
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Cenicriviroc
TAK-652; TBR-652
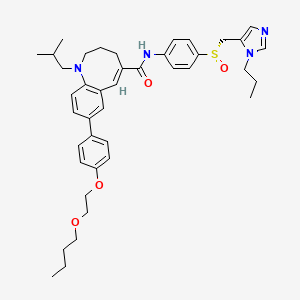
(-)-(S)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-[4-(1-propyl-1H-imidazol-5-ylmethylsulfinyl)phenyl]-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide methanesulfonate
497223-25-3 , Molecular Formula: C41H52N4O4S Molecular Weight: 696.94098
497223-28-6 (mesylate) C41 H52 N4 O4 S . C H4 O3 S, 793.047
Cenicriviroc (TAK-652, TBR-652) is an experimental drug candidate for the treatment of HIV infection.[1] It is being developed by Takeda Pharmaceutical and Tobira Therapeutics.
TBR-652 (formerly TAK-652) is a highly potent and orally active CCR5 antagonist in phase II clinical trials at Takeda for the treatment of HIV infection. Tobira Therapeutics is evaluating the compound in preclinical studies for the treatment of rheumatoid arthritis.
TBR-652 binds CCR5 receptors to interfere with the entry of the HIV-1 virus into macrophages and activated T-cells by inhibiting fusion between viral and cellular membranes. This mechanism of action differs from currently used HIV treatments such as nucleoside reverse transcriptase inhibitors and protease inhibitors.
In 2007, Takeda entered into an agreement with Tobira pursuant to which Tobira obtained exclusive worldwide rights to develop, manufacture and commercialize TBR-652 for the treatment of HIV infection.
Cenicriviroc is an inhibitor of CCR2 and CCR5 receptors,[2] allowing it to function as an entry inhibitor which prevents the virus from entering into a human cell. Inhibition of CCR2 may have an anti-inflammatory effect.
A double-blind, randomized, placebo-controlled clinical study to assess the antiviral activity, safety, and tolerability of cenicriviroc was conducted in 2010. HIV-infected patients taking cenicriviroc had significant reductions in viral load, with the effect persisting up to two weeks after discontinuation of treatment.[3] Additional Phase II clinical trials are underway.[4]
Phase IIb data presented at the 20th Conference on Retroviruses and Opportunistic Infections (CROI) in March 2013 showed similar viral suppression rates of 76% for patients taking 100 mg cenicriviroc, 73% with 200 mg cenicriviroc, and 71% with efavirenz. Non-response rates were higher with cenicriviroc, however, largely due to greater drop-out of patients. A new tablet formulation with lower pill burden may improve adherence. Looking at immune and inflammatory biomarkers, levels of MCP-1 increased and soluble CD14 decreased in the cenicriviroc arms.[5]
Although HIV has been largely rendered a chronic infection, there remains a need for new drugs because of the virus’s propensity to develop resistance to the drugs used to keep it at bay.
Pfizer’s maraviroc was the first drug that acted on the cells to prevent viral entry by antagonising the CCR5 co-receptor. Several others have been investigated and have failed; another that is undergoing clinical trials is Takeda’s cenicriviroc, which has been licensed to Tobira Therapeutics. Unlike maraviroc, the new agent also acts at the CCR2 co-receptor, which is implicated in cardiovascular and metabolic diseases.
In a Phase I double blind, placebo controlled trial designed to study safety, efficacy and pharmacokinetics, treatment-experienced but CCR5 antagonist-naïve patients with HIV-1 were given doses of 25, 50, 75, 100 or 150mg of the drug, or placebo once a day for 10 days.2 The maximum median reductions in HIV-1 RNA values were 0.7, 1.6, 1.8 and 1.7 log10 copies/ml for the respective doses, with a median time to nadir of 10 to 11 days. The effect on CD-4 cell counts was negligible. There was also a significant reduction in levels of monocyte chemotactic protein 1, suggesting that CCR2 was also being blocked. The drug was both generally safe and well tolerated, and no patients withdrew from the trial due to adverse events.
In another Phase I trial, designed to look at pharmacokinetics and pharmacodynamics and carried out in a similar patient population, subjects were given the drug as oral monotherapy for 10 days, again in doses of 25, 50, 75, 100 and 150mg, or placebo.3 The drug was well absorbed into the systemic circulation, and the concentration levels declined slowly, with meant elimination half-lives of one to two days. Potent, dose-dependent reductions in viral load were seen, and again it was generally safe and well tolerated across all levels.
In June 2011, Tobira initiated a multi-centre, double blind, double dummy, 48-week comparative Phase IIb trial in 150 patients with HIV-1 infection. Subjects are being given 100 or 200mg once-daily doses of the drug to evaluate its efficacy, safety and tolerability.
PATENTS
WO 2003014105
WO 2003076411
WO 2005116013
WO 2007144720
WO 2011163389
US 20130079233
WO 2013167743
See also
ancriviroc (formerly known as SCH-C), vicroviroc which has the chemical name (4,6-dimethylprymidine-5-yl){4- [(3S)-4-{(1 R)-2-methoxy-1 -[4-(trifluoromethyl)phenyl]ethyl}-3-methylpiperazin-1 -yl]-4-methylpiperidin-1 – yljmethanone, PRO-140, apliviroc (formerly known as GW-873140, Ono-4128, AK-602), AMD-887, INC- B9471 , CMPD-167 which has the chemical name N-methyl-N-((1R,3S,4S)-3-[4-(3-benzyl-1-ethyl-1H- pyrazol-δ-yOpiperidin-i-ylmethylH-IS-fluorophenyllcyclopent-i-yll-D-valine), methyl1-endo-{8-[(3S)-3- (acetylamino)-3-(3-fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl}-2-methyl-4,5,6,7-tetrahydro-1 H- imidazo[4,5-c]pyridine-5-carboxylate, methyl 3-endo-{8-[(3S)-3-(acetamido)-3-(3-fluorophenyl)propyl]-8- azabicyclo[3.2.1]oct-3-yi}-2-methyl-4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridine-5-carboxylate, ethyl 1- endo-{8-[(3S)-3-(acetylamino)-3-(3-fiuorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl}-2-methyl-4,5,6,7- tetrahydro-1 H-imidazo[4,5-c]pyridine-5-carboxylate and N-{(1S)-3-[3-endo-(5-lsobutyryl-2-methyl-4,5,6,7- tetrahydro-1H-imidazo[4,5-c]pyridin-1-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1-(3-fluorophenyl)propyl}acetamide) and pharmaceutically acceptable salts, solvates or derivatives of the above. The last four compounds are disclosed in WO 03/084954 and WO 05/033107.


http://pubs.acs.org/doi/full/10.1021/jm0509703

Compound (S)-(−)-5b (TAK-652) also inhibited the replication of six macrophage-tropic (CCR5-using or R5) HIV-1 clinical isolates in peripheral blood mononuclear cells (PBMCs) (mean IC90 = 0.25 nM).
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-propyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide ((S)–(−)-5a). The 1 N HCl (160 mL) was added to 1931 (35.68 g, 53.4 mmol), and the mixture was extracted with EtOAc. To the aqueous layer was added 25% aqueous K2CO3 (160 mL), and the mixture was extracted with a mixture of EtOAc and i-PrOH (4:1). The organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo to give (S)-18. To a solution of 16a (18.0 g, 41.1 mmol) and DMF (0.5 mL) in THF (180 mL) was added thionyl chloride (SOCl2) (4.50 mL, 61.7 mmol) at room temperature. After being stirred at room temperature for 1.5 h, the reaction mixture was concentrated in vacuo. A solution of the residue in THF (200 mL) was added dropwise to a mixture of (S)-18 and triethylamine (Et3N) (35.0 mL, 251 mmol) in THF (150 mL) under ice cooling. After being stirred at room temperature for 4 h, water was added to the reaction mixture. The mixture was washed with 10% aqueous AcOH, saturated aqueous NaHCO3, and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on a NH silica gel (hexane/EtOAc = 1:5 → 1:8 → 1:9) to give 21.14 g (75%) of (S)-(−)-5a as a yellow amorphous powder, [α]D = −132.5° (C = 0.507%, EtOH). 1H NMR (300 MHz, CDCl3) δ 0.87−1.03 (9H, m), 1.34−1.49 (2H, m), 1.50−1.85 (8H, m), 2.55−2.65 (2H, m), 3.15−3.25 (2H, m), 3.52−3.58 (4H, m), 3.75−3.83 (4H. m), 4.02 (1H, d, J = 13.8 Hz), 4.08−4.17 (3H, m), 6.56 (1H, d, J = 1.0 Hz), 6.80 (1H, d, J = 8.8 Hz), 6.96 (2H, d, J = 8.8 Hz), 7.31−7.46 (7H, m), 7.55 (1H, s), 7.76 (2H, d, J = 8.8 Hz), 7.98 (1H, s). Anal. (C40H50N4O4S·0.25H2O) C, H, N.
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide methanesulfonate ((S)–(−)-5b). The free base of (S)-(−)-5b was prepared in 80% yield from 16band 19 by a method similar to that described for (S)-(−)-5a. To a solution of the free base of (S)-(−)-5b (64.91 g, 93.1 mmol) in EtOAc (600 mL) was added dropwise a solution of methanesulfonic acid (8.95 g, 93.1 mmol) in EtOAc (160 mL) at room temperature. After being stirred at room temperature for 4 h, the crystals were collected by filtration and washed with EtOAc to give 69.09 g (94%) of (S)-(−)-5b as yellow crystals. The crystals (68.0 g) were purified by recrystallization from 2-butanone to give 58.9 g (85%) of (S)-(−)-5b as yellow crystals, mp 145.5−147.5 °C, [α]D = −191.2° (c = 0.508%, EtOH). 1H NMR (300 MHz, DMSO-d6) δ 0.82−0.97 (12H, m), 1.29−1.39 (2H, m), 1.40−1.55 (4H, m), 1.65−1.85 (2H, m), 2.00−2.25 (1H, m), 2.29 (3H,s), 2.38−2.60 (2H, m), 3.10 (2H, d, J = 7.8 Hz), 3.30−3.60 (4H, m), 3.70 (2H, t, J = 4.8 Hz), 3.98 (2H, t,J = 6.6 Hz), 4.10 (2H, t, J = 4.8 Hz), 4.34 (1H, d, J = 15.0 Hz), 4.68 (1H, d, J = 15.0 Hz), 6.87 (1H, d, J = 8.7 Hz), 6.99 (2H, d, J = 8.7 Hz), 7.16 (1H, s), 7.42−7.60 (8H, m), 7.93 (2H, d, J = 8.7 Hz), 9.05 (1H, s), 10.18 (1H, s). Anal. (C42H56N4O7S2) C, H, N.
…………………
WO 2003014105 OR US20090030032
http://www.google.st/patents/US20090030032?hl=pt-PT&cl=un
EXAMPLE 7 Preparation of Compounds 9 and 10
8-[4-(2-Butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propyl-1H-imidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzazocin-5-carboxamide (317 mg) was resolved by using CHIRAKCEL OJ 50 mm ID×500 mL (hexane/ethanol) to give (−)-8-[4-(2-butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propylimidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzoazocine-5-carboxamide (142 mg) (Compound 9) and (+)-8-[4-(2-butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propylimidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzoazocine-5-carboxamide (143 mg) (Compound 10).
Compound 9
[α]D=−127.4° (C=0.533% in ethanol).
Compound 10
[α]D=+121.0° (C=0.437% in ethanol).
………………………….
WO 2003076411
http://www.google.st/patents/WO2003076411A1?cl=en
http://www.google.st/patents/US20050107606?hl=pt-PT&cl=en

Example 21 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide
To a solution of 8-[4-(2-butoxyethoxy)phenyl]-1-isobutyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid (45 g) in tetrahydrofuran (135 ml) was added N,N-dimethylformamide (230 mg) and added dropwise thionyl chloride (12.45 g) at 10 to 15° C., and the resulting solution was stirred at the same temperature for 40 minutes to prepare an acid chloride.
Separately, to a solution of (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine in tetrahydrofuran (270 ml) was added pyridine (27.59 g), the resulting mixture was adjusted to 5° C. or lower, and then thereto was added dropwise the acid chloride solution at 5° C. or less, and the resulting mixture was stirred at the same temperature for 2 hours. To the mixture were added water (270 ml) and 20% aqueous citric acid solution (180 ml), tetrahydrofuran was distilled off under reduced pressure and the residue was extracted with ethyl acetate. The extract was sequentially washed with water, saturated sodium bicarbonate solution and water, and then the solvent was distilled off. To the residue was added ethyl acetate (360 ml), added heptane (360 ml) at 40° C. and added seed crystals of (−)-8-[4-(2-butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide (10 mg), and the mixture was stirred at 25° C. for 2 hours and stirred at 5° C. for 1 hour. The precipitated crystals were collected by filtration to obtain 63.97 g (yield: 92.1%) of the title compound. Melting point: 120-122° C.
Elemental analysis value: in terms of C41H52N4O4S
Calcd. value: C, 70.66; H, 7.52; N, 8.04.
Analytical value: C, 70.42; H, 7.52; N, 8.01
Industrial Applicability
According to the present invention, an optically active sulfoxide derivative having CCR5 antagonism or an intermediate compound thereof can be prepared without causing side reactions such as racemization and Pummerer rearrangement. In particular, Process 7 is industrially advantageous since it is possible to prepare an optically active Compound (II) by asymmetric oxidization in the presence of an optically active acid.
Example 20 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-propyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide.methanesulfonate
According to the same method as that described in Example 15, the title compound was produced from 8-[4-(2-butoxyethoxy)phenyl]-1-propyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid and (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine.
1H-NMR (CDCl3, δ, 300 MHz) 0.88-1.01 (9H, m), 1.37-1.42 (2H, m), 1.57-1.80 (8H, m), 2.63 (2H, br), 2.77 (3H, s), 3.27 (2H, br), 3.51-3.57 (4H, m), 3.77-3.86 (4H, m), 3.90-4.05 (1H, m), 4.14 (2H, t, J=4.6 Hz), 4.25 (1H, d, J=14.6 Hz), 6.73 (1H, s), 6.84 (1H, d, J=8.7 Hz), 6.93 (2H, d, J=8.8 Hz), 7.21 (2H, d, J=8.7 Hz), 7.40-7.48 (4H, m), 7.61 (1H, s), 7.89 (2H, d, J=8.7 Hz), 8.65 (1H, s), 9.27 (1H, br)
Elemental analysis value: in terms of C41H54N4O7S2
Calcd. value: C, 63.21; H, 6.99; N, 7.19; S, 8.23.
Analytical value: C, 63.00; H, 7.09; N, 7.41; S, 8.25
Example 15 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide.methanesulfonate
8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid (986 mg) was dissolved in tetrahydrofuran (3 ml) and thereto was added N,N-dimethylformamide (one drop). Subsequently, to the resulting solution was added dropwise oxalyl chloride (0.2 ml, 2.29 mmol) under ice-cooling and the mixture was stirred for 80 minutes under ice-cooling to prepare an acid chloride.
Separately, (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine (689 mg) was added to tetrahydrofuran (7 ml) and the resulting solution was cooled to 5° C. To the solution was added dropwise pyridine (0.62 ml) and added dropwise the acid chloride solution at 3 to 5° C., and the mixture was stirred for 2 hours under ice-cooling. To the mixture was added water (20 ml) at 10° C. or lower and the mixture was extracted with ethyl acetate. The organic layer was sequentially washed with water, saturated sodium bicarbonate solution and water, and concentrated under reduced pressure. Thereto was added toluene and the mixture was concentrated under reduced pressure. Thereto was added acetonitrile and the mixture was concentrated under reduced pressure. The residue was dissolved in acetonitrile (7 ml) and acetone (7 ml), thereto was added dropwise methanesulfonic acid (209 mg), and added seed crystals and the mixture was stirred at room temperature for 100 minutes. Subsequently, to the mixture was added acetone-acetonitrile (1:1, 5 ml). After stirring at room temperature overnight, the mixture was stirred for 2.5 hours under ice-cooling. The precipitated crystals were collected by filtration and washed with the ice-cooled acetone (9 ml). The crystals were dried at 40° C. under reduced pressure to obtain 1.51 g (yield: 87%) of the title compound as yellow crystals.
1H-NMR (300 MHz, DMSO-d6, δ): 0.78-0.96 (12H, m), 1.25-1.40 (2H, m), 1.41-1.51 (4H, m), 1.65-1.85 (2H, m), 2.05-2.15 (1H, m), 2.30 (3H, s), 2.35-2.50 (2H, m), 3.05-3.15 (2H, m), 3.30-3.55 (4H, m), 3.65-3.70 (2H, m), 3.90-4.05 (2H, m), 4.05-4.10 (2H, m), 4.30 (1H, d, J=14.73 Hz), 4.65 (1H, d, J=14.73 Hz), 6.85 (1H, d, J=8.97 Hz), 6.97 (1H, d, J=8.79 Hz), 7.17 (1H, s), 7.35-7.75 (6H, m), 7.92 (2H, d, J=8.79 Hz), 9.08 (1H, s), 10.15 (1H, s).
Elemental analysis value: in terms of C41H52N4O4S.CH4SO3
Calcd. value: C, 63.61; H, 7.12; N, 7.06; S, 8.09.
Found value: C, 63.65; H, 7.23; N, 7.05; S, 8.08.
………………………….
References
- Klibanov, Olga M.; Williams, Shannon H.; Iler, Cameron A (2010). “Cenicriviroc, an orally active CCR5 antagonist for the potential treatment of HIV infection”. Current Opinion in Investigational Drugs 11 (8): 940–950. PMID 20721836.
- Baba, Masanori; Takashima, Katsunori; Miyake, Hiroshi; Kanzaki, Naoyuki; Teshima, Koichiro; Wang, Xin; Shiraishi, Mitsuru; Iizawa, Yuji (2005). “TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans”. Antimicrobial Agents and Chemotherapy 49 (11): 4584–4591. doi:10.1128/AAC.49.11.4584-4591.2005. PMC 1280155. PMID 16251299.
- C. Reviriego (2011). Drugs of the Future 36 (7): 511–517. doi:10.1358/dof.2011.36.7.1622066.
- “Tobira Therapeutics Initiates Phase 2b Trial of Cenicriviroc”. The Body. July 5, 2011.
- CROI 2013: CCR5/CCR2 Inhibitor Cenicriviroc Has Both Anti-HIV and Anti-inflammatory Effects. Highleyman, Liz. HIVandHepatitis.com. 7 March 2013.
|
11-26-2012
|
Chemokine receptor antagonists.
|
Journal of medicinal chemistry
|
|
|
6-1-2011
|
Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects.
|
Journal of acquired immune deficiency syndromes (1999)
|
|
|
8-1-2010
|
Cenicriviroc, an orally active CCR5 antagonist for the potential treatment of HIV infection.
|
Current opinion in investigational drugs (London, England : 2000)
|
|
|
3-1-2009
|
The relative activity of “function sparing” HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property?
|
Molecular pharmacology
|
|
|
2-1-2007
|
Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652.
|
Antimicrobial agents and chemotherapy
|
|
|
9-10-2006
|
[Progress in AIDS therapy].
|
Nihon Naika Gakkai zasshi. The Journal of the Japanese Society of Internal Medicine
|
|
|
3-23-2006
|
Highly potent and orally active CCR5 antagonists as anti-HIV-1 agents: synthesis and biological activities of 1-benzazocine derivatives containing a sulfoxide moiety.
|
Journal of medicinal chemistry
|
|
|
11-1-2005
|
TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans.
|
Antimicrobial agents and chemotherapy
|
|
|
1-27-2005
|
Stereoselective synthesis of [L-Arg-L/D-3-(2-naphthyl)alanine]-type (E)-alkene dipeptide isosteres and its application to the synthesis and biological evaluation of pseudopeptide analogues of the CXCR4 antagonist FC131.
|
Journal of medicinal chemistry
|
|
|
1-1-2005
|
TAK-652, a novel CCR5 inhibitor, has favourable drug interactions with other antiretrovirals in vitro.
|
Antiviral therapy
|
……………….
Chemical structures of selected small molecule CCR5 inhibitors. A. Maraviroc (MVC, Selzentry), B. Vicriviroc (VCV), C. Cenicriviroc (TBR-652), D. PF-232798.


