
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
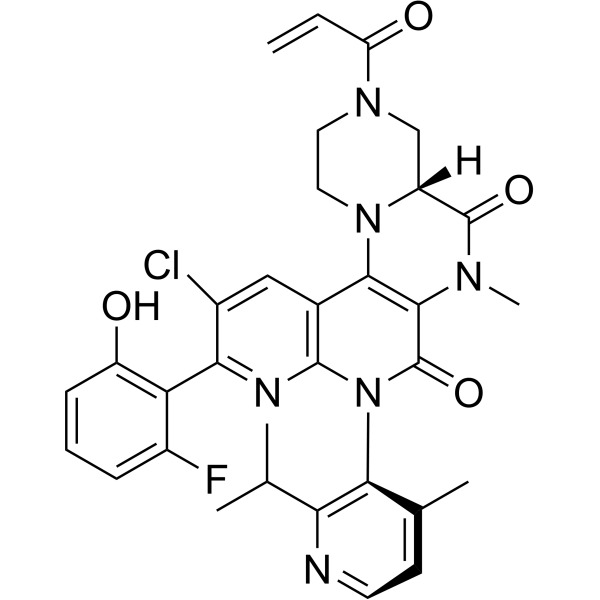
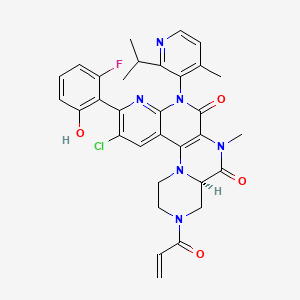
Fulzerasib
GFH925
CAS No. : 2641747-54-6
| Molecular Weight | 617.07 |
|---|---|
| Formula | C32H30ClFN6O4 |
(7R)-16-chloro-15-(2-fluoro-6-hydroxyphenyl)-9-methyl-12-(4-methyl-2-propan-2-ylpyridin-3-yl)-5-prop-2-enoyl-2,5,9,12,14-pentazatetracyclo[8.8.0.02,7.013,18]octadeca-1(10),13,15,17-tetraene-8,11-dione
CHINA 2024, APPROVALS 2024, Innovent Biologics, DUPERT
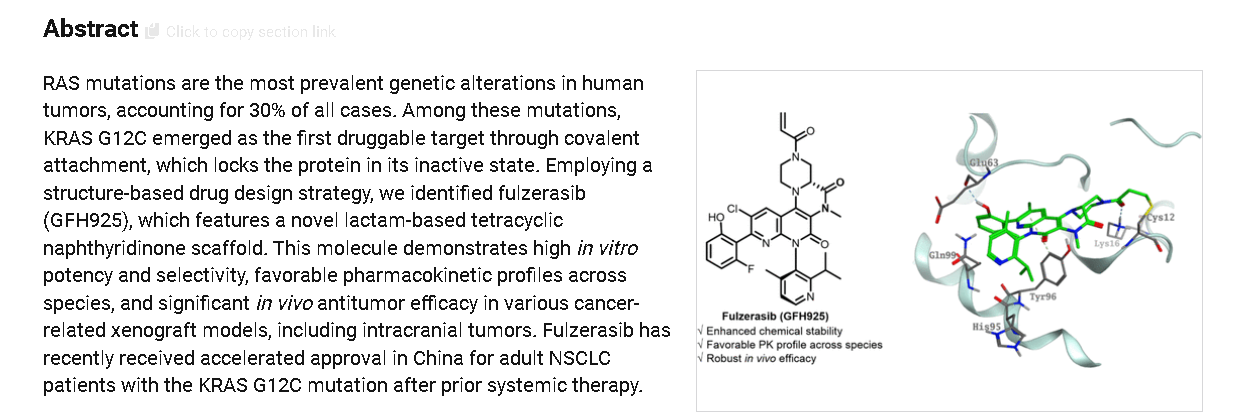
Fulzerasib (Dupert®; Innovent Biologics/GenFleet Therapeutics) is an orally active small molecule inhibitor of the KRAS G12C mutant protein being developed for the treatment of solid tumors harboring the KRAS G12C oncogenic driver mutation, including non-small cell lung cancer (NSCLC) and colorectal cancer. Fulzerasib received its first approval on 21 August 2024 in China, for the treatment of adults with KRAS G12C-mutated advanced NSCLC who have received at least one line of systemic therapy. This conditional approval was based on the positive results of a single-arm, phase II study. This article summarizes the milestones in the development of fulzerasib leading to this first approval for KRAS G12C-mutated advanced NSCLC.
Fulzerasib (GFH925) is an irreversible KRAS G12C inhibitor, has a synergistic anti-cancer effect with cetuximab (HY-P9905)..
PAPER
https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c03183
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021083167&_cid=P20-MEJIF1-91906-1
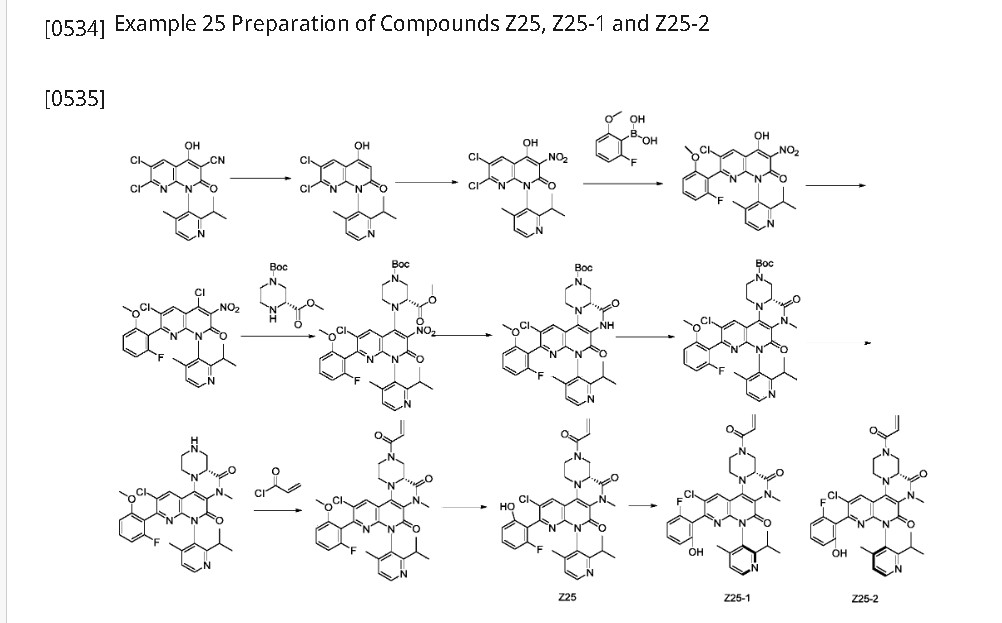
Step 1: Suspend 6,7-dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carbonitrile (30.0 g, 77.319 mmol) in a mixture of 1,4-dioxane (120 mL) and water (120 mL). Slowly add concentrated sulfuric acid (120 mL). Stir at 120°C for 36 hours. Pour the cooled reaction mixture into 200 mL of ice water, adjust the pH to 2-3 with sodium carbonate, and extract with ethyl acetate (1000 mL x 2). Combine the ethyl acetate phases, dry over anhydrous sodium sulfate, filter, and vacuum-dry the filtrate to obtain 6,7-dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-1,8-naphthyridine-2(1H)-one (24 g, Y: 85.7%) as a light brown solid. ES-API: [M+H]
[0537]Step 2: 6,7-Dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-1,8-naphthyridin-2(1H)-one (3.16 g, 8.705 mmol) was dissolved in acetic acid (15 mL). Sodium nitrite (100 mg, 1.58 mmol) and concentrated nitric acid (5.0 mL, 74.52 mmol) were added sequentially. The reaction was stirred at room temperature for 30 minutes. The reaction solution was slowly poured into 100 mL of ice water. The precipitated solid was filtered, and the filter cake was washed with 20 mL of ice water and dried under vacuum to obtain 6,7-dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (3.5 g, Y: 92%) as a yellow solid. ES-API: [M+H]
[0538]Step 3: To a 100 mL three-necked round-bottom flask, add 6,7-dichloro-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (3.5 g, 8.570 mmol), (2-fluoro-6-methoxyphenyl)boronic acid (5.8 g, 34.10 mmol), tetrakistriphenylphosphine palladium (1.15 g, 0.9956 mmol), sodium carbonate (3.5 g, 33.02 mmol), 10 mL of water, and 40 mL of dioxane. Under nitrogen, stir at 100°C for 2-3 hours. After completion, cool the reaction mixture to room temperature, add 80 mL of water and 100 mL of methyl tert-butyl ether, and extract once. The aqueous phase was adjusted to pH 3-5 with 1M hydrochloric acid solution and extracted with ethyl acetate (200 mL x 2). The ethyl acetate phases were combined, dried over anhydrous sodium sulfate, filtered, and the filtrate was dried under vacuum to afford 6-chloro-7-(2-fluoro-6-methoxyphenyl)-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (4.5 g, crude) as a pale yellow solid. ES-API: [M+H]
[0539]Step 4: 6-Chloro-7-(2-fluoro-6-methoxyphenyl)-4-hydroxy-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (4.6 g, 8.57 mmol) was dissolved in acetonitrile (30 mL). Phosphorus oxychloride (7.5 g, 48.92 mmol) and N,N-diisopropylethylamine (10.5 g, 81.24 mmol) were added sequentially. The reaction mixture was gradually heated to 80°C and stirred for 30 minutes. The reaction solution was concentrated, 30 mL of cold acetonitrile was added, and the mixture was added dropwise to 150 mL of saturated sodium bicarbonate solution under an ice-water bath. The mixture was extracted with ethyl acetate (200 mL x 2). The ethyl acetate phases were combined and washed once with 200 mL of saturated brine. The reaction mixture was dried over anhydrous sodium sulfate and filtered. The organic phase was dried and concentrated, and the crude product was purified by flash silica gel column chromatography (EtOAc/PE: 0-50%) to afford 4,6-dichloro-7-(2-fluoro-6-methoxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (3.05 g, Y: 76%) as a yellow solid. ES-API: [M+H]
[0540]Step 5: 4,6-Dichloro-7-(2-fluoro-6-methoxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-1,8-naphthyridin-2(1H)-one (2.5 g, 4.843 mmol) was dissolved in N,N-dimethylacetamide (25 mL). 1-(tert-butyl)-3-methyl(R)-piperazine-1,3-dicarboxylate (3.5 g, 14.34 mmol) and N,N-diisopropylethylamine (2.0 g, 15.47 mmol) were added sequentially. The reaction mixture was stirred at 120°C for 2 hours. 80 mL of ethyl acetate was added to the reaction mixture, and the mixture was washed three times with 80 mL of saturated brine. The ethyl acetate phase was dried and concentrated, and the crude product was purified on a flash silica gel column (EtOAc/PE: 0-80%) to afford 1-(tert-butyl)-3-methyl (3R)-4-(6-chloro-7-(2-fluoro-6-methoxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-2-oxo-1,2-dihydro-1,8-naphthyridin-4-yl)piperazine-1,3-dicarboxylate (2.7 g, Y: 77%) as a yellow solid. ES-API: [M+H]
[0541]Step 6: 1-(tert-Butyl)3-methyl(3R)-4-(6-chloro-7-(2-fluoro-6-methoxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-3-nitro-2-oxo-1,2-dihydro-1,8-naphthyridin-4-yl)piperazine-1,3-dicarboxylate (2.7 g, 3.728 mmol) was dissolved in acetic acid (30 mL), iron powder (835 mg, 14.91 mmol) was added, and the reaction was stirred at 80 °C for 30 minutes. The reaction mixture was concentrated, and 200 mL of ethyl acetate and 100 mL of saturated sodium bicarbonate were added sequentially. The suspension was filtered through celite, and the filter cake was washed with ethyl acetate. The organic phase was separated and washed sequentially with 100 mL of saturated sodium bicarbonate and 150 mL of saturated brine. The mixture was dried and concentrated to give (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylic acid tert-butyl ester (2.70 g, crude) as a yellow solid. ES-API: [M+H]+ = 663.2.
[0542]Step 7: To a 150 mL sealed tube was added tert-butyl (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylate (2.7 g, 3.728 mmol), 30 mL of acetone, anhydrous potassium carbonate (2.2 g, 15.94 mmol), and iodomethane (5.4 g, 38.03 mmol). The tube was sealed and the reaction was stirred at 55°C for 18 hours. The reaction mixture was added with 150 mL of ethyl acetate, washed three times with 100 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column (EtOAc/PE: 0-80%) to obtain (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylic acid tert-butyl ester (2.2 g, Y: 87%) as a yellow solid. ES-API: [M+H]
[0543]Step 8: Tert-butyl (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylate (517 mg, 0.7549 mmol) was dissolved in dichloromethane (8 mL) and trifluoroacetic acid (2 mL) was added. After stirring at room temperature for 2 hours, the reaction mixture was concentrated to afford (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (530 mg, crude), which was used directly in the next reaction. ES-API: [M+H]
[0544]Step 9: (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (530 mg, 0.7549 mmol) was dissolved in dichloromethane (15 mL) and triethylamine (3.0 mL, 21.62 mmol) was added. The reaction mixture was cooled to 0°C and acryloyl chloride (100 mg, 1.1048 mmol) was added dropwise. The reaction was stirred at 0°C for 15 minutes. 80 mL of dichloromethane was added to the reaction solution, and the mixture was washed with 100 mL of saturated aqueous NaHCO₃
and 80 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column (EtOAc/PE: 0-60%) to obtain (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (280 mg, Y: 59%) as a yellow solid. ES-API: [M+H]
[0545]Step 10: In an ice-water bath, (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (280 mg, 0.444 mmol) was added to dry dichloromethane (6.0 mL), and then boron tribromide (5.0 mL, 5.0 mmol) was added. The mixture was warmed to room temperature and reacted overnight. Under ice-water bath conditions, the reaction solution was added dropwise to a saturated sodium bicarbonate solution, extracted twice with dichloromethane (80 mL), dried, and concentrated. The crude product was purified by flash silica gel column chromatography (EtOAc/PE: 0-60%) to give (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-hydroxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-methyl-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (Z25, 233 mg, Y: 85%).
[0546]Step 11: Compound Z25 was separated by preparative chiral HPLC (column type: IA: 10 μm, 30*250 mm, mobile phase: hexane:EtOH = 60:40, flow rate: 25 ml/min, column temperature) to obtain: an atropisomer compound Z25-1 (76.8 mg, peak 1, retention time 2.531 min, Y: 34%).
1 H NMR (500 MHz, DMSO-d
6 )δ10.03(d,J=18.4Hz,1H),8.52(d,J=7.3Hz,1H),8.43(d,J=4.7Hz,1H),7.23(d,J=9.6Hz,2H),7.08(dd,J=16 .6,10.5Hz,1H),6.74–6.62(m,2H),6.15(d,J=16.8Hz,1H),5.75(d,J=10.7Hz,1H),4.73(d,J=14.2Hz,1H),4.4 6 (d, J = 12.9 Hz, 1H), 4.00 (s, 1H), 3.61 (d, J = 10.0 Hz, 1H), 3.51 (s, 1H), 3.34 (s, 3H), 3.22 (s, 1H), 2.64 (t, J = 11.5 Hz, 1H), 2.48–2.42 (m, 1H), 1.98 (d, J = 5.1 Hz, 3H), 1.03 (t, J = 6.9 Hz, 3H), 0.86 (t, J = 7.9 Hz, 3H). ES-API: [M+H]
+ = 617.2. And another atropisomer compound Z25-2 (70 mg, peak 2, retention time 3.683 min, Y: 31%).
1 H NMR (500MHz, CDCl
3 )δ8.64–8.59(m,1H),8.35(s,1H),8.07(s,1H),7.27–7.20(m,2H),7.14–7.02(m,1H),6.75–6.63(m,2H),6.39(dd,J=17.0,2.0Hz,1H),5.88 –5.77(m,1H),4.91(d,J=14.0Hz,1H),4.83(d,J=13.0Hz,1H),3.72–3.58(m,2H),3.50(s,3H),3.43(d,J=12.0Hz,1H),3.1 6(t,J=13.0Hz,1H),2.91(t,J=12.0Hz,1H),2.82-2.73(m,1H),1.93(s,3H),1.24(d,J=7.0Hz,3H),1.12(d,J=7.0Hz,3H). ES-API: [M+H]
+ =617.2. The isomers were detected by analytical chiral HPLC (column type: IA: 5 μm, 4.6*150 mm, mobile phase: hexane:EtOH=60:40, flow rate: 1 ml/min, column temperature=30°C).
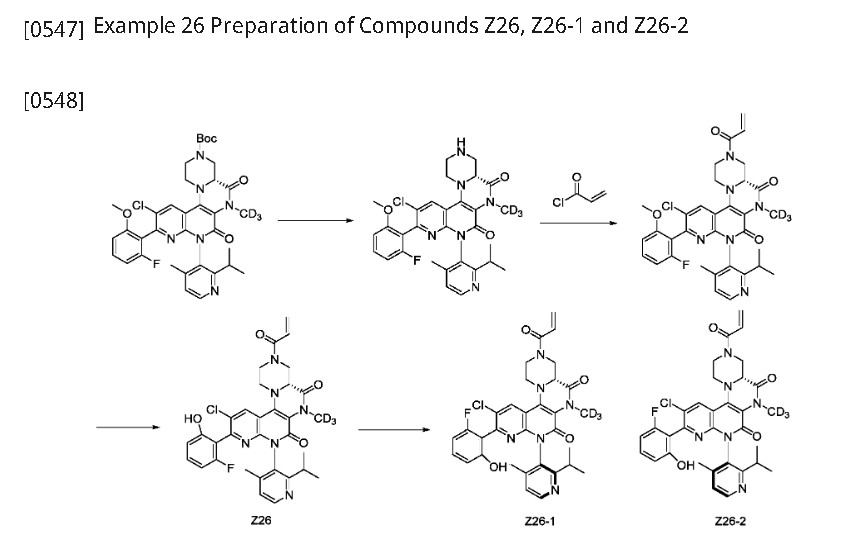
Step 1: (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-5,7-dioxo-1,2,4,4a,5,6,7,8-octahydro-3H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-3-carboxylic acid tert-butyl ester (511 mg, 0.7549 mmol) was dissolved in dichloromethane (8 mL) and trifluoroacetic acid (2 mL) was added. After stirring at room temperature for 2 hours, the reaction mixture was concentrated to give (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (520 mg, crude), which was used directly in the next reaction. ES-API: [M+H]
[0550]Step 2: (4aR)-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (520 mg, 0.7549 mmol) was dissolved in dichloromethane (10 mL) and triethylamine (3.0 mL, 21.62 mmol) was added. The reaction mixture was cooled to 0°C and acryloyl chloride (100 mg, 1.1048 mmol) was added dropwise. The reaction was stirred at 0°C for 15 minutes. 80 mL of dichloromethane was added to the reaction solution, and the mixture was washed with 100 mL of saturated aqueous NaHCO₃
and 80 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column (EtOAc/PE: 0-60%) to obtain (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d₃)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (232 mg, Y: 48%) as a yellow solid. ES-API: [M+H]
[0551]Step 3: Under ice-water bath conditions, (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-methoxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (240 mg, 0.3791 mmol) was added to dry dichloromethane (6.0 mL), and boron tribromide (5.0 mL, 5.0 mmol) was added. The temperature was warmed to room temperature and the reaction was allowed to react overnight. Under ice-water bath conditions, the reaction solution was added dropwise to a saturated sodium bicarbonate solution, extracted twice with dichloromethane (80 mL), dried, and concentrated. The crude product was purified on a flash silica gel column (EtOAc/PE: 0-60%) to give (4aR)-3-acryloyl-11-chloro-10-(2-fluoro-6-hydroxyphenyl)-8-(2-isopropyl-4-methylpyridin-3-yl)-6-(methyl-d3)-2,3,4,4a,6,8-hexahydro-1H-pyrazino[1′,2′:4,5]pyrazino[2,3-c][1,8]naphthyridine-5,7-dione (Z26, 187 mg, Y: 79%). [M+H]
[0552]Step 4: Compound Z26 (187 mg, 0.302 mmol) was separated by preparative chiral HPLC (column type: IA: 10 μm, 30*250 mm, mobile phase: hexane:EtOH = 60:40, flow rate: 25 ml/min, column temperature) to obtain: an atropisomer compound, arbitrarily designated as Z26-1 (68.8 mg, peak 1, retention time 2.525 min, Y: 36.7%).
1 H NMR (500 MHz, DMSO-d
6 )δ10.03(d,J=17.9Hz,1H),8.51(d,J=7.4Hz,1H),8.43(d,J=4.7Hz,1H),7.29–7.18(m,2H),7.08(dd,J =17.0,10.6Hz,1H),6.74–6.61(m,2H),6.15(d,J=16.6Hz,1H),5.75(d,J=11.5Hz,1H),4.73(d,J=13.5 Hz, 1H), 4.46 (d, J = 12.3 Hz, 1H), 4.00 (s, 1H), 3.61 (d, J = 10.5 Hz, 1H), 3.50 (s, 1H), 3.22 (s, 1H), 2.65 (t, J = 12.5 Hz, 1H), 2.49–2.42 (m, 1H), 1.98 (d, J = 5.0 Hz, 3H), 1.02 (d, J = 7.0 Hz, 3H), 0.86 (t, J = 7.9 Hz, 3H). ES-API: [M+H]
+ = 620.3. Another atropisomer, arbitrarily designated Z26-2 (63.2 mg, peak 2, retention time 3.683 min, Y: 33.79%), was obtained.
1 H NMR (400 MHz, CDCl
3 )δ8.62(d,J=4.8Hz,1H),8.35(s,1H),8.07(s,1H),7.24–7.20(m,2H),7.16–7.01(m,1H),6.74–6.6 3(m,2H),6.39(dd,J=16.8,2.0Hz,1H),5.82(dd,J=10.4,2.0Hz,1H),4.91(d,J=13.6Hz,1H),4.83(d δ (d, J = 13.6 Hz, 1H), 3.71–3.57 (m, 2H), 3.42 (d, J = 12.0 Hz, 1H), 3.16 (t, J = 12.8 Hz, 1H), 2.91 (t, J = 12.0 Hz, 1H), 2.81–2.70 (m, 1H), 1.92 (s, 3H), 1.22 (d, J = 6.8 Hz, 3H), 1.10 (d, J = 6.8 Hz, 3H). ES-API: [M+H]
+ = 620.3. The isomeric compounds were detected by analytical chiral HPLC (column type: IA: 5 μm, 4.6*150 mm, mobile phase: hexane:EtOH = 60:40, flow rate: 1 ml/min, column temperature = 30°C).
PAT
- US12054497, Compound Z25-1
- US12054497, Compound Z25-2
- US12054497, Compound Z26-1
- US12054497, Compound Z26-2
PAT
CN112390818
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN319676055&_cid=P20-MEJIKL-96783-1
| Example 25 Preparation of Z25 |
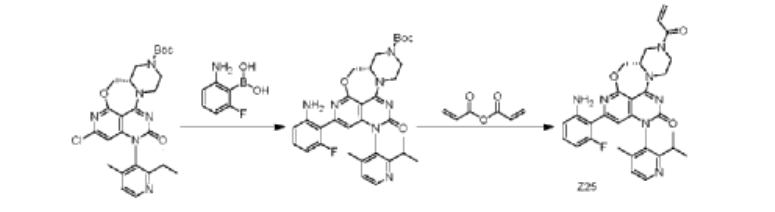
| Step 1: To a 100 mL three-necked round-bottom flask was added (S)-2-chloro-12-(2-isopropyl-4-methylpyridin-3-yl)-11-oxo-5a,6,8,9,11,12-hexahydro-4-oxo-3,7,9a,10,12-pentaazabenzo[4,5]cycloheptyl[1,2,3-de]naphthalene-7(5H)-carboxylic acid tert-butyl ester (1.4 g, 2.66 mmol), (2-amino-6-fluorophenyl)boronic acid (0.6 g, 3.87 mmol), Sphos-Pd-G2 (0.2 g, 0.21 mmol), Sphos (120 mg, 0.29 mmol), potassium phosphate (1.2 g, 5.66 mmol), 10 mL of dioxane, and 2 mL of water. The system was purged with nitrogen three times and then protected with nitrogen. The reaction was continued at 120°C for 2 h. 30 mL of ethyl acetate was added to the reaction solution, which was washed three times with 30 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column to give the target product, (S)-2-(2-amino-6-fluorophenyl)-12-(2-isopropyl-4-methylpyridin-3-yl)-11-oxo-5a,6,8,9,11,12-hexahydro-4-oxa-3,7,9a,10,12-pentaazabenzo[4,5]cyclohepta[1,2,3-de]naphthalene-7(5H)-carboxylic acid tert-butyl ester (845 mg, yield: 41%). ES-API: [M+H]+ = 602.2. |
| Step 2: Dissolve (S)-tert-butyl 2-(2-amino-6-fluorophenyl)-12-(2-isopropyl-4-methylpyridin-3-yl)-11-oxo-5a,6,8,9,11,12-hexahydro-4-oxa-3,7,9a,10,12-pentaazabenzo[4,5]cyclohepta[1,2,3-de]naphthalene 7(5H)-carboxylate (800 mg, 1.33 mmol) in dichloromethane (8 mL), and add trifluoroacetic acid (2 mL). Stir at room temperature for 2 hours. The reaction mixture is concentrated to obtain the target intermediate, which is dissolved in dichloromethane (15 mL) and triethylamine (800 mg, 87.1 mmol) is added. Cool the reaction mixture to 0°C, and add acrylic anhydride (160 mg, 1.27 mmol) dropwise. Stir the reaction mixture at 0°C for 15 minutes. The reaction mixture was added with 40 mL of dichloromethane, washed with 50 mL of saturated aqueous NaHCO₃ and 40 mL of saturated brine, dried, and concentrated. The crude product was purified on a flash silica gel column to obtain the target product, Z25(S)-7-acryloyl-2-(2-amino-6-fluorophenyl)-12-(2-isopropyl-4-methylpyridin-3-yl)-5,5a,6,7,8,9-hexahydro-4-oxa-3,7,9a,10,12-pentaazabenzo[4,5]cyclohepta[1,2,3-de]naphthalen-11(12H)-one (250 mg, yield: 34%). ES-API: [M+H] ⁺ = 556.2. 1 H NMR (500MHz, DMSO) δ8.55 (d, J=4.9Hz, 1H), 7.32 (d, J=4.9Hz, 1H), 7.04 (dd, J= 14.8,8.0Hz,1H),6.95-6.80(m,1H),6.52(d,J=8.3Hz,1H),6.36-6.13(m,4H) ,6.06-5.95(m,1H),5.78(d,J=10.3Hz,1H),4.82-4.04(m,7H),3.56(s,1H),3 .25-3.18(m,1H),2.84-2.70(m,1H),1.98(d,J=5.2Hz,3H),1.15-0.95(m,6H). |
| Example 26 Preparation of Z26 |
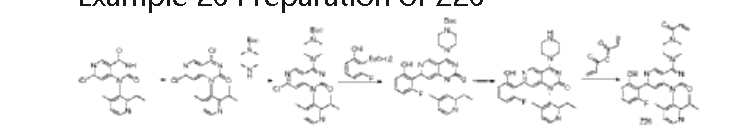
| Step 1: To a solution of 7-chloro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[4,3-d]pyrimidine-2,4(1H,3H)-dione (130 mg, 0.39 mmol) in acetonitrile (3 mL) were added phosphorus oxychloride (1 mL) and N,N-diisopropylethylamine (1 mL) sequentially. The mixture was stirred at 90°C for 2 h. The reaction mixture was concentrated to afford crude 4,7-dichloro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[4,3-d]pyrimidin-2(1H)-one (130 mg). ES-API: [M+H] + = 349.3. |
| Step 2: To a solution of 4,7-dichloro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[4,3-d]pyrimidin-2(1H)-one (130 mg, 0.37 mmol) in acetonitrile (3 mL) was added N,N-diisopropylethylamine (144 mg, 1.12 mmol) and tert-butyl piperazine-1-carboxylate (70 mg, 0.37 mmol) under ice-cooling. The mixture was stirred for 30 minutes. The reaction mixture was poured into 20 mL of water and extracted with ethyl acetate (20 mL x 3). The mixture was dried over anhydrous sodium sulfate and concentrated. The mixture was then purified on a flash silica gel column (0-100% ethyl acetate/petroleum ether) to obtain tert-butyl 4-(7-chloro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[4,3-d]pyrimidin-4-yl)piperazine-1-carboxylate (140 mg) as a white solid. ES-API: [M+H] + = 499.1. |
| Step 3: Under nitrogen protection, a mixture of tert-butyl 4-(7-chloro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[4,3-d]pyrimidin-4-yl)piperazine-1-carboxylate (140 mg, 0.28 mmol), 2-fluoro-6-hydroxyphenylboronic acid (44 mg, 0.42 mmol), chloro(2-dicyclohexylphosphino-2′,6′-dimethoxy-1,1′-biphenyl)(2′-amino-1,1′-biphenyl-2-yl)palladium(II) (13 mg, 0.02 mmol), 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (10 mg, 0.02 mmol) and potassium phosphate (120 mg, 0.84 mmol) in 1,4-dioxane (4 mL) and water (1 mL) was microwaved at 120 ° C for 1 h. The reaction mixture was filtered and washed with ethyl acetate (100 mL). The filtrate was washed with saturated brine (50 mL x 3). The resulting organic phase was dried, concentrated, and purified on a flash silica gel column (0-100% ethyl acetate/petroleum ether) to afford tert-butyl 4-(7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyridinyl[4,3-d]pyrimidin-4-yl)piperazine-1-carboxylate (100 mg, yield: 62%) as a white solid. ES-API: [M+H] + = 575.2. |
| Step 4: To a solution of tert-butyl 4-(7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyridinyl[4,3-d]pyrimidin-4-yl)piperazine-1-carboxylate (100 mg, 0.17 mmol) in dichloromethane (4 mL) was added trifluoroacetic acid (1 mL) under ice-cooling. The mixture was stirred at room temperature for 2 h and concentrated to afford 7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-4-(piperazin-1-yl)pyridin[4,3-d]pyrimidin-2(1H)-one (82 mg, theoretical) as a yellow oil. ES-API: [M+H] + = 475.2. |
| Step 5: Under ice bath, add N,N-diisopropylethylamine (110 mg, 0.85 mmol) to a solution of 7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-4-(piperazin-1-yl)pyridin[4,3-d]pyrimidin-2(1H)-one (82 mg, 0.17 mmol) in dichloromethane (3 mL). After the reaction solution becomes clear, add acrylic anhydride (21 mg, 0.17 mmol) dropwise and stir for 5 minutes. The reaction solution is washed with saturated sodium bicarbonate solution (5 mL). The organic phase is dried, concentrated, and purified by preparative HPLC (ammonium bicarbonate system) to obtain a light yellow solid Z26 (12.44 mg, purity: 100%, yield: 14% ) . NMR (500MHz, DMSO) δ12.86(s,1H),9.26(s,1H),8.59(d,J=4.9Hz,1H),7.35(d,J=4.9Hz,1H),7.29(d d,J=15.0,8.2Hz,1H),6.86(dd,J=16.7,10.4Hz,1H),6.77(d,J=8.3Hz,1H),6.73-6.66(m,2H),6.21( dd,J=16.6,2.3Hz,1H),5.77(dd,J=10.4,2.3Hz,1H),4.07(d,J=5.0Hz,4H),3.88(d,J=36.8Hz,4H),2 .76(dt,J=13.6,6.8Hz,1H),1.96(s,3H),1.10(d,J=6.7Hz,3H),1.04(d,J=6.7Hz,3H).ES-API:[M+H] + =529.2. |
SYN
Fulzerasib is an orally active KRAS G12C inhibitor developed by Innovent Biologics. It selectively targets the KRAS G12C mutation in NSCLC [36,37]. In 2024, the NMPA approved Fulzerasib (brand name: Dupert) for treating adult patients with advanced NSCLC harboring the KRAS G12C mutation who have progressed after prior systemic therapy. Fulzerasib irreversibly binds to the KRAS G12C mutant protein, locking it in an inactive GDP-bound state, thereby inhibiting downstream signaling pathways such as MAPK and PI3K. This action effectively suppresses cancer cell proliferation and survival. The clinical efficacy of Fulzerasib was demonstrated in a Phase II trial (NCT05009303) involving patients with advanced NSCLC and KRAS G12C mutations [38]. In the clinical trial, Fulzerasib demonstrated an ORR of 49.1 % and a disease control rate (DCR) of 90.5 %, with a median PFS of 9.7 months, reflecting robust antitumor efficacy. The agent exhibited favorable tolerability, characterized by manageable toxicity. Treatment-related adverse events were predominantly mild to moderate in severity, with the most frequently reported being diarrhea, nausea, and elevated liver enzymes [38]. The safety profile was consistent with other KRAS G12Cinhibitors, making it a viable therapeutic option.
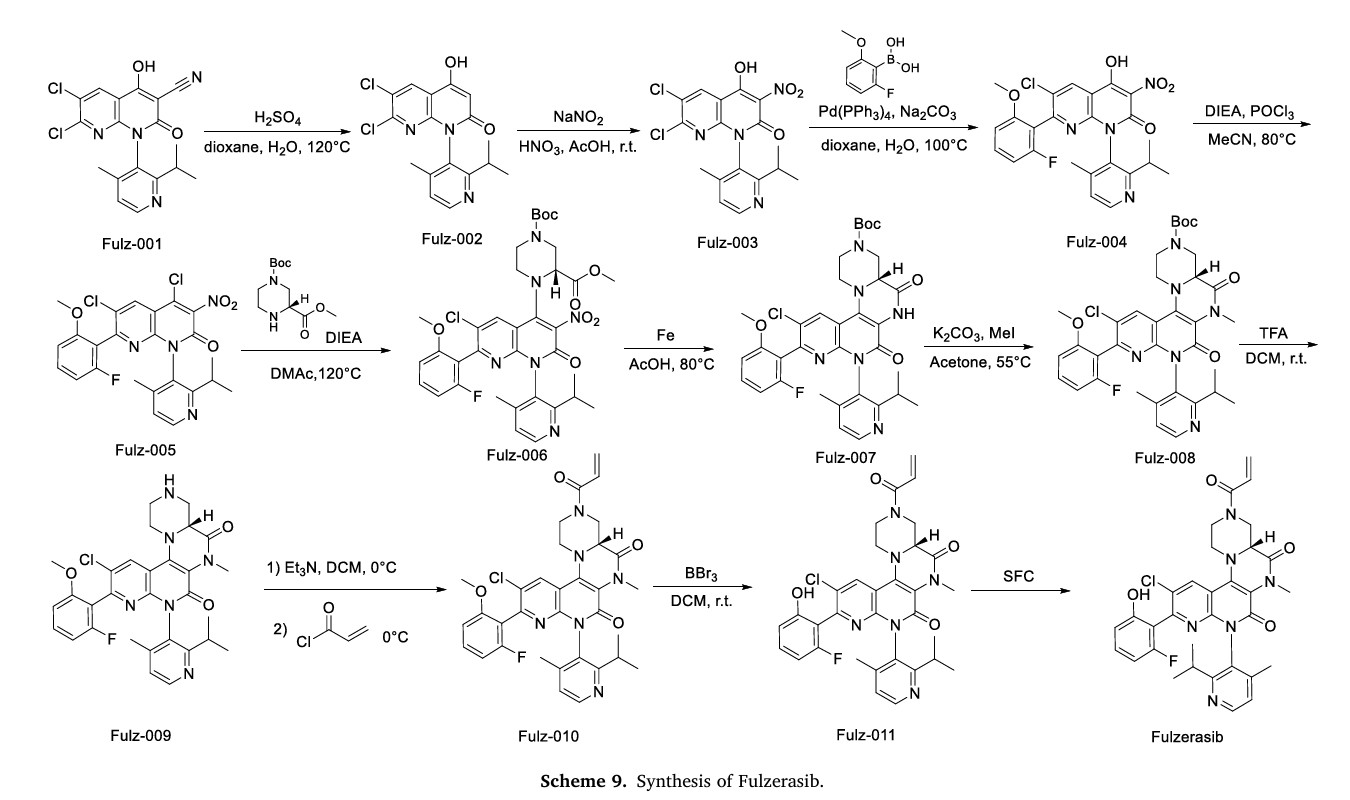
The synthetic route of Fulzerasib, shown in Scheme 9, initiates with H2SO4-mediated decyanation of Fulz-001, affording Fulz-002 [39]. Nitrosation of Fulz-002 with NaNO2 yields Fulz-003, which undergoes
Suzuki-Miyaura coupling with (2-fluoro-6-methoxyphenyl)boronic acid to construct Fulz-004. Phosphochlorination with POCl3 under DIPEA catalysis converts Fulz-004 to Fulz-005. Nucleophilic displacement with methyl (R)-1-N-Boc-piperazine-3-carboxylate assembles Fulz-006. Fe-mediated tandem Mannich cyclization/nitro reduction transforms Fulz-006 into bicyclic amine Fulz-007. Methylation with MeI generates Fulz-008, followed by TFA-mediated Boc cleavage to afford Fulz-009.
Acrylation with acryloyl chloride produces Fulz-010. Selective O-demethylation followed by chiral HPLC resolution delivers Fulzerasib
[36] Y.N. Lamb, Correction: fulzerasib: first approval, Drugs 85 (2025) 281.
[37] Y.N. Lamb, Fulzerasib: first approval, Drugs 84 (2024) 1665–1671.
[38] Q. Zhou, X. Meng, L. Sun, D. Huang, N. Yang, Y. Yu, M. Zhao, W. Zhuang, R. Guo,
Y. Hu, Y. Pan, J. Shan, M. Sun, Y. Yuan, Y. Fan, J. Huang, L. Liu, Q. Chu, X. Wang,
C. Xu, J. Lin, J. Huang, M. Huang, J. Sun, S. Zhang, H. Zhou, Y.L. Wu, Efficacy and
safety of KRASG12C inhibitor IBI351 monotherapy in patients with advanced
NSCLC: results from a phase 2 pivotal study, J. Thorac. Oncol. 19 (2024)
1630–1639.
[39] F. Zhou, T. Jiang, W. He, L. Cai, H. Yang, Z. Liu, J. Lan, Preparation of
Heteroaromatic Ring Dihydropyrimidinone Derivatives as KRAS Gene Mutation
Inhibitors Useful in the Treatment of Cancer, 2021. CN112390818A.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
- [1]. Rafael Rosell, et al. KRAS G12C-mutant driven non-small cell lung cancer (NSCLC). Crit Rev Oncol Hematol. 2024 Mar:195:104228. [Content Brief][2]. Vanesa Gregorc, et al. KROCUS: A phase II study investigating the efficacy and safety of fulzerasib. Free access.June 05, 2024
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Fulzerasib: First ApprovalPublication Name: DrugsPublication Date: 2024-11-26PMID: 39587006DOI: 10.1007/s40265-024-02120-6
- Substituted heterocyclic compounds, their preparation methods and medicinal usesPublication Number: CN-117645614-APriority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: EP-4328229-A2Priority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: US-12054497-B2Priority Date: 2019-10-30Grant Date: 2024-08-06
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: US-2024158417-A1Priority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: EP-4053118-B1Priority Date: 2019-10-30Grant Date: 2024-10-09
- Substituted heterocyclic and cyclic compounds, their preparation and medical usePublication Number: CN-113853373-BPriority Date: 2019-10-30Grant Date: 2022-08-05
- Substituted heterocyclic ring compound, preparation method and medical application thereofPublication Number: CN-115057872-APriority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: EP-4053118-A1Priority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, method for preparing same, and pharmaceutical use thereofPublication Number: KR-20220106765-APriority Date: 2019-10-30
- Substituted heterocyclic fused cyclic compound, preparation method therefor and pharmaceutical use thereofPublication Number: US-2023084095-A1Priority Date: 2019-10-30
- Polymorph of kras inhibitor, preparation method therefor, and use thereofPublication Number: WO-2023116895-A1Priority Date: 2021-12-24
- Polymorph of kras inhibitor, preparation method therefor, and use thereofPublication Number: EP-4455147-A1Priority Date: 2021-12-24
- Polymorph of kras inhibitor, preparation method therefor, and use thereofPublication Number: US-2025051333-A1Priority Date: 2021-12-24
- Key intermediate of kras inhibitor and preparation method thereforPublication Number: WO-2022198904-A1Priority Date: 2021-03-26
- A key intermediate of a KRAS inhibitor and its preparation methodPublication Number: CN-116848111-APriority Date: 2021-03-26
- Pharmaceutical composition, and preparation method therefor and use thereofPublication Number: WO-2024061267-A1Priority Date: 2022-09-21
- Pharmaceutical composition, use thereof, and method for treating cancerPublication Number: WO-2023186075-A1Priority Date: 2022-04-01
- Pharmaceutical composition, use thereof, and method for treating cancerPublication Number: EP-4506004-A1Priority Date: 2022-04-01
- Polymorphs of KRAS inhibitors, methods of making and uses thereofPublication Number: CN-118451081-APriority Date: 2021-12-24
- Polymorph of kras inhibitor and preparation method for polymorph and use thereofPublication Number: TW-202327593-APriority Date: 2021-12-24
////////Fulzerasib, CHINA 2024, APPROVALS 2024, Innovent Biologics, DUPERT, GFH925, GFH 925, IBI351, IBI 351

