
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
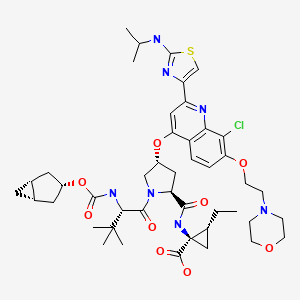
GS 9451, GS-9451, 1098189-15-1 USAN ZZ-81
VEDROPREVIR THERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES 1. Cyclopropanecarboxylic acid, N-[[(1α,3β,5α)-bicyclo[3.1.0]hex-3- yloxy]carbonyl]-3-methyl-L-valyl-(4R)-4-[[8-chloro-2-[2-[(1-methylethyl)amino]- 4-thiazolyl]-7-[2-(4-morpholinyl)ethoxy]-4-quinolinyl]oxy]-L-prolyl-1-amino-2- ethyl-, (1R,2R)-
2. N-{[(1R,3r,5S)-bicyclo[3.1.0]hex-3-yloxy]carbonyl}-3-methyl-L-valyl-(4R)-4-[(8- chloro-2-{2-[(1-methylethyl)amino]thiazol-4-yl}-7-[2-(morpholin-4- yl)ethoxy]quinolin-4-yl)oxy]-L-prolyl-(1R,2R)-1-amino-2- ethylcyclopropanecarboxylic acid
MOLECULAR FORMULA C45H60ClN7O9S
MOLECULAR WEIGHT 910.5 daltons
SPONSOR Gilead Sciences, Inc.
CODE DESIGNATION GS-9451
CAS REGISTRY NUMBER1098189-15-1
WHO NUMBER9745
GS-9451 is a NS3 protease inhibitor in phase II clinical trials at Gilead for the oral treatment of hepatitis C.
…………………………………………………………………………
Discovery of GS-9451: An acid inhibitor of the hepatitis C virus NS3/4A protease Bioorg Med Chem Lett 2012, 22(7): 2629 ……………………………………………………………
PATENTS WO 2012087596 WO 2009005676 WO 2013106631 WO2013101550 ……………………….
WO2012087596A1 Compound 3 can be prepared using synthetic methods and intermediates like those described in USSN 12/215,605 (US 20090257978 A1). Compound 3 can also be prepared described in the following Example. Example 3: Preparation of Compound 3

Compound 315 (12 g, 13 mmol) was dissolved in THF (200 ml), LiOH (11g, 260 mmol) in H20 (200 ml) was added, followed by MeOH (200 ml). The mixture was kept stirring at room temperature for 20 hours. Upon completion of the reaction, 4 N HCI in H20 was added to adjust pH to 7 at 0 °C. The mixture was extracted with EtOAc (2 x 400 ml). The combined organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo to give compound 3 as a yellow solid (11 g, 93%). LC/MS = 911.52(M++1 ). 1H NMR (300MHz, CD3OD)57.95 (d, 1H), 7.90 (s, 1H), 7.48 (s, 1H), 7.31 (d, 1H), 5.42 (s, 1H), 4.37 (dd, 1H), 4.20 (m, 2H), 3.83-3.56 (m, 7H), 3.50 (m, 2H), 3.39 (m, 2H), 2.45 (m, 1H), 2.27(m, 1H), 1.62 (m, 2H), 1.50 (m, 1H), 1.33 (m, 2H), 1.18 (m, 1H), 1.05 (m, 8H), 0.90 (m, 3H), 0.76 (m, 11H), 0.14-0.04 (m, 2H) The intermediate compound 315 was prepared as follows.

301 302 a. Preparation of compound 301. To a dry, argon purged three-neck round bottom flask (1000 mL) were added anhydrous dichloromethane (100 mL) and Et2Zn (28 mL, 273 mmol) at 0 °C. (CAUTION: Source of argon can not be from needle. Use appropriate glass adapter only. A second bubbler can also be attached to the flask to prevent excessive pressure build up.) Cyclopenten-3-ol (10.0 mL, 119 mmol) was then added dropwise (large quantity of ethane gas was produced) to the flask and the reaction mixture was allowed to stir until the evolution of gas had ceased. Diiodomethane (22 mL, 242 mmol) was then added dropwise over a period of 30 minutes. The reaction was allowed to warm to room temperature and continued to stir overnight under a positive flow of argon, at which point TLC analysis had indicated complete disappearance of the starting alcohol. The reaction was then diluted with CH2CI2 and quenched with 2M HCI (white precipitate should be completely dissolved). The biphasic mixture was poured into a separatory funnel and the organic layer was collected. The solvent was removed under reduced pressure until 100 mL of material containing compound 301 remained. b. Preparation of compound 302. Anhydrous dichloromethane (525 mL) was added to the flask followed by the dropwise addition of triethylamine (34 mL, 245 mmol). The reaction continued to stir at room temperature under a positive flow of nitrogen at which point, disuccinimidylcarbonate (40.7 g, 159 mmol) was added to the flask portion wise. The reaction was allowed to stir until TLC analysis indicated complete disappearance of the starting material (2-3 days). Upon completion, the reaction mixture was quenched with 1 M HCI (200 mL x 2) and washed with H20 (200 mL x 2). The desired material was extracted using CH2CI2and the combined organic layers were dried using anhydrous MgS0 and passed through a silica plug. The solvent was removed under reduced pressure and the crude material was purified using flash chromatography (Rf = 0.33, 1 :1 Hex/EtOAc) to provide compound 302 (22 g, 75%): 1H NMR (300 MHz, CDCI3): δ 5. 24 (t, 1 H), 3.82 (s, 4H), 2.24 (m, 2H), 2.03 (d, 2H), 1.38 (m, 2H), 0.48 (m, 1 H), 0.40 (m, 1 H).


c. Preparation of compound 304. N-i-Boc-cis-4-Hydroxy-L-Proline methyl ester 303 (100.0 g, 407.7 mmol) and DABCO (1.5eq, 68.6g, 61 1.6 mmol) were dissolved in anhydrous toluene (200 mL) in a 2 L three necked round bottom flask with a mechanical stirrer and an addition funnel. After cooling the solution to 0 °C under N2, A solution of 4-Bromo-benzenesulfonyl chloride (1.3eq, 135.6g, 530.0 mmol) in 300 mL of toluene was added through addition funnel over 60 minutes. The reaction mixture was stirred and warmed to room temperature overnight (16 hours). The mixture was slowly poured into 2L 1 M Na2C03 (aq.), and the product was extracted with EtOAc (2L). After the organic phase was washed by 0.5 N HCI (2L), H20 (1 L), and brine (1 L), it was dried (MgS04), concentrated to give 195.45 g of a yellow oily brosylate product. To a solution of the above brosylate (407.7 mmol) in dichloromethane (300 mL) was slowly added 4.0 M HCI in dioxane (500 mL, 5eq) and the resulting solution was allowed to stir at room temperature for 2 hours. After ether (500mL) was added to the reaction mixture, the mixture was stirred for 15 minutes and the white precipitate was collected by filtration. The solid was washed with ether and hexane and then dried under vacuum overnight to obtain 153.0 g of the HCI amine salt of compound 304, 381.8 mmol, in 94% yield for two steps. d. Preparation of compound 305. To a solution of Boc-fert-butyl-glycine (97.0g, 420.0 mmol) in DMF (200mL) and DCM (200mL) were added HATU (217.76g, 572.7 mmol) and Hunig’s base (126 mL, 1 145.4 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of the previous HCI salt (153.0 g, 381.8 mmol) and Hunig’s base (126 mL, 1 145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3h, with monitoring by LCMS. The reaction mixture was concentrated to remove dichloromethane under reduced pressure and the white solid that formed was filtered off. The remaining DMF solution was diluted with ethyl acetate (1 L), washed successively with 3% LiCI (aq) (3x650mL), sat’d NH4CI (2x500mL), 0.5N HCI (aq) (2x600ml_), brine (500ml_), sat’d NaHC03 (3x500mL), and brine (500mL). The resulting organic fraction was dried (MgS04) and concentrated to afford compound 305 (111g). e. Preparation of compound 306. To a solution of the methyl ester 305 (120 g, 207.8 mmol) in THF (300 ml_), MeOH (75 mL) was added a solution of LiOH (26.18 g, 623.4 mmol) in H20 (150 ml_). The solution was allowed to stir at room temperature for 4 hours. The mixture was cooled in an ice-bath while acidifying with 3N HCI to pH about 5.5, stirred for 10minut.es, and the resulting white solids were collected by filtration. The solids were washed with more water, ether and hexane. The solids were dried under vacuum at 40°C overnight to give 95.78g (82%) of the acid 306. f. Preparation of compound 307. To a solution of the carboxylic acid 306 (81.4 g, 144.27 mmol) in DMF (200ml_) and dichloromethane (200mL) was added HATU (82.3g, 216.4 mmol) and Hunig’s base (47.5 mL, 432.8 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of amine (158.7 mmol) and Hunig’s base (47.5 mL, 1145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3 hours and monitored by LCMS. After the mixture was concentrated under reduced pressure to remove dichloromethane, the white solids that formed were filtered off. The remaining DMF solution was diluted with ethyl acetate (600mL) and successively washed with 3% LiCI (aq) (2x550mL), sat’d NH4CI (500mL), 1 N HCI (aq) (500mL), sat’d NaHC03(500mL), and brine (300mL). The resulting organic fraction was dried (Na2S04) and concentrated to afford compound 307 (111 g). g. Preparation of compound 308. Compound 307 was dissolved in 4N HCI in dioxane (300 mL) at room temperature and stirred for 2 hours. It was then concentrated under vacuum, and co-evaporated with dichloromethane (2 x 200mL) to dryness. The residue was dissolved in EtOAc (600mL) and sat’d aq. NaHC03 (1 L). It was stirred vigorously. After 10 minutes, carbonic acid bicyclo[3.1.0]hex-3-yl ester 2,5-dioxo-pyrrolidin-1-yl ester 302 (41.4 g, 173.1 mmol) was added in one portion. After the resulting mixture was stirred for another 30 minutes, the organic layer was collected and washed with brine (500mL), dried (Na2S04), and concentrated. The crude product was purified by flash chromatography on silica gel with ethyl acetate/hexane to afford 94.44 g (92%) of compound 308.



h. Preparation of compound 310.1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 309 (70.7 g, 354 mmol) was stirred in 48% aq. HBr (500 mL) at 110 °C for 72 hours. After the mixture was cooled to 0 °C with stirring, the solids were filtered and washed with water. The resulting solids were triturated with a saturated NaHC03 solution (-350 mL), filtered, washed with water, and dried under vacuum to give – 40 g (61%) of crude 310 as a dark brown solid. LC/MS = 186 (M++1). i. Preparation of compound 311. 1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 310 (40 g, 215 mmol) was dissolved in DMF (360 ml). Cesium carbonate (140 g, 430 mmol) was added, followed by bromoacetaldehyde dimethyl acetal (54.5 g, 323 mmol). The mixture was then vigorously stirred at 65 °C for 24 hours. Upon cooling to room temperature, EtOAc (1 L) and H20 (1 L) were added to the mixture. The organic layer was extracted with EtOAc (1 x 400 ml). The combined organic layer was washed with aqueous 3% LiCI solution (2 x 1 L), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give compound 311 as a white solid (39 g, 67%). j. Preparation of compound 312. To a mixture of 1-[2-Amino-3-chloro-4-(2,2-dimethoxy-ethoxy)-phenyl]-ethanone 311 ( 13 g, 47.5 mmol) and isopropylaminothiazole-4-carboxylic acid hydrobromide (12.64 g, 47.5 mmol) in pyridine (150 ml) was slowly added phosphorus oxychloride (9.47 g, 61.8 mmol) at -40 °C. The mixture was then stirred at 0 °C for 4 hours. Upon completion of the reaction, H20 (30 ml) was added dropwise to the mixture. The mixture was then stirred at 0 °C for another 15 minutes. The mixture was concentrated in vacuo. The residue was diluted with EtOAc, washed with a sat. NaHC03 aqueous solution. The organic layer was dried (Na2S04) and concentrated in vacuo. The residue was dissolved in CH2CI2, hexanes were added slowly to the solution, and a yellow solid started to crash out. More hexanes were added until not much product was left in the mother liquid to provide compound 312 (18 g, 85%). k. Preparation of compound 313. 2-lsopropylamino-thiazole-4-carboxylic acid [6-acetyl-2-chloro-3-(2,2-dimethoxy-ethoxy)- phenyl]-amide 312 (18 g, 40.7 mmol) was suspended in toluene (400 ml). NaH (2.4 g, 61 mmol) was added to the vigorously stirred mixture while monitoring H2evolution. The mixture became a clear solution during heating to reflux. The reaction was complete after refluxing for 3 hours. The mixture was cooled to room temperature. A solution of AcOH (69.2 mmol) in H20 (3 vol) was added to the mixture. After vigorous agitation for 1 hour at 0 °C, the solids were collected by filtration, rinsed forward with H20. The wet cake was dried under high vacuum to a constant weight to provide compound 313 ( 5 g, 86%). I. Preparation of compound 314. To a mixture of brosylate intermediate 303 (15 g, 35 mmol) and compound 313 (27.5 g, 38.5 mmol) in NMP (200 ml) was added cesium carbonate (25.1 g, 77 mmol). The mixture was stirred at 65 °C for 5 hours. The reaction was cooled to room temperature and EtOAc (600 ml) and an aqueous solution of 3% LiCI (600 ml) were added to the mixture. The organic layer was washed with aqueous 3% LiCI (1 x 600 ml), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired methyl ester as a yellow solid (23.6 g, 75%). LC/MS = 900. 1 3(M++ 1 ) . m. Preparation of compound 315. Methyl ester 314 (23.6 g, 26 mmol) was dissolved in glacial acetic acid (200 ml), 1.4 N HCI in H20 (75 ml) was added to the solution. The mixture was stirred at 60 °C for 1 hour. Upon completion of the reaction, the mixture was concentrated to remove the solvents, coevaporated with toluene (x 2) to remove residual acetic acid. The residue was then dissolved in EtOAc (500 ml) and sat. NaHC03 aqueous solution (enough to neutralize the mixture) while monitoring C02 evolution. The organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was further dried under high vacuum for 1 h and used as is for the next step. The crude was dissolved in CH2CI2 (360 ml), morpholine (3.4 g, 39 mmol) and sodium triacetoxyborohydride (7.2 g, 34 mmol) were added to the mixture at 0 °C. Then glacial acetic acid (0.47 g, 7.8 mmol) was added dropwise to the mixture. The reaction was complete in 10 minutes at 0 °C. Sat. NaHC03 aqueous solution was added to quench the reaction. After stirring for another 20 minutes, the organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired amine product 315 as a yellow solid (12 g, 50%). LC/MS = 924.63(M++ 1 )
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html

