N-terminus)) . SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 3: Preparation of 5 kDa mPEG-L-Asparaginase Conjugates[0104] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration of 4 mg/mL, in the presence of 150 mg/mL or 22.5 mg/mL 5 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 5 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 5 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) being conjugated corresponding to PEGylation of 84% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (36% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 43% of accessible amino groups (e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 4: Preparation of 2 kDa mPEG-L-Asparaginase Conjugates[0105] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer pH 8.0 at a protein concentration of 4 mg/mL in the presence of150 mg/mL or 22.5 mg/mL 2 kDa mPEG-NHS for 2 hours at 22°C. The resulting crude 2 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 2 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as reference, one corresponding to maximum PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N- terminus) being conjugated corresponding to PEGylation of 86% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (47% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 55% of accessible amino groups {e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 5: Activity of mPEG-r-Crisantaspase Conjugates[0106] L-asparaginase aminohydrolase activity of each conjugate described in the proceeding examples was determined by Nesslerization of ammonia that is liberated from L-asparagine by enzymatic activity. Briefly, 50μL of enzyme solution were mixed with 2OmM of L-asparagine in a 50 mM Sodium borate buffer pH 8.6 and incubated for 10 min at 37°C. The reaction was stopped by addition of 200μL of Nessler reagent. Absorbance of this solution was measured at 450 nm. The activity was calculated from a calibration curve that was obtained from Ammonia sulfate as reference. The results are summarized in Table 2, below:Table 2: Activity of mPEG-r-crisantaspase conjugates

* the numbers “40%” and “100%” indicate an approximate degree of PEGylation of respectively 40-55% and 100% of accessible amino groups (see Examples 2-4, supra).** the ratio mol PEG / mol monomer was extrapolated from data using TNBS assay, that makes the assumption that all amino groups from the protein (e.g., lysine residues and the N-terminus) are accessible.[0107] Residual activity of mPEG-r-crisantaspase conjugates ranged between 483 and 543 Units/mg. This corresponds to 78-87% of L-asparagine aminohydrolase activity of the unmodified enzyme. Example 6: L-Asparagine-Depleting Effect of Unmodified Crisantaspase
PAPER
Biotechnology and Applied Biochemistry (2019), 66(3), 281-289. |
https://iubmb.onlinelibrary.wiley.com/doi/10.1002/bab.1723
Crisantaspase is an asparaginase enzyme produced by Erwinia chrysanthemi and used to treat acute lymphoblastic leukemia (ALL) in case of hypersensitivity to Escherichia coli l-asparaginase (ASNase). The main disadvantages of crisantaspase are the short half-life (10 H) and immunogenicity. In this sense, its PEGylated form (PEG-crisantaspase) could not only reduce immunogenicity but also improve plasma half-life. In this work, we developed a process to obtain a site-specific N-terminal PEGylated crisantaspase (PEG-crisantaspase). Crisantaspase was recombinantly expressed in E. coli BL21(DE3) strain cultivated in a shaker and in a 2-L bioreactor. Volumetric productivity in bioreactor increased 37% compared to shaker conditions (460 and 335 U L−1 H−1, respectively). Crisantaspase was extracted by osmotic shock and purified by cation exchange chromatography, presenting specific activity of 694 U mg−1, 21.7 purification fold, and yield of 69%. Purified crisantaspase was PEGylated with 10 kDa methoxy polyethylene glycol-N-hydroxysuccinimidyl (mPEG-NHS) at different pH values (6.5–9.0). The highest N-terminal pegylation yield (50%) was at pH 7.5 with the lowest poly-PEGylation ratio (7%). PEG-crisantaspase was purified by size exclusion chromatography and presented a KM value three times higher than crisantaspase (150 and 48.5 µM, respectively). Nonetheless, PEG-crisantaspase was found to be more stable at high temperatures and over longer periods of time. In 2 weeks, crisantaspase lost 93% of its specific activity, whereas PEG-crisantaspase was stable for 20 days. Therefore, the novel PEG-crisantaspase enzyme represents a promising biobetter alternative for the treatment of ALL.
ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGEQFSN
MASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTVKSDKPVV
FVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSARYITKTNAST
LDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKVDILYGYQDDPEY
LYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEE
LPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY
Figure S1 – Amino acid sequence of the enzyme crisantaspase without the signal peptide and with the lysines highlighted in red (Swiss-Prot/TrEMBL accession number: P06608|22-348 AA).
……………………………………………………………………………………………………………………………..
As a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in patients who are allergic to E. coli-derived asparaginase products
Press ReleaseFor Immediate Release:June 30, 2021
FDA Approves Component of Treatment Regimen for Most Common Childhood Cancer
Alternative Has Been in Global Shortage Since 2016
Today, the U.S. Food and Drug Administration approved Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) as a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in adult and pediatric patients who are allergic to the E. coli-derived asparaginase products used most commonly for treatment. The only other FDA-approved drug for such patients with allergic reactions has been in global shortage for years.
“It is extremely disconcerting to patients, families and providers when there is a lack of access to critical drugs for treatment of a life-threatening, but often curable cancer, due to supply issues,” said Gregory Reaman, M.D., associate director for pediatric oncology in the FDA’s Oncology Center of Excellence. “Today’s approval may provide a consistently sourced alternative to a pivotal component of potentially curative therapy for children and adults with this type of leukemia.”
Acute lymphoblastic leukemia occurs in approximately 5,700 patients annually, about half of whom are children. It is the most common type of childhood cancer. One component of the chemotherapy regimen is an enzyme called asparaginase that kills cancer cells by depriving them of substances needed to survive. An estimated 20% of patients are allergic to the standard E. coli-derived asparaginase and need an alternative their bodies can tolerate.
Rylaze’s efficacy was evaluated in a study of 102 patients who either had a hypersensitivity to E. coli-derived asparaginases or experienced silent inactivation. The main measurement was whether patients achieved and maintained a certain level of asparaginase activity. The study found that the recommended dosage would provide the target level of asparaginase activity in 94% of patients.
The most common side effects of Rylaze include hypersensitivity reactions, pancreatic toxicity, blood clots, hemorrhage and liver toxicity.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Health Canada, where the application review is pending.
Rylaze received Fast Track and Orphan Drug designations for this indication. Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fulfill an unmet medical need. Orphan Drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted approval of Rylaze to Jazz Pharmaceuticals.
REF
DUBLIN, June 30, 2021 /PRNewswire/ — Jazz Pharmaceuticals plc (Nasdaq: JAZZ) today announced the U.S. Food and Drug Administration (FDA) approval of Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn) for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase.1 Rylaze is the only recombinant erwinia asparaginase manufactured product that maintains a clinically meaningful level of asparaginase activity throughout the entire duration of treatment, and it was developed by Jazz to address the needs of patients and healthcare providers with an innovative, high-quality erwinia-derived asparaginase with reliable supply.
“We are excited to bring this important new treatment to patients who are in critical need, and we are grateful to FDA for the approval of Rylaze based on its established safety and efficacy profile. We are pleased Rylaze was approved before the trial is complete and are diligently working to advance additional clinical trial data. We are committed to quickly engaging with FDA to evolve the Rylaze product profile with additional dosing options and an IV route of administration,” said Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals. “Thank you to our collaborators within the Children’s Oncology Group, the clinical trial investigators, patients and their families, and all of the other stakeholders who helped us achieve this significant milestone.”
Rylaze was granted orphan drug designation for the treatment of ALL/LBL by FDA in June 2021. The Biologics Licensing Application (BLA) approval followed review under the Real-Time Oncology Review (RTOR) program, an initiative of FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The company expects Rylaze will be commercially available in mid-July.
“The accelerated development and approval of Rylaze marks an important step in bringing a meaningful new treatment option for many ALL patients – most of whom are children – who cannot tolerate E. coli-derived asparaginase medicine,” said Dr. Luke Maese, assistant professor at the University of Utah, Primary Children’s Hospital and Huntsman Cancer Institute. “Before the approval of Rylaze, there was a significant need for an effective asparaginase medicine that would allow patients to start and complete their prescribed treatment program with confidence in supply.”
Recent data from a Children’s Oncology Group retrospective analysis of over 8,000 patients found that patients who did not receive a full course of asparaginase treatment due to associated toxicity had significantly lower survival outcomes – regardless of whether those patients were high risk or standard risk, slow early responders.2
About Study JZP458-201
The FDA approval of Rylaze, also known as JZP458, is based on clinical data from an ongoing pivotal Phase 2/3 single-arm, open-label, multicenter, dose confirmation study evaluating pediatric and adult patients with ALL or LBL who have had an allergic reaction to E. coli-derived asparaginases and have not previously received asparaginase erwinia chrysanthemi. The study was designed to assess the safety, tolerability and efficacy of JZP458. The determination of efficacy was measured by serum asparaginase activity (SAA) levels. The Phase 2/3 study is being conducted in two parts. The first part is investigating the intramuscular (IM) route of administration, including a Monday-Wednesday-Friday dosing schedule. The second part remains active to further confirm the dose and schedule for the intravenous (IV) route of administration.
The FDA approval of Rylaze was based on data from the first of three IM cohorts, which demonstrated the achievement and maintenance of nadir serum asparaginase activity (NSAA) greater than or equal to the level of 0.1 U/mL at 48 hours using IM doses of Rylaze 25 mg/m2. The results of modeling and simulations showed that for a dosage of 25 mg/m2 administered intramuscularly every 48 hours, the proportion of patients maintaining NSAA ≥ 0.1 U/mL at 48 hours after a dose of Rylaze was 93.6% (95% CI: 92.6%, 94.6%).1
The most common adverse reactions (incidence >15%) were abnormal liver test, nausea, musculoskeletal pain, fatigue, infection, headache, pyrexia, drug hypersensitivity, febrile neutropenia, decreased appetite, stomatitis, bleeding and hyperglycemia. In patients treated with the Rylaze, a fatal adverse reaction (infection) occurred in one patient and serious adverse reactions occurred in 55% of patients. The most frequent serious adverse reactions (in ≥5% of patients) were febrile neutropenia, dehydration, pyrexia, stomatitis, diarrhea, drug hypersensitivity, infection, nausea and viral infection. Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received Rylaze. Adverse reactions resulting in permanent discontinuation included hypersensitivity (6%) and infection (3%).1
The company will continue to work with FDA and plans to submit additional data from a completed cohort of patients evaluating 25mg/m2 IM given on Monday and Wednesday, and 50 mg/m2 given on Friday in support of a M/W/F dosing schedule. Part 2 of the study is evaluating IV administration and is ongoing. The company also plans to submit these data for presentation at a future medical meeting.
Investor Webcast
The company will host an investor webcast on the Rylaze approval in July. Details will be announced separately.
About Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn)
Rylaze, also known as JZP458, is approved in the U.S. for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase. Rylaze has orphan drug designation for the treatment of ALL/LBL in the United States. Rylaze is a recombinant erwinia asparaginase that uses a novel Pseudomonas fluorescens expression platform. JZP458 was granted Fast Track designation by the U.S. Food and Drug Administration (FDA) in October 2019 for the treatment of this patient population. Rylaze was approved as part of the Real-Time Oncology Review program, an initiative of the FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The full U.S. Prescribing Information for Rylaze is available at: <http://pp.jazzpharma.com/pi/rylaze.en.USPI.pdf>
Important Safety Information
RYLAZE should not be given to people who have had:
- Serious allergic reactions to RYLAZE
- Serious swelling of the pancreas (stomach pain), serious blood clots, or serious bleeding during previous asparaginase treatment
RYLAZE may cause serious side effects, including:
- Allergic reactions (a feeling of tightness in your throat, unusual swelling/redness in your throat and/or tongue, or trouble breathing), some of which may be life-threatening
- Swelling of the pancreas (stomach pain)
- Blood clots (may have a headache or pain in leg, arm, or chest)
- Bleeding
- Liver problems
Contact your doctor immediately if any of these side effects occur.
Some of the most common side effects with RYLAZE include: liver problems, nausea, bone and muscle pain, tiredness, infection, headache, fever, allergic reactions, fever with low white blood cell count, decreased appetite, mouth swelling (sometimes with sores), bleeding, and too much sugar in the blood.
RYLAZE can harm your unborn baby. Inform your doctor if you are pregnant, planning to become pregnant, or nursing. Females of reproductive potential should use effective contraception (other than oral contraceptives) during treatment and for 3 months following the final dose. Do not breastfeed while receiving RYLAZE and for 1 week after the final dose.
Tell your healthcare provider if there are any side effects that are bothersome or that do not go away.
These are not all the possible side effects of RYLAZE. For more information, ask your healthcare provider.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088 (1-800-332-1088).
About ALL
ALL is a cancer of the blood and bone marrow that can progress quickly if not treated.3 Leukemia is the most common cancer in children, and about three out of four of these cases are ALL.4 Although it is one of the most common cancers in children, ALL is among the most curable of the pediatric malignancies due to recent advancements in treatment.5,6 Adults can also develop ALL, and about four of every 10 cases of ALL diagnosed are in adults.7 The American Cancer Society estimates that almost 6,000 new cases of ALL will be diagnosed in the United States in 2021.7 Asparaginase is a core component of multi-agent chemotherapeutic regimens in ALL.8 However, asparaginase treatments derived from E. coli are associated with the potential for development of hypersensitivity reactions.9
About Lymphoblastic Lymphoma
LBL is a rare, fast-growing, aggressive subtype of Non-Hodgkin’s lymphoma, most often seen in teenagers and young adults.8 LBL is a very aggressive lymphoma – also called high-grade lymphoma – which means the lymphoma grows quickly with early spread to different parts of the body.10,11
About Jazz Pharmaceuticals plc
Jazz Pharmaceuticals plc (NASDAQ: JAZZ) is a global biopharmaceutical company whose purpose is to innovate to transform the lives of patients and their families. We are dedicated to developing life-changing medicines for people with serious diseases – often with limited or no therapeutic options. We have a diverse portfolio of marketed medicines and novel product candidates, from early- to late-stage development, in neuroscience and oncology. We actively explore new options for patients including novel compounds, small molecules and biologics, and through cannabinoid science and innovative delivery technologies. Jazz is headquartered in Dublin, Ireland and has employees around the globe, serving patients in nearly 75 countries. For more information, please visit www.jazzpharmaceuticals.com and follow @JazzPharma on Twitter.
About The Children’s Oncology Group (COG)
COG (childrensoncologygroup.org), a member of the NCI National Clinical Trials Network (NCTN), is the world’s largest organization devoted exclusively to childhood and adolescent cancer research. COG unites over 10,000 experts in childhood cancer at more than 200 leading children’s hospitals, universities, and cancer centers across North America, Australia, and New Zealand in the fight against childhood cancer. Today, more than 90% of the 14,000 children and adolescents diagnosed with cancer each year in the United States are cared for at COG member institutions. Research performed by COG institutions over the past 50 years has transformed childhood cancer from a virtually incurable disease to one with a combined 5-year survival rate of 80%. COG’s mission is to improve the cure rate and outcomes for all children with cancer.
Caution Concerning Forward-Looking Statements
This press release contains forward-looking statements, including, but not limited to, statements related to Jazz Pharmaceuticals’ belief in the potential of Rylaze to provide a reliable therapeutic option for adult and pediatric patients to maximize their chance for a cure, plans for a mid-July 2021 launch of Rylaze, the availability of a reliable supply of Rylaze and other statements that are not historical facts. These forward-looking statements are based on Jazz Pharmaceuticals’ current plans, objectives, estimates, expectations and intentions and inherently involve significant risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in such forward-looking statements as a result of these risks and uncertainties, which include, without limitation, effectively launching and commercializing new products; obtaining and maintaining adequate coverage and reimbursement for the company’s products; delays or problems in the supply or manufacture of the company’s products and other risks and uncertainties affecting the company, including those described from time to time under the caption “Risk Factors” and elsewhere in Jazz Pharmaceuticals’ Securities and Exchange Commission filings and reports (Commission File No. 001-33500), including Jazz Pharmaceuticals’ Annual Report on Form 10-K for the year ended December 31, 2020 and future filings and reports by Jazz Pharmaceuticals. Other risks and uncertainties of which Jazz Pharmaceuticals is not currently aware may also affect Jazz Pharmaceuticals’ forward-looking statements and may cause actual results and the timing of events to differ materially from those anticipated. The forward-looking statements herein are made only as of the date hereof or as of the dates indicated in the forward-looking statements, even if they are subsequently made available by Jazz Pharmaceuticals on its website or otherwise. Jazz Pharmaceuticals undertakes no obligation to update or supplement any forward-looking statements to reflect actual results, new information, future events, changes in its expectations or other circumstances that exist after the date as of which the forward-looking statements were made.
Jazz Media Contact:
Jacqueline Kirby
Vice President, Corporate Affairs
Jazz Pharmaceuticals plc
CorporateAffairsMediaInfo@jazzpharma.com
Ireland, +353 1 697 2141
U.S. +1 215 867 4910
Jazz Investor Contact:
Andrea N. Flynn, Ph.D.
Vice President, Head, Investor Relations
Jazz Pharmaceuticals plc
investorinfo@jazzpharma.com
Ireland, +353 1 634 3211
References
- Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) injection, for intramuscular use Prescribing Information. Palo Alto, CA: Jazz Pharmaceuticals, Inc.
- Gupta S, Wang C, Raetz EA et al. Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children’s Oncology Group. J Clin Oncol. 2020 Jun 10;38(17):1897-1905. doi: 10.1200/JCO.19.03024
- National Cancer Institute. Adult Acute Lymphoblastic Leukemia Treatment (PDQ®)–Patient Version. Available at www.cancer.gov/types/leukemia/patient/adult-all-treatment-pdq. Accessed June 29, 2021
- American Cancer Society. Key Statistics for Childhood Leukemia. Available at https://www.cancer.org/cancer/leukemia-in-children/about/key-statistics.html. Accessed June 29, 2021.
- American Cancer Society. Cancer Facts & Figures 2019. www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html. Accessed June 29, 2021.
- Pui C, Evans W. A 50-Year Journey to Cure Childhood Acute Lymphoblastic Leukemia. Seminars in Hematology. 2013;50(3), 185-196.
- American Cancer Society. Key Statistics for Acute Lymphocytic Leukemia (ALL). Available at https://cancerstatisticscenter.cancer.org/?_ga=2.8163506.1018157754.1621008457-1989786785.1621008457#!/data-analysis/NewCaseEstimates. Accessed June 29, 2021.
- Salzer W, Bostrom B, Messinger Y et al. 2018. Asparaginase activity levels and monitoring in patients with acute lymphoblastic leukemia. Leukemia & Lymphoma. 59:8, 1797-1806, DOI: 10.1080/10428194.2017.1386305.
- Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57(4):748–757. DOI: 10.3109/10428194.2015.1101098.
- Leukemia Foundation. Lymphoblastic Lymphoma. Available at https://www.leukaemia.org.au/disease-information/lymphomas/non-hodgkin-lymphoma/other-non-hodgkin-lymphomas/lymphoblastic-lymphoma/. Accessed June 29, 2021.
- Mayo Clinic. Acute Lymphocytic Leukemia Diagnosis. Available at https://www.mayoclinic.org/diseases-conditions/acute-lymphocytic-leukemia/diagnosis-treatment/drc-20369083. Accessed June 29, 2021.
SOURCE Jazz Pharmaceuticals plc
Related Links
CLIP
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4776285/

/////////////asparaginase erwinia chrysanthemi (recombinant)-rywn, Rylaze, Jazz Pharmaceuticals, JZP458-201, JZP458, FDA 2021, APPROVALS 2021, ORPHAN, Fast Track, Acute Lymphoblastic Leukemia, ALL, Antineoplastic Agents
https://chem.nlm.nih.gov/chemidplus/id/1349719227
https://go.drugbank.com/drugs/DB08886

NEW DRUG APPROVALS
ONE TIME
$10.00
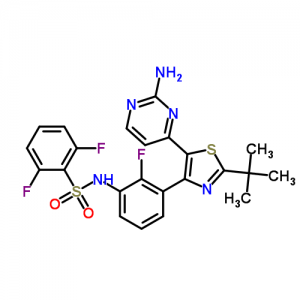
Dabrafenib mesylate, GSK 2118436, ダブラフェニブ 达拉菲尼, An antineoplastic agent that inhibits BRAF kinase


DABRAFENIB
ダブラフェニブ
达拉菲尼,
1195765-45-7 BASE
1195768-06-9 cas of mesylate
Benzenesulfonamide, N-[3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-4-thiazolyl]-2-fluorophenyl]-2,6-difluoro-
MW 519.56 BASE
MF C23 H20 F3 N5 O2 S2 BASE
- Dabarefenib
- Dabrafenib
- GSK 2118436
- Tafinlar
- UNII-QGP4HA4G1B
US FDA APPROVAL….Date of Approval: May 29, 2013
update
| Product details | |
|---|---|
| Name |
Tafinlar
|
| Agency product number |
EMEA/H/C/002604
|
| Active substance |
dabrafenib mesilate
|
| International non-proprietary name (INN) or common name |
dabrafenib
|
| Therapeutic area (MeSH) |
Melanoma
|
| Anatomical therapeutic chemical (ATC) code |
L01EC02
|
| Publication details | |
|---|---|
| Marketing-authorisation holder |
Novartis Europharm Limited
|
| Date of issue of marketing authorisation valid throughout the European Union |
26/08/2013
|
An orally bioavailable inhibitor of B-raf (BRAF) protein with potential antineoplastic activity. Dabrafenib selectively binds to and inhibits the activity of B-raf, which may inhibit the proliferation of tumor cells which contain a mutated BRAF gene. B-raf belongs to the the raf/mil family of serine/threonine protein kinases and plays a role in regulating the MAP kinase/ERKs signaling pathway, which may be constitutively activated due to BRAF gene mutations
Dabrafenib (trade name Tafinlar) is a drug for the treatment of cancers associated with a mutated version of the gene BRAF. Dabrafenib acts as an inhibitor of the associated enzyme B-Raf, which plays a role in the regulation of cell growth. Dabrafenib has clinical activity with a manageable safety profile in clinical trials of phase 1 and 2 in patients with BRAF(V600)-mutated metastatic melanoma.[1][2]
The Food and Drug Administration approved dabrafenib as a single agent treatment for patients with BRAF V600E mutation-positive advanced melanoma on May 30, 2013.[3] Clinical trial data demonstrated that resistance to dabrafinib and other BRAF inhibitors occurs within 6 to 7 months.[4] To overcome this resistance, the BRAF inhibitor dabrafenib was combined with the MEK inhibitor trametinib.[4] As a result of this research, on January 8, 2014, the FDA approved the combination of dabrafenib and trametinib for the treatment of patients with BRAF V600E/K-mutant metastatic melanoma.[5]

Inhibitor of BRAF(V600) mutants
| Active Ingredient: | DABRAFENIB MESYLATE |
| Dosage Form;Route: | CAPSULE;ORAL |
| Proprietary Name: | TAFINLAR |
| Applicant: | GLAXOSMITHKLINE |
| Strength: | EQ 75MG BASE |
| NDA Application Number: | N202806 |
| Product Number: | 002 |
| Approval Date: | May 29, 2013 |
| Reference Listed Drug | Yes |
| RX/OTC/DISCN: | RX |
Patent Data
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
Delist Requested |
|---|---|---|---|---|---|---|---|
| N202806 | 002 | 7994185 | Jan 20, 2030 | Y | Y | U – 1406 | |
| N202806 | 002 | 8415345 | Jan 20, 2030 | Y | Y | U – 1406 |
Exclusivity Data
| NDA Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N202806 | 002 | I – 678 | Jan 8, 2017 |
| N202806 | 002 | ODE | Jan 9, 2021 |
| N202806 | 002 | NCE | May 29, 2018 |
| N202806 | 002 | ODE | May 29, 2020 |
PDF……http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202806s000lbl.pdf
TERMS
I 678, TRAMETINIB, IN COMBINATION WITH DABRAFENIB, FOR THE TREATMENT OF PATIENTS WITH UNRESECTABLE OR METASTATIC MELANOMA WITH BRAF V600E OR V600K MUTATIONS AS DETECTED BY AN FDA-APPROVED TEST
ODE ORPHAN DRUG EXCLUSIVITY
NCE NEW CHEMICAL ENTITY

DABRAFENIB SYNTHESIS
WILL BE UPDATED
—
http://www.google.com/patents/WO2011047238A1?cl=en
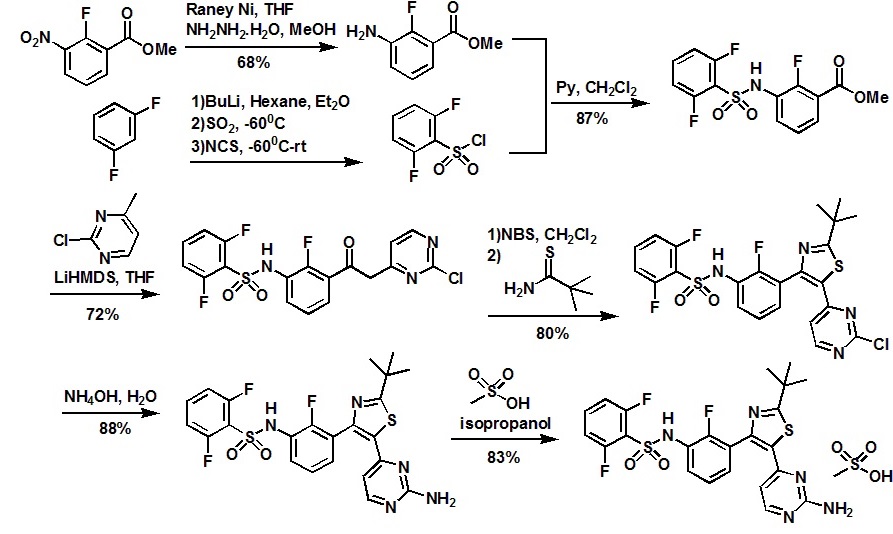
Method 1 : Compound B (first crystal form) – A/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1 dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide

A suspension of A/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]- 2-fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) was heated in a sealed tube to 90 °C for 24 h. The reaction was diluted with DCM and added silica gel and concentrated. The crude product was chromatographed on silica gel eluting with 100% DCM to 1 :1 [DCM:(9:1 EtOAc:MeOH)]. The clean fractions were concentrated to yield the crude product. The crude product was repurified by reverse phase HPLC (a gradient of acetonitrile:water with 0.1 %TFA in both). The combined clean fractions were concentrated then partitioned between DCM and saturated NaHCO3. The DCM layer was separated and dried over Na2SO4. The title compound, /V-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1 H NMR (400 MHz, DMSO-c/6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 – 7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1 .35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Method 2: Compound B (alternative crystal form) – A/-{3-[5-(2-Amino-4-pyrimidinyl)-2- (1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide 19.6 mg of A/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 L of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-40°C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above. Method 3: Compound B (alternative crystal form, large batch) – A/-{3-[5-(2-amino-4- pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

tep A: methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate

Methyl 3-amino-2-fluorobenzoate (50 g, 1 eq) was charged to reactor followed by dichloromethane (250 mL, 5 vol). The contents were stirred and cooled to ~15°C and pyridine (26.2 mL, 1 .1 eq) was added. After addition of the pyridine, the reactor contents were adjusted to ~15°C and the addition of 2,6-diflurorobenzenesulfonyl chloride (39.7 mL, 1 .0 eq) was started via addition funnel. The temperature during addition was kept <25°C. After complete addition, the reactor contents were warmed to 20-25°C and held overnight. Ethyl acetate (150 mL) was added and dichloromethane was removed by distillation. Once distillation was complete, the reaction mixture was then diluted once more with ethyl acetate (5 vol) and concentrated. The reaction mixture was diluted with ethyl acetate (10 vol) and water (4 vol) and the contents heated to 50-55°C with stirring until all solids dissolve. The layers were settled and separated. The organic layer was diluted with water (4 vol) and the contents heated to 50-55° for 20-30 min. The layers were settled and then separated and the ethyl acetate layer was evaporated under reduced pressure to ~3 volumes. Ethyl Acetate (5 vol.) was added and again evaporated under reduced pressure to ~3 volumes.
Cyclohexane (9 vol) was then added to the reactor and the contents were heated to reflux for 30 min then cooled to 0 °C. The solids were filtered and rinsed with cyclohexane (2 x 100 mL). The solids were air dried overnight to obtain methyl 3-{[(2,6- difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (94.1 g, 91 %).
Step B: A/-{3-[(2-chloro-4-pyhmidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (490 g, 1 equiv.), prepared generally in accordance with Step A, above, was dissolved in THF (2.45 L, 5 vols) and stirred and cooled to 0-3 °C. 1 M lithium bis(trimethylsilyl)amide in THF (5.25 L, 3.7 equiv.) solution was charged to the reaction mixture followed addition of 2- chloro-4-methylpyrimidine (238 g, 1 .3 equiv.) in THF (2.45 L, 5 vols). The reaction was then stirred for 1 hr. The reaction was quenched with 4.5M HCI (3.92 L, 8 vols). The aqueous layer (bootom layer) was removed and discarded. The organic layer was concentrated under reduced pressure to ~2L. IPAC (isopropyl acetate) (2.45L) was added to the reaction mixture which was then concentrated to ~2L. IPAC (0.5L) and MTBE (2.45 L) was added and stirred overnight under N2. The solids were filtered. The solids and mother filtrate added back together and stirred for several hours. The solids were filtered and washed with MTBE (~5 vol). The solids were placed in vacuum oven at 50 °C overnight. The solids were dried in vacuum oven at 30 °C over weekend to obtain A/-{3-[(2-chloro-4-pyhmidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (479 g, 72%).
Step C: A/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged /V-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 mL). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated. The dichloromethane layers were concentrated to -120 mL. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 mL. Dimethylacetamide (270 mL) was then added to the reaction mixture and cooled to ~10°C. 2,2- Dimethylpropanethioamide (1 .3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for ~5 minutes between additions. The reaction was warmed to 20-25 °C. After 45 min, the vessel contents were heated to 75°C and held for 1 .75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 30°C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated. Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-25°C. The organic layers were then concentrated under heat and vacuum to 120 mL. The vessel contents were then heated to 50°C and heptanes (120 mL) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for ~2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 30°C to obtain /V-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide (28.8 g, 80%).
Step D: A/-{3-[5-(2-amino-4-pyhmidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide
In 1 gal pressure reactor, a mixture of A/-{3-[5-(2-chloro-4-pyrinnidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 °C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 °C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 °C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HCI to ~pH 5.4-5.5. and added water (1 vol). The aqueous layer was removed and discarded at 60-70 °C. The organic layer was washed with water (1 vol) at 60-70 °C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 °C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 °C for 10 min , then cooled to 20°C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes. The reaction mixture was maintained at ~65-70°C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 °C. The slurry was stirred at 65-70 °C for 2- 3 hours and then cooled slowly to 0-5°C. The product was filtered, washed with
EtOAc/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain A/-{3-[5-(2- amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (102.3 g, 88%).
Method 4: Compound B (mesylate salt) – A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate

To a solution of /V-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide (204 mg, 0.393 mmol) in isopropanol (2 ml_), methanesulfonic acid (0.131 ml_, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield). 1 H NMR (400 MHz, DMSO-c/6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1 .36 (s, 9 H). MS (ESI): 520.0 [M+H]+.
Method 5: Compound B (alternative mesylate salt embodiment) – A/-{3-[5-(2-amino-4- pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide methanesulfonate
A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (as may be prepared according to example 58a) (2.37g, 4.56 mmol) was combined with pre-filtered acetonitrile (5.25 vol, 12.4 ml_). A pre-filtered solution of mesic acid (1 .1 eq., 5.02 mmol, 0.48 g) in H2O (0.75 eq., 1 .78 ml_) was added at 20°C. The temperature of the resulting mixture was raised to 50- 60°C while maintaining a low agitation speed. Once the mixture temperature reached to 50-60°C, a seed slurry of A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate (1 .0 %w/w slurried in 0.2 vol of pre-filtered acetonitrile) was added, and the mixture was aged while agitating at a speed fast enough to keep solids from settling at 50-60°C for 2 hr. The mixture was then cooled to 0-5°C at 0.25°C/min and held at 0-5°C for at 6 hr. The mixture was filtered and the wet cake was washed twice with pre-filtered
acetonitrile. The first wash consisted of 14.2 ml (6 vol) pre-filtered acetonitrile and the second wash consisted of 9.5 ml (4 vol) pre-filtered acetonitrile. The wet solid was dried at 50°C under vacuum, yielding 2.39 g (85.1 % yield) of product. Typically, the salts of the present invention are pharmaceutically acceptable salts.
May 29, 2013 — GlaxoSmithKline plc announced today that the U.S. Food and Drug Administration (FDA) has approved Tafinlar (dabrafenib). Tafinlar is indicated as a single-agent oral treatment for unresectable melanoma (melanoma that cannot be removed by surgery) or metastatic melanoma (melanoma which has spread to other parts of the body) in adult patients with BRAF V600E mutation. Tafinlar is not indicated for the treatment of patients with wild-type BRAF melanoma. The mutation must be detected by an FDA-approved test, such as the companion diagnostic assay from bioMérieux S.A., THxID™-BRAF.
Among those with metastatic melanoma, approximately half have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread
Tafinlar is approved for patients with the BRAF V600E mutation, which accounts for approximately 85 percent of all BRAF V600 mutations in metastatic melanoma.
GSK will be making Tafinlar available for prescription no later than in the early third quarter of 2013.
In 2010, GSK entered a collaboration with bioMérieux to develop a companion diagnostic test to detect BRAF V600 (V600E and V600K) gene mutations found in several cancers, including melanoma. bioMérieux has received FDA pre-market approval of THxID™-BRAF. Currently, it is the only FDA-approved test that detects the V600K mutation.
The primary outcome measure was the estimation of the overall intracranial response rate (OIRR) in each cohort. The OIRR for Cohort A was 18 percent (95% CI: 9.7, 28.2). For Cohort B, the OIRR was also 18 percent (95% CI: 9.9, 30.0). The median duration of response was 4.6 months (95% CI: 2.8, Not Reached) and 4.6 months (95% CI: 1.9, 4.6) in Cohort A and Cohort B, respectively.
Melanoma is the most serious and deadly form of skin cancer. According to statistics from the National Cancer Institute, in 2013 there will be an estimated 9,480 deaths resulting from melanoma in the United States. When melanoma spreads in the body, the disease is called metastatic melanoma.Approximately half of all people with metastatic melanoma have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread.
One in two patients worldwide with metastatic melanoma is expected to survive for a year after diagnosis, while in the U.S., the five-year survival rate was 16 percent (2003-2009).The median age of a newly diagnosed metastatic melanoma patient is almost a decade younger than other cancers.
Tafinlar (dabrafenib) is now approved for the treatment of adult patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. Limitation of use: Tafinlar is not recommended for use in patients with wild-type BRAF melanoma.
Tafinlar is not approved or licensed in Europe and may not be approved in other parts of the world for the treatment of patients with BRAF V600 mutation-positive unresectable melanoma or metastatic melanoma.
Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2•CH4O3S and a molecular weight of 615.68. Dabrafenib mesylate has the following chemical structure:
 |
Dabrafenib mesylate is a white to slightly colored solid with three pKas: 6.6, 2.2, and -1.5. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
TAFINLAR (dabrafenib) capsules are supplied as 50-mg and 75-mg capsules for oral administration. Each 50-mg capsule contains 59.25 mg dabrafenib mesylate equivalent to 50 mg of dabrafenib free base. Each 75-mg capsule contains 88.88 mg dabrafenib mesylate equivalent to 75 mg of dabrafenib free base.
The inactive ingredients of TAFINLAR are colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose. Capsule shells contain hypromellose, red iron oxide (E172), and titanium dioxide (E171).
Dabrafenib mesylate
1195768-06-9 cas of mesylate
N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide;methanesulfonic acid
Chemical structure

………………….
WO2015003571
达拉菲尼甲磺酸盐的新晶型及其制备方法
Deficiencies of the prior art W, the main object of the present invention is to provide as follows has a better stability in an aqueous or aqueous systems Dara Feeney 曱

For purposes of this invention, the present invention provides Dallas Phoenix mesylate Form IV (hereinafter referred to as “Form IV”) and its preparation method. The type IV crystal is a hydrate; preferably each 摩尔达拉菲 Nepal mesylate contains about 1.5 moles of water.
Using Cu- Κα radiation, the crystalline form IV of X-ray powder diffraction pattern at diffraction angles 2Θ of 4.7 ± 0.2 °, 9.2 ± 0.2. , 12.8 ± 0.2. , 13.8 ± 0.2. 15.0 0.2 soil. And 16.3 ± 0.2 ° of the characteristic peaks.
Preferably, the crystalline form IV of the X-ray powder diffraction pattern at diffraction angles 2Θ of 4.7 ± 0.2. , 9.2 ± 0.2. , 12.8 ± 0 · 2. , 13 · 8 ± 0 · 2 o , 15.0 ± 0.2. , 16.3 ± 0 · 2. , 18.0 ± 0.2. , 18 · 6 ± 0.2. , 20 · 6 ± 0.2. , 22.9 ± 0.2 °, 23.8 ± 0.2. And 24.3 ± 0.2. Department characteristic peaks
Form IV of FIG differential scanning calorimetry (DSC) show: sample 151~ 105 ° C there is a large endothermic peak (solvent peak), the sample after dehydration melting range of 132 ~ 148 ° C, then at 200 ° C ~ 245 ° C with a heat transfer crystal peak at 249 ° C and finally melted.
The crystalline form IV has the following advantageous properties:
1) left at room temperature for one month and stable, stable for 1 month at room temperature for -97% RH;
2) known Form I in water suspension was stirred for 15 minutes into the free base monohydrate; and Form IV in water suspension was stirred for 15 minutes remain for 曱 salt Form IV, After stirring overnight converted to the free base monohydrate Form IV described more conducive to maintaining the solubility of the sample is larger than the free base state 曱 sulfonates, Form IV has a better stability in water / aqueous system or sex.
3) 0 to 22 hours compared to the elution amount, any detection points of Form IV of elution volume than the known polymorph I of the elution amount. Description Form IV has a better solubility and bioavailability.
4) 0 to 120 minutes elution amount compared to any detection points of Form IV gum Nang elution volume than the known polymorph I of the dissolution of glue Nang. Description Form IV gum Nang has better dissolution.
The Form IV was prepared using any one of the following methods:
1) The Dallas Feeney known mesylate polymorph I was dissolved in a mixed solution of tetrahydrofuran Yue alcohol, volatile crystallization, and then the precipitated crystals were separated and dried to obtain the Form IV;
The Yue alcohol and tetrahydrofuran in a volume ratio of 0.1 to 100: 1, preferably 0.5~50: 1, more preferably 0.5~5: 1;
2) The Dallas Phoenix Yue sulfonates known polymorph I was dissolved in acetone, volatile crystallization, and the precipitated crystals
Separated, dried, to give the Form IV;
3) The Dallas Phoenix 曱 known polymorph I salt is dissolved in isopropanol, after the addition of polyacrylic acid, volatile crystallization, and then the precipitated crystals were separated and dried to obtain the Form IV;
The polyacrylic acid in an amount of polymorph I of the known amount of 0.1% wt~10% wt, preferably
0.5% wt ~ 10% wt, more preferably 2% wt ~ 5% wt; an average molecular weight of the polyacrylic acid is 2000-5000.
Preparation of the above three methods, the known Dara Feeney 曱 sulfonate polymorph I at room temperature in an amount corresponding to its solubility in a solution of 0.1 to 1 times, preferably 0.5 to 1 times, more preferably 0.8 to 1 times;
The crystallization temperature of room temperature ~ 40 ° C, preferably at room temperature; the crystallization time is 1~14 days, preferably for two days; the dry, you can not vacuum or pressure, the pressure is preferably less than 0.09Mpa; temperature of 30 ° C ~ 120 ° C, preferably 4 (TC ~ 80 ° C, more preferably 40 ° C ~ 60 ° C; for 10 to 72 hours, preferably 10~48 hours, more preferably from 10- 24 hours;
4) The Dallas Phoenix Yue sulfonate polymorph Form II or V is placed to give the Form IV;
The placement of room temperature ~ 40 ° C, preferably room temperature; placement time from 15 minutes to 7 days, preferably
One day;
5) The temperature rise Dara Feeney 曱 sulfonate polymorph II to 120 ° C and then spontaneously cooled to room temperature to obtain the crystalline form
IV;
The preparation of Form I of Preparation Example 1 known
Methods Patent Document WO2009 / 137391 or CN200980126781.6 Example 58a and 58d known polymorph I. Preparation Specifically:
The N- {3- [5- (2- chloro-4-pyrimidinyl) -2- (1,1-Yue-yl-ethyl) -1,3-thiazol-4-yl] – 2-fluorophenyl 2,6-difluorophenyl sulfonamide (196 mg, 0.364mmol) and 7M ammonia in methanol (8ml, 56mmol) was added to a 25 ml autoclave, heated to 90 ° C for 24 hours, TLC showed the starting material the reaction was complete, The reaction system was cooled to room temperature, the solvent was concentrated and the residue was dry column chromatography to obtain N- {3- [5- (2- amino-4-pyrimidinyl) -2- (1,1-dimethylethyl ) -1,3-thiazol-4-yl] -2-fluorophenyl} -2,6-difluorobenzenesulfonamide 90 mg, yield: 45%.
The N- {3- [5- (2- amino-4-pyrimidinyl) -2- (1,1-Yue-yl-ethyl) -1,3-thiazol-4-yl] -2-fluorophenyl 2,6-difluorophenyl sulfonamide (204 mg, 0.393mmol) in isopropanol (2 mL) was added 曱 acid (0.131 ml, 0.393mmol) and the solution was stirred at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and washed with diethyl ether to give N- [3- [5- (2- amino-4-pyrimidinyl) -2- (t-butyl) -4-thiazol-yl] -2-fluoro phenyl] -2,6-difluorobenzenesulfonamide 曱 sulphonates crystalline solid (221 mg, 87% yield) as a white.
1HNM (400MHz, DMSO-d6)5 ppm 10.85(s, lH)7.92-8.05(m, 1H), 7.56-7.72(m, 1H), 6.91-7.50(m, 7H), 5.83-5.98(m, 1H) , 2.18-2.32(m, 3H) , 1.36(s, 9H)。
Preparation of crystal form obtained X-ray powder diffraction pattern shown in Figure 10. Report is consistent with the patent document WO2009 / 137391 or CN200980126781.6.
DSC chart is shown in Fig. Show: Known polymorphs I melt away as 247 ° C~250 ° C.
TGA spectrum shown in Figure 12. Show: decomposition temperature of 261 ° C.
Example 1
Take 10.02 mg polymorph IV (Example 7 Preparation) in 5 ml glass vial, add 0.5 ml of water, ultrasonic resulting suspension stirred at room temperature for 15 minutes, after centrifugation without drying, the present invention is to obtain crystalline form II. The yield was 10.00 mg; 99% yield.
X-ray powder diffraction pattern shown in Figure 6.
TGA pattern shown in Figure 7. Show: Form II at 50 ° C before the weight loss of about 4.6% (about 1.5 water), 50 ° C ~ 155 ° C 1.4% weight loss (about 0.5 water), the decomposition temperature of 287 ° C.
………………….
PATENT
http://www.google.com/patents/WO2009137391A2?cl=en
WO 2009137391
Example 58a: Λ/-{3-r5-(2-Amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Following a procedure analogous to the procedure described in Example 51, Step B using Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) and heating to 90 0C for 24 h, the title compound, Λ/-{3- [5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 –
7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1.35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Example 58b: Λ/-{3-r5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
19.6 mg of Λ/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 μl_ of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-400C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above. Example 58c: Λ/-{3-r5-(2-amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Step A: methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate

Methyl 3-amino-2-fluorobenzoate (50 g, 1 eq) was charged to reactor followed by dichloromethane (250 ml_, 5 vol). The contents were stirred and cooled to ~15°C and pyridine (26.2 ml_, 1.1 eq) was added. After addition of the pyridine, the reactor contents were adjusted to ~15°C and the addition of 2,6-diflurorobenzenesulfonyl chloride (39.7 ml_, 1.0 eq) was started via addition funnel. The temperature during addition was kept <25°C. After complete addition, the reactor contents were warmed to 20-250C and held overnight. Ethyl acetate (150 ml.) was added and dichloromethane was removed by distillation. Once distillation was complete, the reaction mixture was then diluted once more with ethyl acetate (5 vol) and concentrated. The reaction mixture was diluted with ethyl acetate (10 vol) and water (4 vol) and the contents heated to 50- 55°C with stirring until all solids dissolve. The layers were settled and separated.
The organic layer was diluted with water (4 vol) and the contents heated to 50-55° for 20-30 min. The layers were settled and then separated and the ethyl acetate layer was evaporated under reduced pressure to ~3 volumes. Ethyl Acetate (5 vol.) was added and again evaporated under reduced pressure to ~3 volumes. Cyclohexane (9 vol) was then added to the reactor and the contents were heated to reflux for 30 min then cooled to 0 0C. The solids were filtered and rinsed with cyclohexane (2 x 100 ml_). The solids were air dried overnight to obtain methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2- fluorobenzoate (94.1 g, 91 %).
Step B: Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (490 g, 1 equiv.), prepared generally in accordance with Step A, above, was dissolved in THF (2.45 L, 5 vols) and stirred and cooled to 0-3 0C. 1 M lithium bis(trimethylsilyl)amide in THF (5.25 L, 3.7 equiv.) solution was charged to the reaction mixture followed addition of 2-chloro-4- methylpyrimidine (238 g, 1.3 equiv.) in THF (2.45 L, 5 vols). The reaction was then stirred for 1 hr. The reaction was quenched with 4.5M HCI (3.92 L, 8 vols). The aqueous layer (bootom layer) was removed and discarded.
The organic layer was concentrated under reduced pressure to ~2L. IPAC (isopropyl acetate) (2.45L) was added to the reaction mixture which was then concentrated to ~2L. IPAC (0.5L) and MTBE (2.45 L) was added and stirred overnight under N2. The solids were filtered. The solids and mother filtrate added back together and stirred for several hours. The solids were filtered and washed with MTBE (~5 vol). The solids were placed in vacuum oven at 50 0C overnight. The solids were dried in vacuum oven at 30 0C over weekend to obtain Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (479 g, 72%).
Step C: Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 ml_). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated.
The dichloromethane layers were concentrated to -120 ml_. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 ml_. Dimethylacetamide (270 ml.) was then added to the reaction mixture and cooled to -1O0C. 2,2-Dimethylpropanethioamide (1.3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for -5 minutes between additions. The reaction was warmed to 20-25 0C. After 45 min, the vessel contents were heated to 75°C and held for 1.75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 300C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated.
Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-250C. The organic layers were then concentrated under heat and vacuum to 120 ml_. The vessel contents were then heated to 500C and heptanes (120 ml.) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for -2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 300C to obtain Λ/-{3-[5-(2-chloro-4-pyrimidinyl)- 2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (28.8 g, 80%).
Step D:
Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
In 1 gal pressure reactor, a mixture of Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1- dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 0C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 0C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 0C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HCI to ~pH 5.4-5.5. and added water (1vol). The aqueous layer was removed and discarded at 60-70 0C.
The organic layer was washed with water (1 vol) at 60-70 0C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 0C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 0C for 10 min , then cooled to 2O0C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes.
The reaction mixture was maintained at -65-7O0C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 0C. The slurry was stirred at 65-70 0C for 2-3 hours and then cooled slowly to 0-50C. The product was filtered, washed with EtOAc/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain Λ/-{3-[5-(2- amino-4-pyrimidinyl)-2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (102.3 g, 88%).
Example 58d:
Λ/-{3-r5-(2-amino-4-pyrimidinvn-2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate
 MESYLATE
MESYLATETo a solution of Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (204 mg, 0.393 mmol) in isopropanol (2 ml_), methanesulfonic acid (0.131 ml_, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield).
1H NMR (400 MHz, DMSO-d6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1.36 (s, 9 H). MS (ESI): 520.0 [M+H]+.WO2009137391
…………………………………………………………………
PAPER
ACS Medicinal Chemistry Letters (2013), 4(3), 358-362.
http://pubs.acs.org/doi/abs/10.1021/ml4000063

…………………………………………………………….
Patent
http://www.google.com/patents/WO2014158467A1?cl=en

Step C : N- {3-[5-(2-chloro-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)-l ,3-thiazol-4-yl]- 2-fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged N- {3-[(2-chloro-4-pyrimidinyl)acetyl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 mL). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated. The dichloromethane layers were concentrated to -120 mL. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 mL. Dimethylacetamide (270 mL) was then added to the reaction mixture and cooled to ~10°C. 2,2-Dimethylpropanethioamide (1.3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for ~5 minutes between additions. The reaction was warmed to 20-25 °C. After 45 min, the vessel contents were heated to 75°C and held for 1.75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 30°C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated. Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-25°C. The organic layers were then concentrated under heat and vacuum to 120 mL. The vessel contents were then heated to 50°C and heptanes (120 mL) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for ~2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 30°C to obtain N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (28.8 g, 80%).
Compound B is disclosed and claimed, along with pharmaceutically acceptable salts thereof, as being useful as an inhibitor of BRaf activity, particularly in the treatment of cancer, in PCT patent application PCT/US09/42682. Compound B is embodied by Examples 58a through 58e of the application. The PCT application was published on 12 November 2009 as publication WO2009/137391, and is hereby incorporated by reference.
Suitably, Compound B may be prepared according to the methods below:
Method 1 : Compound B (first crystal form) – N-{3-[5-(2-Amino-4-pyrimidinyl)-2- (1,1 -dimethylethyl)- 1 ,3-thiazol-4-yl]- -fluorophenyl} -2,6-difluorobenzenesulfonamide

A suspension of N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) was heated in a sealed tube to 90 °C for 24 h. The reaction was diluted with DCM and added silica gel and concentrated. The crude product was chromatographed on silica gel eluting with 100% DCM to 1 : 1 [DCM: (9: 1 EtOAc:MeOH)]. The clean fractions were concentrated to yield the crude product. The crude product was repurified by reverse phase HPLC (a gradient of acetonitrile: water with 0.1%TFA in both). The combined clean fractions were concentrated then partitioned between DCM and saturated NaHC03. The DCM layer was separated and dried over Na2S04. The title compound, N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)- l,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1H NMR (400 MHz, DMSO- 6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 – 7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1.35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Method 2: Compound B (alternative crystal form) – N-{3-[5-(2-Amino-4- pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide 19.6 mg of N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 L of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-40°C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above.
Method 3: Compound B (alternative crystal form, large batch) – N-{3-[5-(2-amino- 4-pyrimidinyl)-2-(l , 1 -dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6- difluorobenzenesulfonamide

Step D : N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol-4-yl]-
2-fluorophenyl}-2,6-difluorobenzenesulfonamide
In 1 gal pressure reactor, a mixture of N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide ( 120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 °C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 °C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 °C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HC1 to ~pH 5.4-5.5. and added water (lvol). The aqueous layer was removed and discarded at 60-70 °C. The organic layer was washed with water (1 vol) at 60-70 °C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 °C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 °C for 10 min , then cooled to 20°C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes. The reaction mixture was maintained at ~65-70°C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 °C. The slurry was stirred at 65-70 °C for 2-3 hours and then cooled slowly to 0- 5°C. The product was filtered, washed with EtO Ac/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (102.3 g, 88%>).
MESYLATE
Method 4: Compound B (mesylate salt) – N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide methanesulfonate

To a solution of N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (204 mg, 0.393 mmol) in isopropanol (2 mL), methanesulfonic acid (0.131 mL, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield). 1H NMR (400 MHz, DMSO- 6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1.36 (s, 9 H). MS (ESI): 520.0 [M+H]+.
Method 5: Compound B (alternative mesylate salt embodiment) – N-{3-[5-(2- amino-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)-l ,3-thiazol-4-yl]-2-fluorophenyl} -2,6- difluorobenzenesulfonamide methanesulfonate
N- {3-[5-(2-amino-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)- 1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (as may be prepared according to example 58a) (2.37g, 4.56 mmol) was combined with pre-filtered acetonitrile (5.25 vol, 12.4 mL). A pre-filtered solution of mesic acid (1.1 eq., 5.02 mmol, 0.48 g) in H20 (0.75 eq., 1.78 mL) was added at 20°C. The temperature of the resulting mixture was raised to 50-60°C while maintaining a low agitation speed. Once the mixture temperature reached to 50- 60°C, a seed slurry of N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol- 4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate (1.0 %w/w slurried in 0.2 vol of pre-filtered acetonitrile) was added, and the mixture was aged while agitating at a speed fast enough to keep solids from settling at 50-60°C for 2 hr. The mixture was then cooled to 0-5°C at 0.25°C/min and held at 0-5°C for at 6 hr. The mixture was filtered and the wet cake was washed twice with pre-filtered acetonitrile. The first wash consisted of 14.2 ml (6 vol) pre-filtered acetonitrile and the second wash consisted of 9.5 ml (4 vol) pre-filtered acetonitrile. The wet solid was dried at 50°C under vacuum, yielding 2.39 g (85.1% yield) of product
Dara Phoenix (Dabrafenib) by the British GlaxoSmithKline (GSK) has developed Sisu threonine protein kinase (BRAF) inhibitor, as monotherapy ro ー kinds of clothes capsules for carrying BRAF V600E mutation surgical unresectable melanoma or metastatic melanoma treatment of adult patients, Dara Phoenix mesylate in May 2013 was approved by the US Food and Drug Administration (FDA), and is listed on the United States, the trade name Tafinlar (Da Feina). Since the European Medicines Agency (EMA) Committee for Medicinal Products for human use (CHMP) positive evaluation of Tafinlar, making the drug is expected to become after Roche’s Weiluofeini (Vemurafinib) to enter the European market, following a second BRAF inhibitors.
The chemical name Phoenix Dallas: N- [3- [5- (2- amino-4-pyrimidinyl) -2_ (tert-butyl) ~ ~ thiazol-4-yl] _2_ fluorophenyl] – 2,6_-difluorobenzenesulfonamide.

World Patent No. W02009137391, No. W02011047238 and W02012148588 number reported Dallas and Phoenix and its medicinal value synthesis method of the composition. According to the structural characteristics of Dara Phoenix and its analogues, the synthesis of such substances currently have A, B and C are three routes.

A more common route is the synthetic route, by reaction of 3-amino-2-fluorobenzoate (IX) first and 2,6_-difluorobenzene sulfonyl chloride (III) to amidation reaction occurs sulfonamide intermediate ( X); intermediate (X) with 2-chloro-4-methyl pyrimidine (XI) The condensation reaction occurs under the action of a strong base to give the intermediate (XII); intermediate (XII) to give the intermediate bromo
(XIII); intermediate (XIII) with 2,2_ dimethyl thiopropionamide (VI) to give the cyclized intermediate (XIV); and finally, the intermediate (XIV) by ammonolysis to afford the title compound Dallas Phoenix (I).

Different [0009] B is the first route by reaction of 3-amino-2-fluorobenzoate (IX) amino group protection, and thus condensation, cyclization, and bromo; then be obtained by deprotection of the amino group and the sulfonamide Intermediate (XIV); similarly, the intermediate
(XIV) obtained by ammonolysis target compound Dara Phoenix (I).

c route design features that first aminolysis reaction, and then give the desired product by deprotection and amino sulfonamide reaction. Clearly, this design is suitable for the route of these substituted amino ー aminolysis reaction, and for compounds such as Dallas Phoenix having pyrimidinylamino structure is not applicable. The reason is that if there are two aromatic amino groups will make the final sulfonamide ー reaction step to lose selectivity.

Example IV: the reaction flask was added N- [3- (5- formyl-2-t-butyl-ko -4_ thiazolyl) -2_ fluorophenyl] -2,6_ difluoro benzenesulfonamide (VIII) (5.4g, 11.5mmol), N, N- dimethylformamide dimethyl acetal (DMF-DMA) (2.74g, 23mmol) and xylene 50mL, heated to 140 ° C. About every four hours methanol was distilled out of the resulting reaction system, the reaction takes about 24 hours in total, the end of the reaction was detected by TLC. Cool, add hexane 40mL, have produced a yellow solid, filtered, and dried solids obtained after January nitrate melon (1.36,11.5mmol), sodium hydroxide (0.46g, 11.5mmol) and n-Ding enjoy 5OmL, warmed to 120 ° C, The reaction for 12 inches, TLC the reaction was complete. Cooling, with a crystal precipitated crystallized slowly for 3 inches, and filtered. The filter cake starched water, filtered and dried to yield an off-white solid Dara Phoenix (I) 3.58g, yield 60%.
References
- Gibney, G. T.; Zager, J. S. (2013). “Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies”. Expert Opinion on Drug Metabolism & Toxicology 9 (7): 1. doi:10.1517/17425255.2013.794220. PMID 23621583.
- Huang, T.; Karsy, M.; Zhuge, J.; Zhong, M.; Liu, D. (2013). “B-Raf and the inhibitors: From bench to bedside”. Journal of Hematology & Oncology 6: 30. doi:10.1186/1756-8722-6-30. PMC 3646677. PMID 23617957.
- “GSK melanoma drugs add to tally of U.S. drug approvals”. Reuters. May 30, 2013.
- “Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations” 367 (18). New England Journal of Medicine. November 1, 2012. pp. 1694–703. doi:10.1056/NEJMoa1210093. PMC 3549295. PMID 23020132.
“Dabrafenib/Trametinib Combination Approved for Advanced Melanoma”. OncLive. January 9, 2013.
Updates
Dabrafenib prediction
1H NMR PREDICT

![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_1h.png)
13C NM PREDICT
![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_13c.png)
COSY NMR PREDICT

HMBC, HSBC NMR PREDICT
INTERMEDIATES
INT 1
 Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate;Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, 1195768-23-0
Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate;Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, 1195768-23-0
 methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester;
methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester; methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3
methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, CAS No. 1195768-19-4
Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, CAS No. 1195768-19-4 CAS No. 1042055-86-6 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate
CAS No. 1042055-86-6 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoateSYN 1
![]()
 DABARAFENIB
DABARAFENIB
GLAXOSMITHKLINE LLC; HOOS, Axel; GRESHOCK, Joel Patent: WO2014/66606 A2, 2014 ; Location in patent: Page/Page column 20; 24 ;
SYN 2

![]()
WO2011/47238 A1, ;
SYN 3

![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 4
![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 5
![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 6
WO2011/47238 A1, ;
SYN 7
methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3WO2011/47238
WO2015003571
达拉菲尼甲磺酸盐的新晶型及其制备方法
Brief Description
Figure 1 Form IV of the present invention an X-ray powder diffraction pattern.
Figure 2 is a schematic diagram Form IV of DSC language.
Figure 3 Form IV of the present invention TGA profiles.
Figure 4 is a dynamic water adsorption of Form IV of the invention, FIG.
Figure 5 Form IV of the present invention 1HNMR spectrum.
Figure 6 of the present invention, Form II X-ray powder diffraction pattern.
Figure 7 of the present invention, Form II TGA profiles.
Figure 8 Form III of the present invention an X-ray powder diffraction pattern.
Figure 9 Form V of the present invention, an X-ray powder diffraction pattern.
Figure 58a and in Example 10 in accordance with Patent Document WO2009 / 137391 or in CN200980126781.6
58d method described for the preparation of polymorph I of the known X-ray powder diffraction pattern.
Figure 11 is in accordance with Patent Document WO2009 / 137391 or CN200980126781.6 in the method of Example 58a and 58d described for the preparation of polymorph I of the known DSC pattern.
12 is in accordance with Patent Document WO2009 / 137391 or CN200980126781.6 in the method of Example 58a and 58d described for the preparation of polymorph I of the known TGA profiles.
Figure 13 is a known polymorph I in Comparative Example 1 in various stages XRPD comparison chart with the sample from top to bottom in the order of: Dara Phoenix free base hydrate, a known polymorph I in water was stirred for 15 After minutes to obtain a sample, and a known polymorph I.
Figure 14 Form IV in the present invention Comparative Example 1 each stage XRPD comparison chart with the sample from top to bottom in the order of: Form IV, Form IV in water with stirring for 15 minutes to obtain a sample, Form IV After stirring overnight in water to obtain a sample, as well as the free base of the hydrate Dara Phoenix. Figure 15 is a Comparative Form IV polymorph I of the known elution compared to the situation in Figure 1 (A to Form IV, ■ known Form 1).
Figure 16 is known in the polymorph I of Comparative Example 2 in various stages XRPD comparison chart (figure from top to bottom as follows: Form I is known API by “wet granulation” process of granulation (excluding section 3-step tablet) obtained by the sample, the known polymorphs I and amount of excipients formulated physically mixed formulation obtained sample, lactose monohydrate and microcrystalline cellulose according to Formulation physical sample after mixing, Dara Feeney free base hydrate, as well as known Form 1).
17 is a crystalline form IV according to the present invention in Comparative Example 2 in various stages XRPD comparison chart (from top to bottom as follows: In Form IV according to API “wet granulation” process of granulation (not included in Step 3 tableting) after the sample obtained, Form IV and excipients Formulation amount by physically mixing the obtained sample, the sample lactose monohydrate and microcrystalline cellulose according to Formulation after physical mixing, and Form IV).
 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-{3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide | |
| Clinical data | |
| Trade names | Tafinlar |
|
|
| Legal status | |
| Identifiers | |
| CAS number | 1195765-45-7 |
| ATC code | L01XE23 |
| PubChem | CID 44462760 |
| ChemSpider | 25948204 |
| ChEBI | CHEBI:75045 |
| ChEMBL | CHEMBL2028663 |
| Chemical data | |
| Formula | C23H20F3N5O2S2 |
| Molecular mass | 519.56 g/mol |
