 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

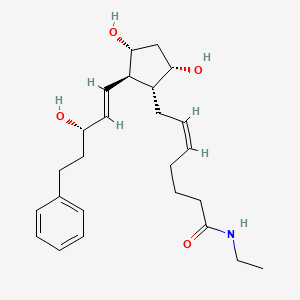
BIMATOPROST
155206-00-1
Bimatoprost ophthalmic solution is a topical medication used for controlling the progression of glaucoma or ocular hypertension, by reducing intraocular pressure. It is a prostaglandin analogue that works by increasing the outflow of aqueous fluid from the eyes. It binds to the prostanoid FP receptor.
Allergan reported Lumigan® sales of US$625 million and Latisse® sales of US$100 million in 2013
| Systematic (IUPAC) name | |
|---|---|
| 7-[3,5-dihydroxy-2- (3-hydroxy-5-phenyl-pent-1-enyl)- cyclopentyl]-N-ethyl-hept-5-enamide | |
| Clinical data | |
| Trade names | Lumigan |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602030 |
| Licence data | US Daily Med:link |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Identifiers | |
| CAS number | 155206-00-1 |
| ATC code | S01EE03 |
| PubChem | CID 5311027 |
| IUPHAR ligand | 1958 |
| DrugBank | DB00905 |
| ChemSpider | 4470565 |
| UNII | QXS94885MZ |
| Chemical data | |
| Formula | C25H37NO4 |
| Mol. mass | 415.566 g/mol |
Bimatoprost (marketed in the U.S., Canada and Europe by Allergan, under the trade name Lumigan) is a prostaglandinanalog/prodrug used topically (as eye drops) to control the progression of glaucoma and in the management of ocular hypertension. It reduces intraocular pressure (IOP) by increasing the outflow of aqueous fluid from the eyes.[1] In December 2008, the indication to lengthen eyelashes was approved by the U.S. Food and Drug Administration (FDA); the cosmetic formulation of bimatoprost is sold as Latisse /ləˈtiːs/.[2] In 2008-2011, at least three case series suggested that bimatoprost has the ability to reduce adipose (fat) tissue.[3][4][5]
Allergen originally developed Bimatoprost and marketed it as Lumigan® for the treatment of elevated intraocular pressure (IOP), with open-angle glaucoma or ocular hypertension. Bimatoprost was later reformulated as a topical formulation and marketed as Latisse® for use in the treatment of hypotrichosis of the eyelashes.
Cosmetic use
In patients using ophthalmic prostaglandins such as travoprost and latanoprost, it has been anecdotally noted[by whom?] that there had been an increase in diameter, density and length of eyelashes. Allergan has initiatedclinical trials investigating the usage of Lumigan as a cosmetic drug.[6] On December 5, 2008, the FDA Dermatologic and Ophthalmic Drugs Advisory Committee voted to approve bimatoprost for the cosmetic use of darkening and lengthening eyelashes.The medical term for this is treatment of hypotrichosis, however, the FDA approval is for purely cosmetic purposes.[7]
For cosmetic purposes, it is administered once daily by applying the solution to the skin at the base of the eyelash using an applicator device “Application Guide”, where it has its effect upon the hair follicle.
Bimatoprost activates prostamide alpha F2 receptors found in the hair follicle to stimulate its growth rate. Research led by Professor Randall and the University of Bradford found that it may also offer a treatment for scalp hair regrowth in trials conducted on samples taken from men undergoing hair transplants.[8]
According to Allergan’s package labeling, users of its Latisse cosmetic product didn’t develop darker irises in clinical studies; however, “patients should be advised about the potential for increased brown iris pigmentation which is likely to be permanent.”[9]
Several cosmetics companies have released products based on prostaglandin analogs, as non-drug cosmetics.
- Age Intervention Eyelash by Jan Marini Skin Research
- RevitaLash by Athena Cosmetics Corp.
These companies have been sued by Allergan for patent infringement.[6] The FDA has seized Age Intervention Eyelash as an “unapproved and misbranded drug” because Jan Marini Skin Research promoted it as something that increases eyelash growth[10] and because it is “adulterated” with bimatoprost.[11]
Fat-reducing properties
Reductions in orbital fat (i.e., fat around the eye) have been observed in patients using bimatoprost as glaucoma therapy.[12] Of particular interest, the loss of orbital fat was unilateral in patients who used bimatoprost on only one eye.[13] The effect appears reversible upon cessation of bimatoprost use. The effect is likely to explain deepening of the lid sulcus described in a series of three patients on bimatoprost.[14] The mechanism for the apparent fat reduction remains unclear. However, bimatoprost is chemically analogous to prostaglandin F2α (PGF2α), a compound which is known to reduce fat by inhibition of adipocyte differentiation and survival.[15]
Formulations
Lumigan is a 0.03% solution of bimatoprost, and contains benzalkonium chloride as a preservative. Contact lenses should therefore be removed before use, and replaced no less than 15 minutes later;[1] other eye drops or ointments should be given no less than five minutes before or after bimatoprost.[1]
Efficacy
Studies have shown once-daily bimatoprost 0.03% ophthalmic solution to be more effective than timolol twice daily in reduction of intraocular pressure (IOP) and as effective as or more effective than the prostaglandin analogues latanoprost and travoprost in reducing IOP.[16]
Side effects
Possible side effects of this medication are:
- May cause blurred vision.
- May cause eyelid redness.
- May permanently darken eyelashes.
- May cause eye discomfort.
- May eventually cause permanent darkening of the iris to brown.
- May cause a temporary burning sensation during use.
- May cause thickening of the eyelashes.
- It may cause unexpected growth of hair if applied inappropriately, on the cheek, for example.
- It may cause infection if the one-time applicators which come with the genuine product are reused.
- Lashes may grow so long that they become ingrown and scratch the cornea.
- May cause darkening of the eyelid or of the area beneath the eye.[17]
On November 19, 2007, the FDA issued a warning during the seizure of a bimatoprost-containing cosmetic.[18] The warning stated that “the extra dose of bimatoprost may decrease the prescription drug’s effectiveness. Damage to the optic nerve may lead to decreased vision and possibly blindness.”
PATENTS
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2585691 | 2009-05-19 | 2026-03-14 |
| Canada | 2144967 | 2003-11-11 | 2013-09-09 |
| United States | 7351404 | 2004-05-25 | 2024-05-25 |
| United States | 6403649 | 1992-09-21 | 2012-09-21 |
Jiang Xing Chen, “Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost.” U.S. Patent US20090287003, issued November 19, 2009.
US20090287003
LUMIGAN® 0.01% and 0.03% (bimatoprost ophthalmic solution) is a synthetic prostamide analog with ocular hypotensive activity. Its chemical name is (Z)-7-[(1R,2R,3R,5S)-3,5Dihydroxy-2-[(1E,3S)-3-hydroxy-5-phenyl-1-pentenyl]cyclopentyl]-5-N-ethylheptenamide, and Its molecular weight is 415.58. Its molecular formula is C24H37NO4. Its chemical structure is:
 |
Bimatoprost is a powder, which is very soluble in ethyl alcohol and methyl alcohol and slightly soluble in water. LUMIGAN® 0.01% and 0.03% is a clear, isotonic, colorless, sterile ophthalmic solution with an osmolality of approximately 290 mOsmol/kg.
LUMIGAN® 0.01% contains Active: bimatoprost 0.1 mg/mL; Preservative: benzalkonium chloride 0.2 mg/mL; Inactives: sodium chloride; sodium phosphate, dibasic; citric acid; and purified water. Sodium hydroxide and/or hydrochloric acid may be added to adjust pH. The pH during its shelf life ranges from 6.8-7.8.
LUMIGAN® 0.03% contains Active: bimatoprost 0.3 mg/mL; Preservative: benzalkonium chloride 0.05 mg/mL; Inactives: sodium chloride; sodium phosphate, dibasic; citric acid; and purified water. Sodium hydroxide and/or hydrochloric acid may be added to adjust pH. The pH during its shelf life ranges from 6.8-7.8.

SYNTHESIS


……………………………….
http://www.google.com/patents/EP2495235A1?cl=en

-
PGF2α prostaglandin synthetic routes so far reported may comprise the following two major steps:

-
The starting compound is generally protected on the secondary hydroxyl group:

where Y stands for p-phenylbenzoyl (PPB) (Ia) or benzoyl (Bz) (Ib) or an analogous substituted aryl group.
-
For example, EP0364417B1 (Kabi Pharmacia AB) describes the following synthetic sequence:

-
The product is obtained as a mixture of epimers where the 15-OH can be in α-position or β-position. The removal of unwanted β-isomer and of other impurities represents one of the main difficulties in the preparation of prostaglandins and many methods have been proposed to reduce the amount of the undesired diastereoisomers formed during the preparation.
-
In EP0544899B1 (Pharmacia AB) the reduction of the α,β-unsaturated ketone is performed with lithium-tri(sec-butyl)borohydride at ―130°C.
-
In this kind of processes the low selectivity in the reduction of the keto group leads to a tedious separation of the diastereomers and generally decreases the global yield of the synthesis.
-
A different method of stereoselective reduction of the ketone is proposed in US7674921 B1 (Cayman Chemical Co.) where the reaction with lithium aluminium hydride in the presence of (S)-binaphtol in tetrahydrofuran at -78°C is reported.
-
An alternative is presented in US6927300B2 and in US 7157590B2(Finetech Lab. Ltd.) where the critical step of the reduction of the α,β-unsaturated keto-group is performed on the compound reported below

where R1 is an aryl carbonyl group in the presence of (-)β-chlorodiisopinocampheylborane in tetrahydrofuran at ―25°C which allows to obtain a ratio of 95/5 of the two diastereoisomers (R)-(IV)/(S)-(IV).

-
A further improvement in the preparation of PGF2α is directed to the reduction of lactone to lactol in order to introduce the α-chain by subsequent Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide.
-
The reaction was usually carried out using diisobutylaluminum hydride at very low temperatures, namely between -80°C and ―40°C. Unfortunately, in these conditions the partial removal of the protecting group PPB or benzoyl (Bz) is possible.
-
To avoid the inconvenient of working with mixtures of products, the p-phenylbenzoyl group or the benzoyl group R is removed by basic hydrolysis and a suitable protecting group is introduced at the two hydroxyl groups of the so obtained compound according to the following scheme:

-
In this way two additional steps of protection and deprotection are introduced possibly leading to a decrease of the global yield of the process at an advanced stage of the synthesis.
-
Moreover when P’ is a silylated group as reported in US7268239B2(Resolution Chemical Ltd.), a mixture of products is obtained because of the migration of the silyl group as shown in scheme 3

-
The same approach of hydrolysis of the aroyl group R followed by diprotection of the two hydroxyl groups with trialkylsilyl group or triaryl silyl group or tetrahydropiranyl group is disclosed in US7642370B2(Daichii Fine Chemical Co.). The protection of the two hydroxyl groups coming before the reduction of the lactone ring to lactol is described also in US7674921B1 (Cayman Chemical Co.) and in US6689901B2(Pharmacia and Upjohn Company).
-
It follows that one important issue in the synthesis of PGF2α is the involvement of intermediates with the most appropriate hydroxyl protecting group. As a consequence the use of the starting Corey lactone carrying a protection able to survive to the conditions of the subsequent reactions allows to form intermediates easy to isolate and to purify. Moreover the protection should be selectively removed in mild conditions.
-
In US5359095 (Pharmacia AB) the preparation of PGF2α prostaglandins is described starting from the commercially available (-)-Corey lactone (Ia) which is oxidized to the corresponding aldehyde (IIa)

-
The reaction is carried out in the presence of dicyclohexylcarbodiimide in dimethylsulfoxide and 1,2-dimethoxyethane; after quenching with orthophosphoric acid the aldehyde is obtained. In the same application it is reported that the crude aldehyde (II) is particularly unstable and must be used within a short period after preparation.
-
In US2010/0010239A1 (Sandoz AG) compound (I) is preferably oxidized in dichloromethane with oxalyl chloride and DMSO; the aldehyde (II) is not isolated and is processed directly in the solution where it is obtained or, when necessary, stored in solution at a temperature between ―20 and 0°C.
-
In US7268239B2 (Resolution Chem. Ltd.) the aldehyde (II)

where Z is (C6-C10)-aryl optionally substituted with one to three substituents independently selected from the group consisting of halo, C1 to C6 alkyl and unsubstituted C6 to C10 aryl, is formed by oxidation of the alcohol with sodium hypochlorite and 2,2,6,6-tetramethtyl-1-piperidinyloxy free radical (TEMPO); in order to avoid the risk of degradation, the aldehyde is directly used in the organic solution where it is synthesized.
-
According to US2009/0287003A1 (Eastar Chem. Corp.) five steps can be performed to prepare the aldehyde to be used as starting material for the synthesis of latanoprost. The five steps are reported in the scheme below:

-
In WO2010/097672 (Sifavitor) the starting material is the Corey lactone where the hydroxyl group is protected astert-butyldimethyl silyl derivative and during the synthesis a second protection is introduced according the scheme reported below referred to the preparation of Bimatoprost:

-
In case Bimatoprost is the end product, compound 9 with A=benzyl (in the following scheme, compound 9a) is directly converted by Wittig reaction to the acid 10 which is then converted in one step to ethylamide affording Bimatoprost, according to the following scheme:
-
In a preferred embodiment, the conversion of compound 10a to Bimatoprost is performed by reaction with ethylamine in an organic solvent, which is chosen among amides, ethers, ketones and chlorinated solvents, more preferably N,N-dimethylformamide, at a temperature between ―35 and 25°C, more preferably between -20°C and -10°C, in the presence of triethylamine and a suitable coupling reagent, which is preferably 1-(methylsulfonyloxy)benzotriazole. The molar ratio between compound 10a and ethylamine is comprised between 1 and 5, more preferably is 3.5, while the molar ratio between compound 10a and the coupling reagent is comprised between 1 and 2.5, more preferably is 1.8.
-
When Latanoprost is the desired product the double bond on the side chain of compound 9a is hydrogenated to form compound 11, then by Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide compound 11 is converted into Latanoprost acid 12. By conversion of the carboxylic acid into isopropyl ester, the final product Latanoprost is obtained:

-
In the case of Travoprost, compound 9 with A=3-(trifluoromethyl)phenoxy (in the following scheme, compound 9b) is converted into 10b, which in turn is converted into Travoprost by esterification of the carboxylic acid by reaction with 2-iodopropane, according to scheme 10:

-














EXAMPLE 13
(Z)-7-((1R,2R,3R,5S)-3,5-dihydroxy-2-((S,E)-3-hydroxy-5-phenylpent-1-enyl)cyclopentyl)hept-5-enoic acid (Bimatoprost free acid)
-

-
4-Carboxybutyltriphenylphosphonium bromide 15 (65.4 g, 0.148 mol) was suspended in tetrahydrofuran (150.0 mL) at 0°C under nitrogen atmosphere. A solution of potassium tert-butoxide in tetrahydrofuran (296.0 mL, 0.296 mol) was added dropwise and the mixture turned into orange. After stirring for 45 minutes at 0°C the system was cooled to ―15°C. A solution of (3aR,4R,5R,6aS)-4-((S,E)-3-hydroxy-5-phenylpent-1-enyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (10.0 g, 0.033 mol) in tetrahydrofuran (46.0 mL) was added dropwise at a temperature lower than – 10°C. After three hours at -15°C no more starting material was visible on TLC and water (200 mL) was added. The mixture was extracted with diisopropyl ether (144 mL) and the layers were separated. The aqueous phase was treated with 0.6 N HCl to pH 6.0. Two extractions with ethyl acetate (2x 250 mL) were then performed and the combined organic layers were concentrated under vacuum at 40°C. An oil (26.80 g) was obtained which was purified on silica gel (eluent: dichloromethane:methanol from 97.5:2.5 to 85:15). The fractions of interest were combined and concentrated at 35°C under reduced pressure affording a colorless oil (11.7 g, 0.030 mol, 91%).
-
1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.29-7.25 (m, 2H, Ph), 7.19-7.15 (m, 3H, Ph), 5.60 (dd, J=7.2, 15.2 Hz, 1H, vinyl), 5.51-5.41 (m, 2H, H vinyl), 5.38-5.31 (m, 1H, vinyl), 4.5-3.8 (m, 7H), 2.67 (m, 2H, -CH 2-Ph), 2.37-1.43 (m, 14H).
-
13C-NMR {400 MHz, CDCl3, δ (ppm)}: 177.2 (C), 141.9 (C), 134.9 (CH), 133.0 (CH), 129.6 (CH), 129.1 (CH), 128.4 (2xCH, arom.), 128.3 (2xCH arom.),125.8 (CH), 77.4 (CH), 72.3 (CH), 72.2 (CH), 55.1 (CH), 50.5 (CH), 42.7 (CH2), 38.5 (CH2), 32.9 (CH2), 31.8 (CH2), 26.3 (CH2), 25.2 (CH2), 24.4 (CH2).
-
HPLC-MS (ESI): [M-H2O+1]+= 371; [M+Na]+ = 411; [2M+Na]+ = 799.

EXAMPLE 14
(5Z)-7-[(2R)-3,5-Dihydroxy-2-[(1E)-3-hydroxy-5-phenylpent-1-en-1-yl]cyclopentyl]-N-ethylhept-5-enamide (Bimatoprost)
-

-
Bimatoprost acid (11.50 g, 0.030 mol) was dissolved in dimethylformamide (92.0 mL), and stirred at -15°C. Triethylamine (7.25 mL, 5.26 g, 0.052 mol) was then added over 5 minutes followed by the portionwise addition of 1-(methylsulfonyloxy)benzotriazole 16 (prepared according to Bulletin of the Chemical Society of Japan, 1978, 51(11), 3320-3329) (11.27 g, 0.053 mol). The mixture was then stirred for one hour at -15°C and an aqueous solution of ethylamine (70% weight, 8.4 mL, 0.104 mol) was added dropwise over 5 minutes. The temperature was allowed to reach 0°C and the reaction was checked by TLC. The mixture was washed with water (172.0 mL) and extracted four times with ethyl acetate (4x 230.0 mL). The combined organic layers were washed with 5% sodium bisulfate solution (100 mL, 50 mL, 50 mL). The bisulfate aqueous phases were extracted with ethyl acetate (50.0 mL). The organic layers were concentrated at 40°C under reduced pressure affording the crude product as an oil (16.98 g). Treatment with dichloromethane and diisopropylether at 0°C for one hour followed by filtration afforded a solid which was then recrystallized from ethyl acetate (6.79 g, 0.016 mol, 55%).
-
1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.26 (m, 2H, Ph), 7.17 (m, 3H, Ph), 6.07 (t, J=5.6Hz, 1H, -NH-), 5.57 (dd, J=7.6, 15.2 Hz, 1H, H-14), 5.45 (dd, J=8.8, 15.2 Hz, 1H, H-13), 5.35 (m, 2H, H-5+H-6), 4.32 (d, J=4.8 Hz, 1H, OH-11), 4.08 (m, 2H, H-9+H-15), 3.90 (m, 1H, H-11), 3.73 (m, 2H, OH-9+OH-15), 3.20 (m, 2H, -N-CH 2-CH3), 2.64 (m, 2H, -CH2-17), 2.30 (m, 1H, H-12), 2.18 (m, 2H, H-7+H-10), 2.11 (m, 3H, CH2-2+H-7), 2.03 (m, 2H, H-4), 1.89 (m, 1H, H-16), 1.77 (m, 2H, H-10+H-16), 1.64 (m, 2H, H-3), 1.45 (m, 1H, H-8), 1.09 (t, J=6.8Hz, 3H, -N-CH2-CH3).
-
13C-NMR {400 MHz, CDCl3, δ (ppm)}: 173.4 (C), 142.0 (C), 135.0 (CH), 133.2 (CH), 129.6 (CH), 129.1 (CH), 128.4 (2xCH arom), 128.3 (2xCH arom), 125.7 (CH arom), 77.5 (CH), 72.22 (CH), 72.20 (CH), 55.4 (CH), 50.1 (CH), 42.8 (CH2), 38.7 (CH2), 35.8 (CH2), 34.3 (-N-CH2), 31.8 (CH2), 26.6 (CH2), 25.6 (CH2), 25.3 (CH2), 14.7 (CH3).
-
HPLC-MS (ESI): [M+Na]+ = 438, [(M-H2O) +H]+=398, [(M-2H2O) +H]+=380.

………………………………..
http://www.google.com.ar/patents/US20090287003
Bimatoprost refers to (Z)-7-[(1R,2R,3R,5S)-3,5-Dihydroxy-2-[1E,3S)-3-hydroxy-5-phenyl-1-pentenyl]cyclopentyl]-5-N-ethylheptenamide, and its molecular weight is 415.58. Its molecular formula is C25H37NO4. Its chemical structure is:


……………………………
http://www.google.com/patents/EP2454227A1?cl=en
Example 1
Synthesis of Bimatoprost

9a-iso

Bimatoprost
Scheme 3. Synthesis of Bimatoprost.
An exemplary synthesis of bimatoprost, a prostaglandin analog, is shown in Scheme 3. The synthesis is scalable, highly convergent and includes a conjugate addition between two chiral synthons, cyclopentenone derivative 6 and vinyl iodide 7a to form ketone 8a. 7a and similar vinyl halides can be prepared in a manner analogous to that shown for the corresponding THP- protected vinyl iodide in U.S. Patent No. 7,109,371 to Clissold et al., or by other methods known in the art. Ketone 8a is reduced to the corresponding isomeric alcohols 9a and 9a-iso, followed by esterification with 5-hexenoic acid to produce ester intermediate 10a. A single isomer of the alcohol can be produced, if desired, by using a stereoselective reducing agent, such as a SELECTRIDE™ (Sigma-Aldrich, St. Louis, Missouri, United States of America) reducing agent. Ring-closure metathesis (RCM) of 10a produced 10- membered ring lactone 11a, which was subsequently deprotected to form lactone 12a. Ring-opening of lactone 12a with ethylamine produced Bimatoprost. The overall yield of Bimatoprost starting from 6 and 7a was good, with each step having a yield of about 60% or greater.
Individual steps in the synthesis of Bimatoprost are described further hereinbelow in Examples 5-10. An alternative step for the synthesis of ketone 8a, using an alkyne reagent, is also shown hereinbelow in Example 13. Ring- opening of 11a prior to deprotection to form a hydroxy-protected Bimatoprost and its subsequent deprotection are described in Examples 11 and 12. As shown below in Schemes 4-6 of Examples 2-4, other exemplary prostanoids were prepared via analogous routes as that shown in Scheme 3. Details regarding individual steps in these syntheses are also shown hereinbelow in Examples 5-10.
Synthesis of Bimatoprost from 12a:
As shown in Scheme 3 in Example 1 , a 250 ml_ 3-necked round-bottom flask equipped with a magnetic bar, a temperature probe, rubber septa, and a nitrogen gas inlet was charged at room temperature with 4.1 g (11.1 mmol) of deprotected lactone 12a in 20 ml_ of THF, 22.2 mi_( 44.3 mmol) of 2 M trimethylaluminum in THF, and 67 ml_ (133 mmol) of 2 M ethylamine in THF. The reaction mixture was heated at 40 °C for 18 h and TLC analysis indicated complete reaction. The mixture was diluted with 50 mL of water and the pH was adjusted to 6 with 1 N HCI. The layers were separated and the aqueous layer was back extracted with 20 mL of ethyl acetate for two times. The combined organic layers were washed with 40 mL of brine, dried over sodium sulfate, filtered, and concentrated.
The crude product was triturated with 20 mL of MTBE at 35 0C for 3 h, cooled to room temperature, and filtered to obtain 3.1 g (67.3% yield) of Bimatoprost, confirmed by 1H NMR.
Example 11
Synthesis of Protected Bimatoprost

Protected Bimatoprost
Scheme 7. Synthesis of Protected Bimatoprost.
As shown in Scheme 7 above, a 250 mL 3-necked round-bottom flask, equipped with a magnetic stirring bar, a temperature probe, rubber septa, and nitrogen inlet, was charged at room temperature, under nitrogen, with 3.0 g (5.01 mmol) of compound 11a in 3O mL of THF, and 15mL ethylamine, 2.0 M in THF. The mixture heated at 40 °C for 24 h and then reflux for another 3 h. TLC analysis (hexanes/ethyl acetate, 10:1) indicated complete reaction. The mixture was cooled to room temperature and diluted with 30 mL of MTBE and 25 mL of water. The layers were separated and the aqueous layer was washed with 15 mL of MTBE. The combined organic extracts were washed with 25 mL of brine, dried over sodium sulfate, filtered, concentrated, and chromatographically purified to afford 2.90 g (90.0% yield) of bis-silylated bimatoprost, confirmed by 1H NMR.
Example 12
Deprotection of Protected Bimatoprost

(8)
Protected Bimatoprost
Bimatoprost Scheme 8. Deprotection of Protected Bimatoprost.
As shown in Scheme 8 above, a 250 ml_ 3-necked round-bottom flask, equipped with a magnetic stirring bar, a temperature probe, rubber septa, and nitrogen inlet, was charged at room temperature, under nitrogen, with 3.Og (3.11 mmol) of protected bimatoprost from Example 12, 30 ml. of THF1 and 0.9 g (90.8 mmol) of ammonium hydrogen difluoride. The reaction mixture was heated at 40 0C for 24 h and TLC analysis (hexanes/ethyl acetate, 1 :1) indicated complete reaction. The mixture was then diluted with 30 ml. of MTBE and 25 ml_ of water followed by layer separation. The aqueous layer was back extracted with 15 ml_ of MTBE. The combined organic extracts were washed with 25 mL of brine, dried over sodium sulfate, filtered, concentrated, and chromatographically purified to afford 1.02 g (80.0% yield) of bimatoprost confirmed by 1H NMR.
………………………………
http://www.google.com/patents/WO2013186550A1?cl=en

Example 7 – Experimental procedures for the synthesis of bimatoprost
A synthesis of bimatoprost is shown and described below.

bimatoprost (97) 7A. (±)-5-Phenyl-l-(trimethylsilyl)pent-l-yn-3-ol, 81

Following a modified procedure of Matsuda (Matsuda, F. Et al., C em. Eur. J. 5, 3252-3259 (1999)); n-butyllithium (1.6 M in hexanes, 8.8 ml, 14.1 mmol, 1.0 eq.) was added dropwise to a solution of ethynyltrimethylsilane (2.0 ml, 14.1 mmol, 1.0 eq.) in THF (6 ml) at -78 °C. After addition, the mixture was allowed to warm slowly to 0 °C and stirred for 1 h. The mixture was cooled to -78 °C and a solution of hydrocinnamaldehyde (2.2 ml, 17.0 mmol, 1.2 eq.) in THF (3 ml) was added dropwise. The mixture was then allowed to slowly warm to -10 °C and stirred for 1 h before being quenched by the addition of saturated NH4CI solution (10 ml) followed by EtOAc (10 ml). The aqueous layer was extracted with EtOAc (3 x 10 ml), the combined organic phases were washed with brine (15 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/EtOAc (100:5), giving the title product 81 as a clear, colourless oil (3.1 g, 94 %). Analytical data consistent with the literature (Matsuda, F. Et al., Chem. Eur. J. 5, 3252-3259 (1999)). max (filmVcm-1 3326 (broad), 2951, 2172, 1604, 1496, 1454, 1249, 1045, 838
*H NMR (400 MHz; CDCI3) δΗ = 0.23 (s, 9H, 3 x CH3), 2.07 (m, 3H, CH2, OH), 2.84 (t, J = 7.8 Hz, 2H, CH2), 4.40 (t, J = 6.6 Hz, 1H, CHO ), 7.25 (m, 3H, ArCH’s), 7.33 (app t, J = 7.3 Hz, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 0.0 (3 x CH3), 31.6 (CH2), 39.3 (CH2), 62.3 (CH), 90.0 (C), 106.7 (C), 126.1 (ArCH), 128.6 (2 x ArCH), 128.7 (2 x ArCH), 141.4 (ArC)
m/z (ESI+) 255.1 [MNa]+, 215.1
7B. Methyl 3-phenylpropanoate, 82

82 Hydrocinnamic acid (10.0 g, 66.6 mmol, 1 eq.) was dissolved in methanol (90 ml), cone. H2S04 (1 ml) added dropwise with stirring and the reaction mixture was stirred under reflux for 5 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in water (100 ml) and extracted with EtOAc (3 x 50 ml). The combined organic phases were washed with 10 % NaHC03 aq. (2 x 50 ml), brine (50 ml) before being dried (MgS04), filtered and concentrated to give the title compound 82 (10.8 g, 99 %) as a clear, colourless oil. Analytical data consistent with the literature (Black, P. J. et al., Eur. J. Org. Chem. 4367-4378 (2006)). vmax (filmVcm-1 3028, 2952, 1734, 1436, 1194, 1160, 749, 697
*H NMR (400 MHz; CDCI3) δΗ = 2.68 (t, J = 7.8 Hz, 2H, CH2), 3.00 (t, J = 7.8 Hz, 2H,
CH2), 3.71 (s, 3H, CH3), 7.25 (m, 3H, ArCH’s), 7.33 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 31.1 (CH2), 35.8 (CH2), 51.7 (CH3), 126.4 (2 x ArCH),
128.4 (ArCH), 128.6 (2 x ArCH), 140.6 (ArC), 173.5 (C=0)
m/z (CI+) 165.1 [MH]+ (20%), 133.1 (100%), 105.1 (55%), 93.1 (51%), 85.0 (57%)
7C. /V-Methoxy-/V-methyl-3-phenylpropanamide, 83

Following a procedure of Trost (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754
(2006)); to a slurry of Λ/,Ο-dimethylhydroxylamine hydrochloride (4.91 g, 50.4 mmol, 2.1 eq.) in toluene (50 ml) at -10 °C was added AIMe3 (2 M in hexanes, 25.2 ml, 50.4 mmol, 2.1 eq.) dropwise. After addition, the mixture was allowed to warm to r.t. and stirred for 1 h. The mixture was cooled to -5 °C and a solution of methyl 3-phenylpropanoate 82 (3.94 g, 24.0 mmol, 1 eq.) in toluene (40 ml) was added dropwise. The reaction mixture was then allowed to warm slowly to r.t. and stirred for 3 h. The solution was cooled to 0 °C and quenched carefully by dropwise addition of HCI and the reaction mixture was extracted with EtOAc (4 x 70 ml). The combined organic phases were washed with brine (50 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/EtOAc (80:20 to 75:25), giving the Weinreb amide 83 as a clear, colourless oil (4.45 g, 97 %). Analytical data consistent with the literature (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754 (2006); Murphy, J. A. et al., Org. Lett. 7, 1427-1429 (2005)). max (film cm-1 3017, 2937, 1659, 1453, 1414, 1383, 1176, 988, 750
*H NMR (400 MHz; CDCI3) δΗ = 2.74 (t, J = 7.8 Hz, 2H, CH2), 2.97 (t, J = 7.8 Hz, 2H, CH2), 3.18 (s, 3H, CH3), 3.60 (s, 3H, CH30), 7.25 (m, 5H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 30.7 (CH3), 32.2 (CH2), 33.8 (CH2), 61.2 (CH30), 126.1
(ArCH), 128.4 (4 x ArCH), 141.3 (ArC), 173.7 (C=0)
m/z (CI+) 194.1 [MH]+ (100%), 164.1 (20%), 133.1 (12%)
7D. 5-Phenyl-l-(tri/sopropylsilyl)pent-l-yn-3-one, 84

Following a procedure of Trost (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754
(2006)); /7-Butyllithium (2.5 M in hexanes, 10.4 ml, 26 mmol, 1.7 eq.) was added dropwise to a solution of tri/sopropylsilyl acetylene (5.8 ml, 26 mmol, 1.7 eq.) in THF (53 ml) at -78 °C. After addition, the mixture was allowed to warm slowly to 0 °C and stirred for 1 h. The mixture was cooled to -78 °C and a solution of 83 (3.0 g, 15.3 mmol, 1 eq.) in THF (20 ml) was added dropwise. The mixture was then allowed to slowly warm to -10 °C and stirred for 1 h before being quenched by the addition of saturated aq. NH4CI (50 ml). The mixture was extracted with EtOAc (3 x 30 ml) and the combined organic phases were washed with brine (50 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (100:0 to 98:2), giving the title product 84 as a clear, colourless oil (4.61 g, 96 %). max (filmycnr1 2944, 2866, 1675, 1462, 1445, 1103, 881, 696
*H NMR (400 MHz; CDCI3) δΗ = 1.12 (m, 21 H, 6 x CH3, 3 x CH), 2.92 (m, 2H, CH2), 3.03 (m, 2H, CH2), 7.22 (m, 3H, ArCH’s), 7.31 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 11.1 (3 x CH), 18.6 (6 x CH3), 30.2 (CH2), 47.4 (CH2), 96.2 (C), 104.2 (C), 126.4 (ArCH), 128.4 (2 x ArCH), 128.7 (2 x ArCH), 140.4 (ArC), 186.7 (C=0) m/z (CI+) 315.3 [MH]+ (100%), 271.2 52%), 157.2 (27%)
HRMS (CI+) calcd for C20H3iOSi [MH]+ 315.2144, found 315.2139
7E. (S)-5-Phenyl-l-(tri/sopropylsilyl)pent-l-yn-3-ol, 85

Following a modified procedure of Trost (Trost, B. M. et al., J. Am. C em. Soc. 128, 6745- 6754 (2006)); Potassium hydroxide (8.5 mg, 0.15 mmol, 1.2 mol%) and RuCI(p- cymene)[(S,S)-Ts-DPEN] (80.7 mg, 0.127 mmol, 1 mol%) were added to /PrOH (110 ml) and the resultant mixture was stirred for 2 min at r.t.. 5-Phenyl-l-(tri/sopropylsilyl)pent-l-yn-3-one 84 (4.0 g, 12.7 mmol, 1 eq.) was added via syringe and the mixture was stirred at r.t. for 45 min. The mixture was concentrated in vacuo to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (20:1 to 10:1), giving the title product 85 as a clear, colourless oil (4.0 g, 99 %). The enantiomeric excess was determined to be 99 % via HPLC analysis of its derivative 87. Analytical data consistent with the literature (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754 (2006)). max (filmVcm-1 3322 (broad), 2942, 2864, 1462, 1045, 1011, 996, 882, 697, 675
*H NMR (300 MHz; CDCI3) δΗ = 1.07-1.11 (m, 21 H, 6 x CH3, 3 x CH), 1.83 (d, J = 5.5 Hz, 1H, OH), 2,03 (m, 2H, CH2), 2.82 (t, J = 7.9 Hz, 2H, CH2), 4.40 (dd, J = 6.4, 5.5 Hz, 1H, CHOH), 7.17-7.32 (m, 5H, ArCH’s)
13C NMR (75 MHz; CDCfe) 5C = 11.2 (3 x CH), 18.7 (6 x CH3), 31.6 (CH2), 39.7 (CH2), 62.4 (CH), 86.1 (C), 108.5 (C), 126.1 (ArCH), 128.6 (4 x ArCH), 141.5 (ArC)
m/z (ESI+) 339.2 [MNa]+, 299.2., 225.0
HRMS (ESI+) calcd for C20H32OSiNa [MNa]+ 339.2114, found 339.2127
[a]D 22 + 28.5 (c. 2.0, CHCI3) (lit., +27.17 (c. 2.14, CH2CI2)) 7F. (S)-5-Phenylpent-l-yn-3-ol, 86

Following a modified procedure of Trost (Trost, B. M. et al., J. Am. C em. Soc. 128, 6745- 6754 (2006)); Tetrabutylammonium fluoride (1.0 M in THF, 25 ml, 25 mmol, 2.5 eq.) was added to a solution of (S)-5-phenyl-l-(tri/sopropylsilyl)pent-l-yn-3-ol 85 (3.165 g, 10 mmol, 1 eq.) in THF (95 ml). The reaction mixture was stirred for 1 h at r.t. and then quenched by addition of saturated aq. NH4CI (50 ml). The mixture was extracted with Et20 (3 x 40 ml), the combined organic phases were washed with brine (50 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (10: 1 to 9:1), giving the title product 86 as a clear, colourless oil (1.6 g, 99 %). max (filmVcm-1 3289 (broad), 1603, 1496, 1454, 1300 (broad), 1040, 1010, 744
*H NMR (400 MHz; CDCI3) δΗ = 2.10 (m, 3H, CH2, OH), 2.55 (d, J = 2.2 Hz, 1H, C≡CH), 2.85 (t, J = 7.8 Hz, 2H, CH2), 4.40 (m, 1H, CHOH), 7.25 (m, 3H, ArCH’s), 7.33 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 31.4 (CH2), 39.2 (CH2), 61.7 (CHOH), 73.5 (C≡CH), 84.8 (C≡CH), 126.2 (2 x ArCH), 128.6 (3 x ArCH), 141.2 (ArC)
m/z (CI+) 161.1 [MH]+ (15%), 155.1 (37%), 143.1 (88%), 119.1 (42%), 91.1 (100%) HRMS (CI+) calcd for CnH130 [MH]+161.0966, found 161.0972
[a]D 22 + 20.0 (c. 2.0, CHC )
7G. (S)-5-Phenyl-l-(trimethylsilyl)pent-l-yn-3-ol, 87
1) nBuLi, THF
-78 °C to 0 °C
OH 2) Me3SiCI OH
3) Citric acid
86 87 Following a procedure of Trost (Trost, B. M. et al., J. Am. C em. Soc. 128, 6745-6754 (2006)); /7-butyllithium (2.5 M in hexanes, 474 pL, 1.18 mmol, 3 eq.) was added dropwise to a solution of (S)-5-phenylpent-l-yn-3-ol 86 (63.3 mg, 0.39 mmol, 1 eq.) in THF (1 ml) at -78 °C. The mixture was allowed to warm to 0 °C and stirred 30 min before being cooled to -78 °C. TMSCI (148.2 pL, 1.18 mmol, 3 eq.) was added dropwise and the mixture was allowed to warm to r.t. and stirred for 2 h. Citric acid (65 mg) in methanol (0.7 ml) was added and the mixture stirred for 1 h. The mixture was poured into a mixture of brine (3 ml) and Et20 (5 ml). The aqueous layer was extracted with Et20 (3 x 10 ml) and the combined organic phases were washed with brine (10 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (9:1), giving the title product 87 as a clear, colourless oil (18.5 mg, 15.5 %). Analytical data consistent with the literature (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754 (2006)). vmax(filmVcm-1 3344 (broad), 2956, 2172, 1496, 1454, 1249, 1046, 838
*H NMR (400 MHz; CDCI3) δΗ = 0.19 (s, 9H, 3 x CH3), 1.80 (d, J = 5.6 Hz, 1H, OH), 2.02 (m, 2H, CH2), 2.80 (t, J = 7.8 Hz, 2H, CH2), 4.36 (dd, J = 6.3, 5.6 Hz, 1H, CHOH), 7.20 (m, 3H, ArCH’s), 7.29 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = -0.1 (3 x CH3), 31.4 (CH2), 39.2 (CH2), 62.2 (CHOH), 89.9 (C), 106.4 (C), 125.9 (ArCH), 128.4 (2 x ArCH), 128.5 (2 x ArCH), 141.3 (ArC)
m/z (ESI+) 255.1 [MNa]+
HRMS (ESI+) calcd for Ci4H20OSiNa [MNa]+ 255.1175, found 255.1185
[a]D 22 + 28.6 (c. 0.735, CHCI3) (lit., +32.91 (c. 2.37, CH2CI2))
Chiral-HPLC data: ee = >99 % (Chiralcel OD column, 210 nm, hexane/2-propanol: 90/10, flow rate: 0.7 mlVmin, room temperature; ¾: minor 15.5 min, major 10.7 min)
7H. (S)-tert-Butyldimethyl(5-phenylpent-l-yn-3-yloxy)silane, 88

Following a procedure of Noyori (Suzuki, M. et al., J. Med. Chem. 41, 3084-3090 (1998)); Imidazole (919.1 mg, 13.5 mmol, 1.8 eq.) and t-butylchlorodimethylsilane (1.35 g, 9.0 mmol) were added to a solution of (S)-5-phenylpent-l-yn-3-ol 86 (1.2 g, 7.5 mmol, 1 eq.) in CH2CI2 (18 ml), cooled to 0 °C. The reaction mixture was then stirred at room temperature for 14 h before being poured into 1 M HCI (50 ml). The mixture was extracted with 40/60 petroleum ether (3 x 50 ml). The combined organic phases were washed with brine (50 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (99:1), giving the title product 88 as a clear, colourless oil (1.77 g, 86 %). Analytical data consistent with the literature (Kiyotsuka, Y. et al., Org. Lett. 10, 1719-1722 (2008).; Sato, F. et al., EP 1211241 Al, Taisho Pharmaceutical co., LTD (2002)). max (filmVcm-1 3309, 2954, 2929, 2886, 2857, 1251, 1091, 834, 776
*H NMR (400 MHz; CDCI3) δΗ = 0.15 (s, 3H, CH3), 0.17 (s, 3H, CH3), 0.95 (s, 9H, 3 x CH3), 2.04 (m, 2H, CH2), 2.46 (d, J = 2.1 Hz, C≡CH), 2.81 (m, 2H, CH2), 4.41 (dt, J = 6.3, 2.1 Hz, CHOTBDMS), 7.24 (m, 3H, ArCH’s), 7.32 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = -4.9 (CH3), -4.4 (CH3), 18.3 (C), 25.9 (3 x CH3), 31.4
(CH2), 40.3 (CH2), 62.2 (CHOTBDMS), 72.5 (C≡CH), 85.4 (C≡CH), 125.9 (ArCH), 128.5 (2 x ArCH), 128.6 (2 x ArCH), 141.7 (ArC)
m/z (CI+) 275.2 [MH]+ (12%), 259.1 (37%), 217.1 (50%), 189.1 (38%), 143.1 (100%) HRMS (CI+) calcd for Ci7H27OSi [MH]+275.1831, found 275.1825
[a]D 22 -17.0 (c. 2.0, CHCI3)
71. (S,£)-teif-Butyl(l-iodo-5-phenylpent-l-en-3-yloxy)dimethylsila

A flame dried Schlenk flask, evacuated and purged with nitrogen, was charged with alkyne 88 (1.50 g, 5.46 mmol, 1 eq.). Anhydrous CH2CI2 (35 ml) was added and the reaction stirred at r.t. Zr(Cp)2HCI (2.82 g, 10.9 mmol, 2 eq.) was added as a solid, in portions. The yellow suspension was stirred at r.t. for 1 h. The resulting yellow solution was cooled to 0 °C and iodine (1.52 g, 6.01 mmol, 1.1 eq.) added as a solid, in one portion. The cooling bath was removed and the reaction mixture stirred at room temperature for 1 h. The reaction mixture was poured into water (100 ml) and extracted with 40/60 petroleum ether (4 x 50 ml). The combined organic phases were washed with water (100 ml), saturated Na2S203 solution (2 x 100 ml) and brine (100 ml) before being dried (MgS04), filtered, and concentrated to give the crude material. This was purified by flash chromatography, eluting with 40/60 petroleum ether. The fractions containing product were combined and washed with saturated Na2S203 solution (20 ml), dried (MgS04), filtered, and concentrated to give the title compound 89 (1.98 g, 90%) as a clear, colourless oil. Analytical data consistent with the literature (Sato, F. et al., EP 1211241 Al, Taisho Pharmaceutical co., LTD (2002)). max (filmVcm-1 2952, 2928, 2856, 1604, 1360, 1251, 1086, 942, 833, 774
*H NMR (400 MHz; CDCI3) δΗ = 0.04 (s, 3H, CH3), 0.06 (s, 3H, CH3), 0.92 (s, 9H, 3 x CH3), 1.82 (m, 2H, CH2), 2.65 (m, 2H, CH2), 4.15 (dq, J = 6.0, 1.2 Hz, 1H, C /-/OTBDMS), 6.25 (dd, J = 14.3, 1.2 Hz, 1H, CH=CHI), 6.57 (dd, J = 14.3, 6.0 Hz, 1H, CH=CHI), 7.18 (m, 3H, ArCH’s), 7.29 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = -4.9 (CH3), -4.5 (CH3), 18.2 (C), 25.8 (3 x CH3), 31.0 (CH2), 39.1 (CH2), 74.6 (CHOTBDMS), 76.1 (CH=CHI), 125.8 (ArCH), 128.3 (2 x ArCH), 128.4 (2 x ArCH), 141.8 (ArC), 148.8 (CH=CHI)
m/z (ESI+) 425.1 [MNa]+
HRMS (ESI+) calcd for Ci7H27IOSiNa [MNa]+ 425.0768, found 425.0754
[a]D 22 -4.5 (c. 2.0, CHCI3)
73. tert-Butyl((S,l£)-l-((3aR,4R,6aS)-2-methoxy-5- ((trimethylsilyloxy)methylene)hexahydro-2H-cyclopenta[d]furan-4-yl)-5- phenylpent-l-en-3-yloxy)dimethylsilane, 92

90 92 Vinyl iodide 89 (1.45 g, 3.6 mmol, 1.2 eq.) was added via syringe to a flame dried Schlenk flask (evacuated and purged with nitrogen several times and allowed to cool). Anhydrous Et20 (14.5 ml) was added via syringe and the resulting solution cooled to -78 °C. t-BuLi (1.6 M, 4.5 ml, 7.2 mmol, 2.4 eq.) was added dropwise and the reaction mixture stirred at -78 °C for 2 h and -40 °C for 2 h before being cooled back to -78 °C. Meanwhile, thiophene (303 mg, 288 μΙ, 3.6 mmol, 1.2 eq.) was added via syringe to a flame dried Schlenk flask (evacuated and purged with nitrogen several times and allowed to cool). Anhydrous THF (14.5 ml) was added via syringe and the resulting solution cooled to -30 °C. n-BuLi (1.6 M, 2.25 ml, 3.6 mmol, 1.2 eq.) was added dropwise and the solution stirred at -30 °C for 30 min. The solution was then cooled to -78 °C and CuCN (322.4 mg, 3.6 mmol, 1.2 eq.) added as a solid, in one portion. The cooling bath was removed and the suspension allowed to warm to r.t. The resulting tan/brown solution of cuprate was added dropwise via syringe to the Schlenk flask containing the vinyl lithium and anhydrous THF (14.5 ml) added. The mixture was stirred at -20 °C for 1 h to allow formation of mixed cuprate 90. This was cooled to -78 °C and a solution of enal 24 (504.6 mg, 3.0 mmol, 1.0 eq.) in anhydrous THF (14.5 ml) was added dropwise. The mixture was stirred at -78 °C for 1 h and then allowed to warm slowly to -20 °C. TMSCI (2.2 ml) was added via syringe followed by NEt3 (2.8 ml). The reaction was quenched by the addition of saturated NH4CI solution (80 ml) and extracted with Et20 (3 x 80 ml). The combined organic phases were washed with saturated NH4CI solution (40 ml) before being dried (MgS04), filtered, and concentrated to give the crude material as a yellow oil. This was used directly in the next step.
7K. (3aR 4R,5R,6aS)-4-((S,£)-3-(tert-Butyldimethylsilyloxy)-5-phenylpent-l- enyl)-2-methoxyhexahydro-2H-cyclopenta[d]furan-5-ol, 93

92 93
The crude material from the conjugate addition / trapping experiment, containing 92, was dissolved in C^Cb/MeOH (3:1) (32 ml) and cooled to -78 °C. A stream of ozone was passed through the stirred solution. The reaction was monitored periodically by TLC in order to judge completion of the ozonolysis (judged by consumption of silyl enol ether). At this point, the flask was purged with a stream of nitrogen during 10 min and NaBH4 (204 mg, 5.4 mmol) was added in one portion. The reaction mixture was stirred at -78 °C for 2 h before the cooling bath was removed and the reaction allowed to warm to r.t.. The reaction was stirred at r.t. for 1 h. The reaction mixture was poured into saturated NaCI solution (20 ml) and extracted with EtOAc (3 x 40 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the crude product as a pale yellow oil. This was purified by column chromatography on silica, eluting with petrol/EtOAc (9:1 to 8:2), giving the alcohol 93 (as a mixture of diastereoisomers) as a clear, colourless oil (731.0 mg, 56.0% (2 steps from enal 24)). max (filmVcm-1 3434 (broad), 2928, 1496, 1471, 1454, 1360, 1250, 1098, 1044, 1003, 970, 834, 774
*H NMR (400 MHz; CDCI3)
δΗ = (mixture of 2 diastereoisomers, signals of minor diastereoisomer indicated by *) 0.05 (s, 3H, CH3), 0.06* (s, 3H, CH3), 0.07 (s, 3H, CH3), 0.08* (s, 3H, CH3), 0.93 (s, 9H, 3 x CH3), 0.94* (s, 9H, 3 x CH3), 1.74-2.52* (m, 8H, 3 x CH2, 2 x CH), 1.74-2.52 (m, 7H, 3 x CH2, CH), 2.56-2.76* (m, 2H, CH2), 2.56-2.76 (m, 3H, CH2, CH), 3.35 (s, 3H, OCH3), 3.39* (s, 3H, OCH3), 3.81* (m, 1H, CHOH), 3.94 (m, 1H, CHOH), 4.15* (m, 1H, C/-OTBDMS), 4.15 (m, 1H, CHOTBDMS), 4.53 (app td, J = 6.6, 3.2 Hz, 1H, CH), 4.63* (app td, J = 7.5, 4.6 Hz, 1H, CH), 5.09* (app d, J = 5.6 Hz, 1H, CH), 5.14 (app d, J = 4.4 Hz, 1H, CH), 5.48* (m, 1H, =CH), 5.48 (m, 1H, =CH), 5.60* (m, 1H, =CH), 5.60 (m, 1H, =CH), 7.20* (m, 3H, ArCH’s), 7.20 (m, 3H ArCH’s), 7.30* (m, 2H, ArCH’s), 7.30 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = (mixture of 2 diastereoisomers, signals of minor diastereoisomer indicated by *) -4.6 (CH3), -4.6* (CH3), -4.0* (CH3), -3.9 (CH3), 18.4 (C), 18.4* (C), 26.0 (3 x CH3), 26.0* (3 x CH3), 31.8 (CH2), 31.9 (CH2), 38.0 (CH2), 39.3 (CH2), 39.7 (CH2), 40.2 (CH2), 40.3 (CH2), 42.5 (CH2), 45.8 (CH), 46.1* (CH), 54.6* (CH30), 55.0 (CH30), 56.7* (CH), 57.6 (CH), 72.8 (CHOTBDMS), 72.9* (CHOTBDMS), 77.9* (CHOH), 78.9 (CHOH), 81.2 (CH), 83.3* (CH), 106.6 (CH), 107.7* (CH), 125.7 (ArCH), 125.8 (ArCH), 126.0 (ArCH), 128.4 (ArCH), 128.5 (ArCH), 128.6 (ArCH), 130.3* (=CH), 131.1 (=CH), 135.1* (=CH), 135.2 (=CH), 142.2* (ArC), 142.5 (ArC)
m/z (ESI+) 455.1 [MNa]+
HRMS (ESI+) calcd for C25H4o04SiNa [MNa]+ 455.2588, found 455.2587 7L. (3a ?,4 ?,5 ?,6aS)-4-((S,E)-3-Hydroxy-5-phenylpent-l-enyl)hexahyd cyclopenta[b]furan-2,5-diol, 94

93 94
Alcohol 93 (210 mg, 0.485 mmol) was stirred with 1.5% aqueous HCI / THF (3:2) (10 ml) at r.t. for 16 h. The mixture was extracted with CH2CI2 (5 x 15 ml) and the combined organic phases were dried (MgS04), filtered, and concentrated to give the triol 94 and silanol byproduct as a clear, colourless oil. This material was taken forward for the subsequent transformation without purification.
7M. 5-Bromo-/V-ethylpentanamide, 95
1) SOCI2, DMF cat.
Toluene, 55 °C

95
Dimethylformamide (116 μΙ_, 1.5 mmol, 0.1 eq.) and thionyl chloride (1.63 ml, 22.5 mmol, 1.5 eq.) were added to a solution of 5-bromovaleric acid (2.71 g, 15 mmol, 1 eq.) in toluene (20 ml) and the reaction mixture was stirred at 50 °C for 4 h. The volatiles were removed under high vacuum, THF (130 ml) was added and the mixture was cooled to 0 °C. N,N- Diisopropylethylamine (4.2 ml, 24 mmol, 1.6 eq.) and ethylamine (2 M solution in THF, 9.0 ml, 18 mmol, 1.2 eq.) were added dropwise. The reaction mixture was stirred for 1 h at 0 °C before being quenched by the addition of saturated aq. NH4CI (100 ml). The reaction mixture was extracted with Et20 (4 x 75 ml) and the combined organic phases were washed with brine (100 ml) before being dried (MgS04), filtered, and concentrated to give the crude material. This was purified by flash chromatography, eluting with petrol/EtOAc (4:6), giving the title product 95 as a clear, colourless oil (2.75 g, 88 %). Analytical data consistent with the literature (Halazy, S. et al., WO 9612713, Pierre Fabre Medicament (1996)). max (filmVcm-1 3275 (broad), 2970, 2933, 1640, 1543, 1439, 1276, 1150, 643
*H NMR (400 MHz; CDCI3) δΗ = 1.13 (t, J = 7.2 Hz, 3H, CH3), 1.78 (m, 2H, CH2), 1.89 (m, 2H, CH2), 2.19 (t, J = 7.3 Hz, CH2CO), 3.27 (dt, J = 7.2, 5.5 Hz, 2H, NHCH2), 3.41 (t, J = 6.5 Hz, 2H, BrCH2), 5.79 (broad s, 1H, NH)
13C NMR (100 MHz; CDCI3) 5C = 15.0 (CH3), 24.4 (CH2), 32.2 (CH2Br), 33.4 (CH2), 34.4 (NHCH2), 35.7 (CH2C=0), 172.3 (C=0)
m/z (CI+) 210.0 [M81BrH]+ (80%), 208.0 [M79BrH]+ (80%), 128.1 (100%), 107.1 (60%) HRMS (CI+) calcd for C7Hi5 81BrNO [M81BrH]+ 210.0317, found 210.0320; calcd for
C7H15 79BrNO [M79BrH]+ 208.0337, found 208.0341
(5-(Ethylamino)-5-oxopentyl)triphenylphosphonium bromide, 96

Triphenylphosphine (2.88 g, 11 mmol, 1.1 eq.) was added to a solution of 5-bromo-A/- ethylpentanamide 95 (2.08 g, 10 mmol, 1 eq.) in MeCN (5 ml) and the mixture was stirred for 14 h at 80 °C. The mixture was allowed to cool to r.t. and concentrated under vacuum. The residue was added dropwise into Et20 (100 ml) and stirred vigourously for 10 min. The resulting solid was filtered, washed with Et20 (2 x 10 ml) and dissolved in CH2CI2 (15 ml). This solution was then added dropwise into Et20 (200 ml) and stirred for 10 min. Solids were filtered, washed with Et20 (2 x 10 ml) and dried under high vacuum to give the title product 96 as a white powder (3.87 g, 82 %).
Mp 177-179 °C
max (filmVcm“1 3275 (broad), 2970, 2933, 1640, 1543, 1439, 1276, 1150, 643
*H NMR (400 MHz; CDCI3) δΗ = 1.06 (t, J = 7.3 Hz, 3H, CH3), 1.63 (m, 2H, CH2), 1.91 (quin., J = 6.5 Hz, CH2), 2.57 (t, J = 6.5 Hz, 2H, CH2C=0), 3.15 (q, J = 7.3, 5.6 Hz, 2H, NHCH2), 3.66 (m, 2H, CH2P+), 7.66 (m, 6H, ArCH’s), 7.75-7.82 (m, 9H, ArCH’s), 8.26 (t, J = 5.6 Hz, 1H, NH)
13C NMR (100 MHz; CDCI3) 5C = 14.8 (s, CH3), 21.1 (d, J = 4.6 Hz, CH2), 22.4 (d, J = 49.9 Hz, CH2P+), 26.1 (d, J = 17.7 Hz, CH2), 34.1 (s, CH2), 34.2 (s, CH2NH), 118.2 (d, J = 86.1 Hz, 3 x ArC), 130.6 (d, J = 12.3 Hz, 6 x ArCH), 133.8 (d, J = 10.0 Hz, 6 x ArCH), 135.1 (d, J = 3.1 Hz, 3 x ArCH), 172.7 (C=0) m/z (ESI+) 390.2 [M-Br]+
HRMS (ESI+) calcd for C25H29NOP [M-Br]+ 390.1981, found 390.1971
70. (Z)-7-((lR,2R,3R,5S)-3,5-Dihydroxy-2-((S,£)-3-hydroxy-5-phenylpent-l- enyl)cyclopentyl)-/V-ethylhept-5-enamide, bimatoprost, 97

(5-(Ethylamino)-5-oxopentyl)triphenylphosphonium bromide 96 (1.37 g, 2.91 mmol, 6 eq.) was added to a flame dried Schlenk flask, under N2, and anhydrous THF (10 ml) added. The resulting suspension was cooled to 0 °C. KOt-Bu (653.0 mg, 5.82 mmol, 12 eq.) was added in one portion and the resulting orange mixture stirred at 0 °C for 40 min. A solution of crude triol 94 (0.485 mmol, 1 eq.) in anhydrous THF (2.5 ml) was added dropwise via syringe. After complete addition the mixture was stirred at r.t. for 1 h. The reaction was quenched with saturated aq. NH4CI (10 ml) and extracted with EtOAc (5 x 10 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the crude material as solids. These were triturated with EtOAc (10 ml) and the solids filtered and washed with EtOAc (4 x 10 ml). The filtrate was concentrated under vacuum and purified by column chromatography on silica, eluting with EtOAc/MeOH (97.5:2.5 to 95:5) to give 97 (99.2 mg) as a yellowish oil which was further purified by preparative TLC (EtOAc/MeOH 5%) to give BIMATOPROST97 (82.6 mg, 41% over 2 steps) as a clear, colourless oil. Analytical data consistent with the literature (Zanoni, G. et al., Tetrahedron 66, 7472-7478 (2010); Gutman, A. et al., US 20090163596 (2009)).
max (filmVcm-1 3300 (broad), 2930, 1643, 1550, 1453, 1332, 1293, 1048, 1029, 968, 729, 698
*H NMR (400 MHz; CDCI3) δΗ = 1.09 (t, J = 7.1 Hz, 3H, CH3), 1.42-2.40 (m, 14H, 6 x CH2, 2 x CH), 2.67 (m, 2H, CH2), 3.22 (dq, J = 7.1, 6.3 Hz, 2H, CH2NH), 3.41 (broad s, 3H, 3 x OH), 3.80-4.30 (broad m, 3H, 3 x CHOH), 5.37 (m, 2H, 2 x =CH), 5.47 (dd, J = 15.2, 7.9 Hz, 1H, =CH), 5.59 (dd, J = 15.2, 7.9 Hz, 1H, =CH), 5.90 (broad s, 1H, NH), 7.17 (m, 3H, ArCH’s), 7.26 (m, 2H, ArCH’s)
13C NMR ( 100 MHz; CDCI3) 5C = 14.8 (CH3), 25.4 (CH2), 25.6 (CH2), 26.7 (CH2), 31.9 (CH2), 34.4 (CH2NH), 35.8 (CH2C=0), 38.8 (CH2), 42.9 (CH2), 50.2 (CH), 55.5 (CH), 72.3 (CHOH), 72.4 (CHOH), 77.7 (CHOH), 125.8 (ArCH), 128.4 (2 x ArCH), 128.5 (2 x ArCH), 129.1 (=CH), 129.7 (=CH), 133.7 (=CH), 135.1 (=CH), 142.0 (ArC), 173.4 (C=0)
m/z (ESI+) 438.2 [MNa]+
HRMS (ESI+) calcd for Q^IV^Na [MNa]+ 438.2614, found 438.2615
[a]D 22 +41.1 (c. 0.35, CH2CI2) (lit. – Zanoni, G. et al., Tetrahedron 66, 7472-7478 (2010), +32.7 (c. 0.33, CH2CI2)) (lit. – Gutman, A. et al., US 20090163596 (2009), +36 (c. 1, MeOH))
References
- “Bimatoprost Ophthalmic”. MedlinePlus. January 1, 2003. Archived from the original on 2007-10-05. Retrieved 2007-11-19.
- “Allergan gets FDA approval for eyelash treatment”. BusinessWeek. Associated Press. December 26, 2008. Retrieved December 26, 2008.
- Park J, Cho HK, Moon JI (2011). “Changes to upper eyelid orbital fat from use of topical bimatoprost, travoprost, and latanoprost.”. Japanese Ophthalmological Society 55 (1): 22–27.doi:10.1007/s10384-010-0904-z. PMID 21331688.
- Jayaprakasam A, Ghazi-Nouri S. (2010). “Periorbital fat atrophy – an unfamiliar side effect of prostaglandin analogues.”. Orbit 29 (6): 357–359. doi:10.3109/01676830.2010.527028.PMID 21158579.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL. (2008). “Periorbital changes associated with topical bimatoprost.”. Ophthalmology Plastic and Reconstructive Surgery 24 (4): 302–307. doi:10.1097/IOP.0b013e31817d81df. PMID 18645437.
- Rundle, Rhonda L. (2007-11-19). “Drug That Lengthens Eyelashes Sets Off Flutter”. The Wall Street Journal. Retrieved 2007-11-19.
- The Pink Sheet: [1] Lauren Smith December 15, 2008; Volume 70, Number 050,Page[verification needed]
- Federation of American Societies for Experimental Biology Journal “The prostamide-related glaucoma therapy, bimatoprost, offers a novel approach for treating scalp alopecias” Randall et all. October 26, 2012.
- Latisse prescribing information: “Important Safety Information”
- MSNBC: FDA Seizes $2 Million Of Potentially Harmful SJ Eye Product KNTV-TV November 17, 2007[dead link]
- Reuters: “U.S. seizes discontinued eyelash product”. Jim Wolf. November 16, 2007.
- Tappeiner C, Perren B, Iliev ME, Frueh BE, Goldblum D (May 2008). “Orbitale Fettgewebsatrophie bei lokaler Bimatoprost-Therapie – Kann Bimatoprost einen Enophthalmus verursachen?” [Orbital fat atrophy in glaucoma patients treated with topical bimatoprost–can bimatoprost cause enophthalmos?]. Klinische Monatsblätter für Augenheilkunde (in German)225 (5): 443–5. doi:10.1055/s-2008-1027362. PMID 18454393.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL (2008). “Periorbital changes associated with topical bimatoprost”. Ophthalmic Plastic and Reconstructive Surgery 24 (4): 302–7. doi:10.1097/IOP.0b013e31817d81df. PMID 18645437.
- Peplinski LS, Albiani Smith K (August 2004). “Deepening of lid sulcus from topical bimatoprost therapy”. Optometry and Vision Science 81 (8): 574–7.doi:10.1097/01.opx.0000141791.16683.4a. PMID 15300114.
- Serrero G, Lepak NM (April 1997). “Prostaglandin F2alpha receptor (FP receptor) agonists are potent adipose differentiation inhibitors for primary culture of adipocyte precursors in defined medium”. Biochemical and Biophysical Research Communications 233 (1): 200–2. doi:10.1006/bbrc.1997.6433. PMID 9144422.
- Curran MP (2009). “Bimatoprost: a review of its use in open-angle glaucoma and ocular hypertension”. Drugs Aging 26 (12): 1049–71. doi:10.2165/11203210-000000000-00000.PMID 19929032.
- “Long Lashes Without Prescription, but With Risks”. Catherine Saint Louis. The New York Times. May 1, 2010
- “Potentially Harmful “Cosmetic” Eye Product Seized” (Press release). U.S. Food and Drug Administration (FDA). November 19, 2007. Retrieved 2007-12-05.
Citations
- Chen M, Cheng C, Chen Y, Chou C, Hsu W (2006). “Effects of bimatoprost 0.03% on ocular hemodynamics in normal tension glaucoma.”. J Ocul Pharmacol Ther 22 (3): 188–93. doi:10.1089/jop.2006.22.188. PMID 16808680.
- Kruse P, Rieck P, Sherif Z, Liekfeld A (2006). “Cystoid macular edema in a pseudophakic patient after several glaucoma procedures. Is local therapy with bimatoprost the reason?”. Klinische Monatsblätter für Augenheilkunde 223 (6): 534–7. doi:10.1055/s-2005-858992. PMID 16804825.
- Steinhäuser S (2006). “Decreased high-density lipoprotein serum levels associated with topical bimatoprost therapy.”. Optometry 77 (4): 177–9.doi:10.1016/j.optm.2006.02.001. PMID 16567279.
- Park J, Cho HK, Moon JI (2011). “Changes to upper eyelid orbital fat from use of topical bimatoprost, travoprost, and latanoprost.”. Japanese Ophthalmological Society 55 (1): 22–27. doi:10.1007/s10384-010-0904-z. PMID 21331688.
- Jayaprakasam A, Ghazi-Nouri S. (2010). “Periorbital fat atrophy – an unfamiliar side effect of prostaglandin analogues.”. Orbit 29 (6): 357–359.doi:10.3109/01676830.2010.527028. PMID 21158579.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL. (2008). “Periorbital changes associated with topical bimatoprost.”. Ophthalmology Plastic and Reconstructive Surgery 24 (4): 302–307. doi:10.1097/IOP.0b013e31817d81df. PMID 18645437.
External links
- Medical News Today: FDA Seizes $2 Million Of Cosmetic Eye Product Which Contains Drug Ingredient And Makes Unapproved Drug Claims. Christian Nordqvist. 18 November 2007
- Wired Science: FDA Seizes Cosmetic That Can Blind. Brandon Keim. November 19, 2007
- Eye Drops: [The generic name of the Latisse eye drop is Bimatoprost Ophthalmic Solution 0.03%]. Crazzy Paul. Aug 01, 2013
|
1-25-2012
|
Method of enhancing hair growth
|
|
|
1-25-2012
|
NITRIC OXIDE DONATING PROSTAMIDES
|
|
|
11-25-2011
|
BIMATOPROST CRYSTALLINE FORM I
|
|
|
10-19-2011
|
Method of Enhancing Hair Growth
|
|
|
7-22-2011
|
IMPROVED PROCESS FOR THE PRODUCTION OF BIMATOPROST
|
|
|
6-3-2011
|
Process for the Preparation of Prostaglandin Analogues and Intermediates Thereof
|
|
|
5-27-2011
|
COMPOSITIONS AND METHODS FOR STIMULATING HAIR GROWTH
|
|
|
5-27-2011
|
ENHANCED BIMATOPROST OPHTHALMIC SOLUTION
|
|
|
5-25-2011
|
Bimatoprost crystalline form I
|
|
|
5-13-2011
|
COMPOSITIONS FOR ENHANCING HAIR GROWTH
|
|
3-2-2011
|
Process for the Preparation of Prostaglandin Analogues and Intermediates Thereof
|
|
|
12-24-2010
|
METHOD FOR THE PURIFICATION OF PROSTAGLANDINS
|
|
|
12-15-2010
|
Enhanced bimatoprost ophthalmic solution
|
|
|
9-17-2010
|
Compositions and Methods for Reducing Body Fat
|
|
|
5-28-2010
|
COMPLEXES OF PROSTAGLANDIN DERIVATIVES AND MONOSUBSTITUTED, CHARGED BETA-CYCLODEXTRINS
|
|
|
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
|
|
|
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
|
|
|
2-24-2010
|
Compositions and methods for reducing body fat
|
|
|
1-27-2010
|
10-HYDROXY-11-DIHYDROPROSTAGLANDIN ANALOGS AS SELECTIVE EP4 AGONISTS
|
|
|
1-15-2010
|
Process for the Production of Prostaglandins and Prostaglandin Analogs
|
|
11-20-2009
|
Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost
|
|
|
11-4-2009
|
Enzymatic transformation of a prostaglandin (bimatoprost) intermediate
|
|
|
10-16-2009
|
Method for preparing prostaglandin F analogue
|
|
|
8-14-2009
|
METHOD OF ENHANCING HAIR GROWTH
|
|
|
6-12-2009
|
Enhanced Bimatoprost Ophthalmic Solution
|
|
|
4-3-2009
|
METHOD FOR SCREENING OF PROSTAGLANDIN COMPOUNDS COMPRISING AN OPTIMAL FORMULATION FOR THE ENHANCEMENT OF HAIR GROWTH AND THE STIMULATION OF FOLLICULAR ANAGEN AND FORMULATIONS RESULTING THEREFROM
|
|
|
10-15-2008
|
5-Thiopiperdinyl prostaglandin e analogs
|
|
|
2-20-2008
|
COMPOSITIONS AND METHODS COMPRISING PROSTAGLANDIN-RELATED COMPOUNDS AND TREFOIL FACTOR FAMILY PEPTIDES FOR THE TREATMENT OF GLAUCOMA WITH REDUCED HYPEREMIA
|
|
|
2-15-2008
|
Novel Prostamides For The Treatment Of Glaucoma And Related Diseases
|
|
|
11-23-2007
|
COMPOSITIONS AND METHODS COMPRISING PROSTAGLANDIN-RELATED COMPOUNDS AND TREFOIL FACTOR FAMILY PEPTIDES FOR THE TREATMENT OF GLAUCOMA WITH REDUCED HYPEREMIA
|
|
7-18-2007
|
Compositions and methods comprising prostaglandin related compounds and trefoil factor family peptides for the treatment of glaucoma with reduced hyperemia
|
|
|
5-18-2007
|
NOVEL PROSTAMIDES FOR THE TREATMENT OF GLAUCOMA AND RELATED DISEASES
|
|
|
4-27-2007
|
Compositions comprising benzo (g) quinoline derivatives and prostaglandin derivatives
|
|
|
3-7-2007
|
Prostamides for the treatment of glaucoma and related diseases
|
|
|
1-31-2007
|
10-Hydroxy-11-dihydroprostaglandin analogs as selective EP4 agonists
|
|
|
1-24-2007
|
Process for the preparation of prostaglandin derivatives
|
|
|
1-10-2007
|
Protected diols for prostaglandin synthesis
|
|
|
1-3-2007
|
Process for the preparation of 17-phenyl-18,19,20-thinor-pgf 2a and its derivatives
|
|
|
11-29-2006
|
Protected and unprotected triols for prostaglandin synthesis
|
|
|
9-20-2006
|
Prostaglandin synthesis
|
|
9-6-2006
|
Cyclopentane heptan(ENE)OIC acid, 2-heteroarylalkenyl derivatives as therapeutic agents
|
|
|
7-5-2006
|
Method for imparting artificial tan to human skin
|
|
|
8-24-2005
|
Inhibition of irritating side effects associated with use of a topical ophthalmic medication
|
|
|
3-18-2005
|
Methods for the treatment of gray hair using cyclopentane(ene) heptan(en)oic acid amides
|
|
|
3-9-2005
|
9,11-cycloendoperoxide pro-drugs of prostaglandin analogues for treatment of ocular hypertension and glaucoma
|
|
|
2-11-2005
|
Compositions for delivery of therapeutics into the eyes and methods for making and using same
|
|
|
5-29-2002
|
Ocular hypotensive lipids
|
| EP0364417A1 | Sep 6, 1989 | Apr 18, 1990 | Pharmacia AB | Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension |
| EP0364417B1 | Sep 6, 1989 | Feb 9, 1994 | Pharmacia AB | Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension |
| EP0544899B1 | Jun 19, 1992 | Sep 6, 1995 | CHINOIN Gyogyszer és Vegyészeti Termékek Gyára RT. | Prostaglandins |
| US5359095 | Feb 8, 1994 | Oct 25, 1994 | Pharmacia Ab | Hydrogenation of doulbe bond in intermediate compound without deoxygenatio of allytic alcohol |
| US6689901 | Jun 25, 2002 | Feb 10, 2004 | Pharmacia & Upjohn Company | Process and intermediates to prepare latanoprost |
| US6927300 | Jan 26, 2001 | Aug 9, 2005 | Finetech Laboratories Ltd | Process for the preparation of Latanoprost |
| US7157590 | May 3, 2002 | Jan 2, 2007 | Finetech Laboratories Ltd. | Chemical intermediate for vision defect drug |
| US7268239 | Jul 27, 2005 | Sep 11, 2007 | Resolution Chemicals Limited | Process for the preparation of prostaglandins and analogues thereof |
| US7642370 | Mar 20, 2007 | Jan 5, 2010 | Daiichi Fine Chemical Co., Ltd. | Method for preparing prostaglandin derivative |
| US7674921 | Feb 25, 2008 | Mar 9, 2010 | Cayman Chemical Company, Inc. | cloprostenol or latanoprost; topical use; highly crystalline structures that are easy to formulate into ophthalmic solutions; hydrolysis of these analogs releases only the active PGF2 alpha analog free acid, without the production of toxic and irritant small aliphatic alcohol coproducts |
| US20090259066 * | Jul 3, 2008 | Oct 15, 2009 | Everlight Usa, Inc. | Method for preparing prostaglandin F analogue |
| US20090287003 | Sep 29, 2006 | Nov 19, 2009 | Jiang Xing Chen | Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost |
| US20100010239 | Jul 10, 2009 | Jan 14, 2010 | Sandoz Ag | Process for the Production of Prostaglandins and Prostaglandin Analogs |
| WO2010097672A1 | Feb 18, 2010 | Sep 2, 2010 | Sifavitor S.R.L. | Process for the preparation of prostaglandin derivatives |
Organic spectroscopy should be brushed up and you get confidence
read my blog
ORGANIC SPECTROSCOPY INTERNATIONAL is the blog
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com
http://orgspectroscopyint.blogspot.in/ is the link
 amcrasto@gmail.com
amcrasto@gmail.com


Woah! I’m really digging the template/theme of this website. It’s simple, yet effective.
A lot of times it’s difficult to get that “perfect balance” between user friendliness and visual appeal. I must say that you’ve done a great job
with this. In addition, the blog loads extremely fast for me on Chrome.
Outstanding Blog!