
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

DASATINIB
ダサチニブ水和物
BMS 354825
863127-77-9 HYDRATE, USAN, BAN INN, JAN
UNII: RBZ1571X5H
302962-49-8 FREE FORM Dasatinib anhydrous USAN, INN
Molecular Formula, C22-H26-Cl-N7-O2-S.H2-O, Molecular Weight, 506.0282T6N DNTJ A2Q D- DT6N CNJ B1 FM- BT5N CSJ DVMR BG F1[WLN]X78UG0A0RNдазатиниб [Russian] [INN]دازاتينيب [Arabic] [INN]达沙替尼 [Chinese] [INN]1132093-70-9[RN]302962-49-8[RN]5-Thiazolecarboxamide, N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-87129966762[Beilstein]
A pyrimidine and thiazole derived ANTINEOPLASTIC AGENT and PROTEIN KINASE INHIBITOR of BCR-ABL KINASE. It is used in the treatment of patients with CHRONIC MYELOID LEUKEMIA who are resistant or intolerant to IMATINIB.
An orally bioavailable synthetic small molecule-inhibitor of SRC-family protein-tyrosine kinases. Dasatinib binds to and inhibits the growth-promoting activities of these kinases. Apparently because of its less stringent binding affinity for the BCR-ABL kinase, dasatinib has been shown to overcome the resistance to imatinib of chronic myeloid leukemia (CML) cells harboring BCR-ABL kinase domain point mutations. SRC-family protein-tyrosine kinases interact with a variety of cell-surface receptors and participate in intracellular signal transduction pathways; tumorigenic forms can occur through altered regulation or expression of the endogenous protein and by way of virally-encoded kinase genes. (NCI Thesaurus)
5-Thiazolecarboxamide, N-(2-chloro-6-methylphenyl)-2-((6-(4-(2-hydroxyethyl)-1-piperazinyl)-2-methyl-4-pyrimidinyl)amino)-, monohydrate
Synthesis ReferenceUS6596746
DASATINIB ANHYDROUS
- KIN 001-5
- NSC 759877
- Sprycel
- 302962-49-8 Dasatinib anhydrous
- 5-THIAZOLECARBOXAMIDE, N-(2-CHLORO-6-METHYLPHENYL)-2-((6-(4-(2-HYDROXYETHYL)-1-PIPERAZINYL)-2-METHYL-4-PYRIMIDINYL)AMINO)-
- BMS-354825
- DASATINIB [INN]
- DASATINIB [MI]
- DASATINIB [WHO-DD]
- DASATINIB ANHYDROUS
| No. | NDA No. | Major Technical Classification | Patent No. | Estimated Expiry Date | Drug Substance Claim | Drug Product Claim | Patent Use Code (All list) |
| 1 | N021986 | Formula | 6596746 | 2020-06-28 | Y | Y | U – 748 |
| 2 | N021986 | Formula | 6596746 | 2020-06-28 | Y | Y | U – 780 |
| 3 | N021986 | Uses(Indication) | 7125875 | 2020-04-13 | U – 779 | ||
| 4 | N021986 | Uses(Indication) | 7125875 | 2020-04-13 | U – 780 | ||
| 5 | N021986 | Uses(Indication) | 7153856 | 2020-04-28 | U – 780 | ||
| 6 | N021986 | Crystal | 7491725 | 2026-03-28 | Y | Y | |
| 7 | N021986 | Formulation | 8680103 | 2025-02-04 | Y |



SPRYCEL (dasatinib) is an inhibitor of multiple tyrosine kinases.
The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2- methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate).
The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure: Dasatinib is a white to off-white powder and has a melting point of 280°–286° C.
The drug substance is insoluble in water and slightly soluble in ethanol and methanol. SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol
| DASATINIBDASATINIB (DASATINIB) | ANDA #202103 | TABLET;ORAL | Discontinued | APOTEX INC |
| SPRYCELSPRYCEL (DASATINIB) | NDA #021986 | TABLET;ORAL | Prescription | BRISTOL MYERS SQUIBBSPRYCEL (DASATINIB) | NDA #022072 | TABLET; ORAL | Prescription | BRISTOL MYERS SQUIBB |
Clip
https://www.pharmainbrief.com/files/2017/09/A-106-17-20170918-Reasons.pdfhttps://www.accessdata.fda.gov/drugsatfda_docs/appletter/2016/202103Orig1s000ltr.pdfU.S. Patent Number Expiration Date 6,596,746 (the ‘746 patent) June 28, 20207,125,875 (the ‘875 patent) April 13, 20207,153,856 (the ‘856 patent) April 28, 20207,491,725 (the ‘725 patent) March 28, 20268,680,103 (the ‘103 patent) February 4, 2025
Drug Name:Dasatinib HydrateResearch Code:BMS-354825Trade Name:Sprycel®MOA:Kinase inhibitorIndication:Acute lymphoblastic leukaemia (ALL); Chronic myeloid leukemia (CML )Status:ApprovedCompany:Bristol-Myers Squibb (Originator)Sales:$1,620 Million (Y2015);
$1,493 Million (Y2014);
$1,280 Million (Y2013);
$1,019 Million (Y2012);
$803 Million (Y2011);ATC Code:L01XE06Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2006-06-28 | Marketing approval | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | Eq. 20 mg/50 mg/70 mg/80 mg/100 mg/140 mg Dasatinib | Bristol-Myers Squibb | Priority; Orphan |
| 2006-06-28 | Additional approval | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | 70 mg | Bristol-Myers Squibb | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2006-11-20 | Marketing approval | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | 20 mg/50 mg/70 mg/80 mg/100 mg/140 mg | Bristol-Myers Squibb | Orphan |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-06-16 | Modified indication | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | 20 mg/50 mg | Bristol-Myers Squibb, Otsuka | |
| 2009-01-21 | Marketing approval | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | 20 mg/50 mg | Bristol-Myers Squibb, Otsuka |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-09-17 | Marketing approval | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 20 mg | 南京正大天晴制药 | ||
| 2013-09-17 | Marketing approval | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 50 mg | 南京正大天晴制药 | ||
| 2013-09-17 | Marketing approval | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 70 mg | 南京正大天晴制药 | ||
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 50 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 50 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 50 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 20 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 20 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 20 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 70 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 70 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 70 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 100 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 100 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 100 mg | Bristol-Myers Squibb |
SPRYCEL (dasatinib) is a kinase inhibitor. The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate). The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure:
 |
Dasatinib is a white to off-white powder. The drug substance is insoluble in water and slightly soluble in ethanol and methanol.
SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol.
Dasatinib hydrate was first approved by the U.S. Food and Drug Administration (FDA) on June 28, 2006, then approved by European Medicine Agency (EMA) on Nov 20, 2006, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 21, 2009. It was developed and marketed as Sprycel® by Bristol Myers Squibb in the US.
Dasatinibhydrate is a kinase inhibitor.It is indicated for the treatment ofchronic myeloid leukemia and acutelymphoblastic leukemia.
Sprycel® is available as film-coatedtabletfor oral use, containing 20, 50, 70, 80, 100 or 140 mg offreeDasatinib. The recommended dose is 100 mg once daily forchronic myeloid leukemia. Another dose is 140 mg once daily for accelerated phase chronic myeloid leukemia, myeloid or lymphoid blast phase chronic myeloid leukemia, or Ph+ acutelymphoblastic leukemia.
Dasatinib, also known as BMS-354825, is an orally bioavailable synthetic small molecule-inhibitor of SRC-family protein-tyrosine kinases. Dasatinib binds to and inhibits the growth-promoting activities of these kinases. Apparently because of its less stringent binding affinity for the BCR-ABL kinase, dasatinib has been shown to overcome the resistance to imatinib of chronic myeloid leukemia (CML) cells harboring BCR-ABL kinase domain point mutations.
Dasatinib, sold under the brand name Sprycel among others, is a targeted therapy medication used to treat certain cases of chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL).[3] Specifically it is used to treat cases that are Philadelphia chromosome-positive (Ph+).[3] It is taken by mouth.[3]
Common adverse effects include low white blood cells, low blood platelets, anemia, swelling, rash, and diarrhea.[3] Severe adverse effects may include bleeding, pulmonary edema, heart failure, and prolonged QT syndrome.[3] Use during pregnancy may result in harm to the baby.[3] It is a tyrosine-kinase inhibitor and works by blocking a number of tyrosine kinases such as Bcr-Abl and the Src kinase family.[3]
Dasatinib was approved for medical use in the United States and in the European Union in 2006.[3][2] It is on the World Health Organization’s List of Essential Medicines.
Medical uses
Dasatinib is used to treat people with chronic myeloid leukemia and people with acute lymphoblastic leukemia who are positive for the Philadelphia chromosome.[5]
In the EU dasatinib is indicated for children with
- newly diagnosed Philadelphia chromosome-positive chronic myelogenous leukaemia in chronic phase (Ph+ CML CP) or Ph+ CML CP resistant or intolerant to prior therapy including imatinib.[2]
- newly diagnosed Ph+ acute lymphoblastic leukaemia (ALL) in combination with chemotherapy.[2]
- newly diagnosed Ph+ CML in chronic phase (Ph+ CML-CP) or Ph+ CML-CP resistant or intolerant to prior therapy including imatinib.[2]
and adults with
- newly diagnosed Philadelphia-chromosome-positive (Ph+) chronic myelogenous leukaemia (CML) in the chronic phase;[2]
- chronic, accelerated or blast phase CML with resistance or intolerance to prior therapy including imatinib mesilate;[2]
- Ph+ acute lymphoblastic leukaemia (ALL) and lymphoid blast CML with resistance or intolerance to prior therapy.[2]
Adverse effects
The most common side effects are infection, suppression of the bone marrow (decreasing numbers of leukocytes, erythrocytes, and thrombocytes),[6] headache, hemorrhage (bleeding), pleural effusion (fluid around the lungs), dyspnea (difficulty breathing), diarrhea, vomiting, nausea (feeling sick), abdominal pain (belly ache), skin rash, musculoskeletal pain, tiredness, swelling in the legs and arms and in the face, fever.[2] Neutropenia and myelosuppression were common toxic effects. Fifteen people (of 84, i.e. 18%) in the above-mentioned study developed pleural effusions, which was a suspected side effect of dasatinib. Some of these people required thoracentesis or pleurodesis to treat the effusions. Other adverse events included mild to moderate diarrhea, peripheral edema, and headache. A small number of people developed abnormal liver function tests which returned to normal without dose adjustments. Mild hypocalcemia was also noted, but did not appear to cause any significant problems. Several cases of pulmonary arterial hypertension (PAH) were found in people treated with dasatinib,[7] possibly due to pulmonary endothelial cell damage.[8]
On October 11, 2011, the U.S. Food and Drug Administration (FDA) announced that dasatinib may increase the risk of a rare but serious condition in which there is abnormally high blood pressure in the arteries of the lungs (pulmonary hypertension, PAH).[9] Symptoms of PAH may include shortness of breath, fatigue, and swelling of the body (such as the ankles and legs).[9] In reported cases, people developed PAH after starting dasatinib, including after more than one year of treatment.[9] Information about the risk was added to the Warnings and Precautions section of the Sprycel drug label.[9]
Pharmacology

Crystal structure[10] (PDB 2GQG) of Abl kinase domain (blue) in complex with dasatinib (red).
Dasatinib is an ATP-competitive protein tyrosine kinase inhibitor. The main targets of dasatinib are BCR/Abl (the “Philadelphia chromosome”), Src, c-Kit, ephrin receptors, and several other tyrosine kinases.[11] Strong inhibition of the activated BCR-ABL kinase distinguishes dasatinib from other CML treatments, such as imatinib and nilotinib.[11][12] Although dasatinib only has a plasma half-life of three to five hours, the strong binding to BCR-ABL1 results in a longer duration of action.[12]
History
See also: Discovery and development of Bcr-Abl tyrosine kinase inhibitors
Dasatinib was developed by collaboration of Bristol-Myers Squibb and Otsuka Pharmaceutical Co., Ltd,[13][14][15] and named for Bristol-Myers Squibb research fellow Jagabandhu Das, whose program leader says that the drug would not have come into existence had he not challenged some of the medicinal chemists‘ underlying assumptions at a time when progress in the development of the molecule had stalled.[16]
Society and culture
Legal status
Dasatinib was approved for used in the United States in June 2006 and in the European Union in November 2006[17][2]
In October 2010, dasatinib was approved in the United States for the treatment of newly diagnosed adults with Philadelphia chromosome positive chronic myeloid leukemia in chronic phase (CP-CML).[18]
In November 2017, dasatinib was approved in the United States for the treatment of children with Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in the chronic phase.[19]
Approval was based on data from 97 pediatric participants with chronic phase CML evaluated in two trials—a Phase I, open-label, non-randomized, dose-ranging trial and a Phase II, open-label, non-randomized trial.[19] Fifty-one participants exclusively from the Phase II trial were newly diagnosed with chronic phase CML and 46 participants (17 from the Phase I trial and 29 from the Phase II trial) were resistant or intolerant to previous treatment with imatinib.[19] The majority of participants were treated with dasatinib tablets 60 mg/m2 body surface area once daily.[19] Participants were treated until disease progression or unacceptable toxicity.[19]
Economics
The Union for Affordable Cancer Treatment objected to the price of dasatinib, in a letter to the U.S. trade representative. The average wholesale price in the U.S. is $367 per day, twice the price in other high income countries. The price in India, where the average annual per capita income is $1,570, and where most people pay out of pocket, is Rs6627 ($108) a day. Indian manufacturers offered to supply generic versions for $4 a day, but, under pressure from the U.S., the Indian Department of Industrial Policy and Promotion refused to issue a compulsory license.[20]
Bristol-Myers Squibb justified the high prices of cancer drugs with the high R&D costs, but the Union of Affordable Cancer Treatment said that most of the R&D costs came from the U.S. government, including National Institutes of Health funded research and clinical trials, and a 50% tax credit. In England and Wales, the National Institute for Health and Care Excellence recommended against dasatinib because of the high cost-benefit ratio.[20]
The Union for Affordable Cancer Treatment said that “the dasatinib dispute illustrates the shortcomings of US trade policy and its impact on cancer patients”[20]
Brand names
In Bangladesh dasatinib is available under the trade name Dasanix by Beacon Pharmaceuticals.In India, It is marketed by brand name NEXTKI by EMCURE PHARMACEUTICALS[medical citation needed]
Research
Dasatinib has been shown to eliminate senescent cells in cultured adipocyte progenitor cells.[21] Dasatinib has been shown to induce apoptosis in senescent cells by inhibiting Src kinase, whereas quercetin inhibits the anti-apoptotic protein Bcl-xL.[21] Administration of dasatinib along with quercetin to mice improved cardiovascular function and eliminated senescent cells.[22] Aged mice given dasatinib with quercetin showed improved health and survival.[22]
Giving dasatinib and quercetin to mice eliminated senescent cells and caused a long-term resolution of frailty.[23] A study of fourteen human patients suffering from idiopathic pulmonary fibrosis (a disease characterized by increased numbers of senescent cells) given dasatinib and quercetin showed improved physical function and evidence of reduced senescent cells.[21]Route 1
Reference:1. WO2005077945A2 / US2012302750A1.Route 2
Reference:1. WO0062778A1 / US6596746B1.Route 3
Reference:1. J. Med. Chem. 2004, 47, 6658-6661.
2. J. Med. Chem. 2006, 49, 6819-6832.Route 4
Reference:1. CN104292223A.Route 5
Reference:1. CN103420999A.
Syn 1

Reference
Balaji, N.; Sultana, Sayeeda. Trace level determination and quantification of potential genotoxic impurities in dasatinib drug substance by UHPLC/infinity LC. International Journal of Pharmacy and Pharmaceutical Sciences. Department of Chemistry. St. Peter’s University. Tamil Nadu, India 600054. Volume 8. Issue 10. Pages 209-216. 2016
SYN 2

Reference
Zhang, Shaoning; Wei, Hongtao; Ji, Min. Synthesis of dasatinib. Zhongguo Yiyao Gongye Zazhi. Dept. of Pharmaceutical Engineering, School of Chemistry & Chemical Engineering. Southeast University. Nanjing, Jiangsu Province, Peop. Rep. China 210096. Volume 41. Issue 3. Pages 161-163. 2010
SYN 3

Reference
Suresh, Garbapu; Nadh, Ratnakaram Venkata; Srinivasu, Navuluri; Yennity, Durgaprasad. A convenient new and efficient commercial synthetic route for dasatinib (Sprycel). Synthetic Communications. Division of Chemistry, Department of Science and Humanities. Vignan’s Foundation for Science Technology and Research University. Guntur, India. Volume 47. Issue 17. Pages 1610-1621. 2017
SYN 4

Reference
Chen, Bang-Chi; Zhao, Rulin; Wang, Bei; Droghini, Roberto; Lajeunesse, Jean; Sirard, Pierre; Endo, Masaki; Balasubramanian, Balu; Barrish, Joel C. A new and efficient preparation of 2-aminothiazole-5-carbamides: applications to the synthesis of the anticancer drug dasatinib. ARKIVOC (Gainesville, FL, United States). Discovery Chemistry. Bristol-Myers Squibb Research and Development. Princeton, USA 08543. Issue 6.Pages 32-38. 2010
SYN 5

Reference
An, Kang; Guan, Jianning; Yang, Hao; Hou, Wen; Wan, Rong. Improvement on the synthesis of Dasatinib. Jingxi Huagong Zhongjianti. College of Science. Nanjing University of Technology. Nanjing, Jiangsu Province, Peop. Rep. China 211816. Volume 41. Issue 2. Pages 42-44. 2011
PATENT
https://patents.google.com/patent/US7491725B2/en
EXAMPLESExample 1Preparation of Intermediate:
(S)-1-sec-Butylthiourea

To a solution of S— sec-butyl-amine (7.31 g, 0.1 mol) in chloroform (80 mL) at 0° C. was slowly added benzoyl isothiocyanate (13.44 mL, 0.1 mol). The mixture was allowed to warm to 10° C. and stirred for 10 min. The solvent was then removed under reduced pressure, and the residue was dissolved in MeOH (80 mL). An aqueous solution (10 mL) of NaOH (4 g, 0.1 mol) was added to this solution, and the mixture was stirred at 60° C. for another 2 h. The MeOH was then removed under reduced pressure, and the residue was stirred in water (50 mL). The precipitate was collected by vacuum filtration and dried to provide S-1-sec-butyl-thiourea (12.2 g, 92% yield). mp 133-134° C.; 1H NMR (500 MHz, DMSO-D6) δ 7.40 (s, 1H), 7.20 (br s, 1H), 6.76 (s, 1H), 4.04 (s, 1H), 1.41 (m, 2H), 1.03 (d, J=6.1 Hz, 3H), 0.81 (d, J=7.7 Hz, 3H); 13C NMR (125 MHz, DMSO-D6) δ 182.5, 50.8, 28.8, 19.9, 10.3; LRMS m/z 133.2 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.49; H, 8.88; N, 21.32; S, 24.27.
Example 2Preparation of Intermediate:
(R)-1-sec-Butylthiourea

(R)-1-sec-Butylthiourea was prepared in 92% yield according to the general method outlined for Example 1. mp 133-134° C.; 1H NMR(500 MHz, DMSO) δ 0.80(m, 3H, J=7.7), 1.02(d, 3H, J=6.1), 1.41(m, 2H), (3.40, 4.04)(s, 1H), 6.76(s, 1H), 7.20(s, br, 1H), 7.39(d, 1H, J=7.2); 13C NMR (500 MHz, DMSO) δ: 10.00, 19.56, 28.50, 50.20, 182.00; m/z 133.23 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.32; H, 9.15; N, 21.14; S, 24.38.
Example 3Preparation of:

To a solution of 3-amino-N-methyl-4-methylbenzamide hydrochloride (1.0 g, 5 mmol) in acetone (10 mL) at 0° C. was added pyridine (1.2 mL, 15 mmol) dropwise via syringe. 3-Methoxyacryloyl chloride (0.72 mL. 6.5 mmol) was added and the reaction stirred at room temperature for 1 h. The solution was cooled again to 0° C. and 1N HCl (1.5 mL) was added dropwise via pipet. The reaction mixture was stirred for 5 min, then water (8.5 mL) was added via an addition funnel. The acetone was removed in vacuo and the resulting solution stirred for 4h. Crystallization began within 15 min. After stirring for 4 h, the vessel was cooled in an ice bath for 30 min, filtered, and rinsed with ice cold water (2×3 mL) to give compound 3A (0.99 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.12 (br s, 1H), 7.76 (s, 1H), 7.29 (m, 2H), 7.05 (d, J=7.9 Hz, 1H), 5.47 (d, J=12.3 Hz, 1H), 3.48 (s, 3H), 2.54 (d, J=4.7 Hz, 3H), 2.03 (s, 3H); HPLC rt 2.28 min (Condition A).
3B. Example 3To a 50 mL RBF containing the above compound 3A (0.5 g, 2.0 mmol) was added THF (2.5 mL) and water (2 mL), followed by NBS (0.40 g, 2.22 mmol), and the solution was stirred for 90 min. R-sec-butylthiourea (Ex. 2) (267 mg), was added, and the solution was heated to 75° C. for 8 h. Conc. NH4OH was added to adjust the pH to 10 followed by the addition of EtOH (15 mL). Water (15 mL) was added and the slurry stirred for 16 h, filtered, and washed with water to give Example 3 as a light brown solid (0.48 g, 69% yield, 98% purity). MS 347.1; HPLC 2.59.
Example 4Preparation of:

Example 4 is prepared following the methods of Example 3 but using the appropriate acryl benzamide and Example 1.
Example 5Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (The Compound of Formula (IV))

5A. 1-(6-Chloro-2-methylpyrimidin-4-yl)thiourea

To a stirring slurry of 4-amino-5-chloro-2-methylpyrimidine (6.13 g, 42.7 mmol) in THF (24 mL) was added ethyl isothiocyanatoformate (7.5 mL, 63.6 mmol), and the mixture heated to reflux. After 5h, another portion of ethyl isothiocyanato formate (1.0 mL, 8.5 mmol) was added and after 10h, a final portion (1.5 mL, 12.7 mmol) was added and the mixture stirred 6h more. The slurry was evaporated under vacuum to remove most of the solvent and heptane (6 mL) added to the residue. The solid was collected by vacuum filtration and washed with heptane (2×5 mL) giving 8.01 g (68% yield) of the intermediate ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate.A solution of ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate (275 mg, 1.0 mmol) and 1N sodium hydroxide (3.5 eq) was heated and stirred at 50° C. for 2h. The resulting slurry was cooled to 20-22° C. The solid was collected by vacuum filtration, washed with water, and dried to give 185 mg of 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea (91% yield). 1H NMR (400 MHz, DMSO-d6): δ2.51 (S, 3H), 7.05 (s, 1H), 9.35 (s,1H), 10.07 (s, 1H), 10.91 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 25.25, 104.56, 159.19, 159.33, 167.36, 180.91.
5B. (E)-N-(2-Chloro-6-methylphenyl)-3-ethoxyacrylamide

To a cold stirring solution of 2-chloro-6-methylaniline (59.5 g 0.42 mol) and pyridine (68 ml, 0.63 mol) in THF (600 mL) was added 3-ethoxyacryloyl chloride (84.7 g, 0.63 mol) slowly keeping the temp at 0-5° C. The mixture was then warmed and stirred for 2 h. at 20° C. Hydrochloric acid (1N, 115 mL) was added at 0-10° C. The mixture was diluted with water (310 mL) and the resulting solution was concentrated under vacuum to a thick slurry. The slurry was diluted with toluene (275 mL) and stirred for 15 min. at 20-22° C. then 1 h. at 0° C. The solid was collected by vacuum filtration, washed with water (2×75 mL) and dried to give 74.1 g (73.6% yield) of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide). 1H NMR (400 Hz, DMSO-d6) δ 1.26 (t, 3H, J=7 Hz), 2.15 (s, 3H), 3.94 (q, 2H, J=7 Hz), 5.58 (d, 1H, J=12.4 Hz), 7.10-7.27 (m, 2H, J=7.5 Hz), 7.27-7.37 (d, 1H, J=7.5 Hz), 7.45(d, 1H, J=12.4 Hz), 9.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 14.57, 18.96, 67.17, 97.99, 126.80, 127.44, 129.07, 131.32, 132.89, 138.25, 161.09, 165.36.
5C. 2-Amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide

To a mixture of compound 5B (5.00 g, 20.86 mmol) in 1,4-dioxane (27 mL) and water (27 mL) was added NBS (4.08 g, 22.9 mmol) at −10 to 0° C. The slurry was warmed and stirred at 20-22° C. for 3h. Thiourea (1.60 g, 21 mmol) was added and the mixture heated to 80° C. After 2h, the resulting solution was cooled to 20-22° and conc. ammonium hydroxide (4.2 mL) was added dropwise. The resulting slurry was concentrated under vacuum to about half volume and cooled to 0-5° C. The solid was collected by vacuum filtration, washed with cold water (10 mL), and dried to give 5.3 g (94.9% yield) of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6) δ δ 2.19 (s, 3H), 7.09-7.29 (m, 2H, J=7.5), 7.29-7.43 (d, 1H, J=7.5), 7.61 (s, 2H), 7.85 (s, 1H), 9.63 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 18.18, 120.63, 126.84, 127.90, 128.86, 132.41, 133.63, 138.76, 142.88, 159.45, 172.02.
5D. 2-(6-Chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide

To a stirring solution of compound 5C (5.00 g, 18.67 mmol) and 4,6-dichloro-2-methylpyrimidine (3.65 g 22.4/mmol) in THF (65 mL) was added a 30% wt. solution of sodium t-butoxide in THF (21.1 g, 65.36 mmol) slowly with cooling to keep the temperature at 10-20° C. The mixture was stirred at room temperature for 1.5 h and cooled to 0-5° C. Hydrochloric acid, 2N (21.5 mL) was added slowly and the mixture stirred 1.75 h at 0-5° C. The solid was collected by vacuum filtration, washed with water (15 mL) and dried to give 6.63 g (86.4% yield) of compound 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).
5E. Example 5To a mixture of compound 5D (4.00 g, 10.14 mmol) and hydroxyethylpiperazine (6.60 g, 50.69 mmol) in n-butanol (40 mL) was added DIPEA (3.53 mL, 20.26 mmol). The slurry was heated at 118° C. for 4.5 h, then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with n-butanol (5 mL), and dried. The product (5.11 g) was dissolved in hot 80% EtOH—H2O (80 mL), and the solution was clarified by filtration. The hot solution was slowly diluted with water (15 mL) and cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with 50% ethanol-water (5 mL) and dried affording 4.27 g (83.2% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide as monohydrate. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.40 (s, 3H), 2.42 (t, 2H, J=6), 2.48 (t, 4H, J=6.3), 3.50 (m, 4H), 3.53 (q, 2H, J=6), 4.45 (t, 1H, J=5.3), 6.04 (s, 1H), 7.25 (t, 1H, J=7.6), 7.27 (dd, 1H, J=7.6, 1.7), 7.40 (dd, 1H, J=7.6, 1.7), 8.21 (s, 1H), 9.87 (s, 1H), 11.47.
Example 6Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide

To a slurry of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide 5B (120 mg, 0.50 mmol) in THF (0.75 ml) and water (0.5 mL) was added NBS (98 mg, 0.55 mmol) at 0° C. The mixture was warmed and stirred at 20-22° C. for 3h. To this was added 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea 5A (100 mg, 0.49 mmol), and the slurry heated and stirred at reflux for 2h. The slurry was cooled to 20-22° C. and the solid collected by vacuum filtration giving 140 mg (71% yield) of 2-(6-chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).Compound 5D was elaborated to N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide, following Step 5E.
Example 7Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide7A. 2-[4-(6-Chloro-2-methyl-pyrimidin-4-yl)-piperazin-1-yl]-ethanol
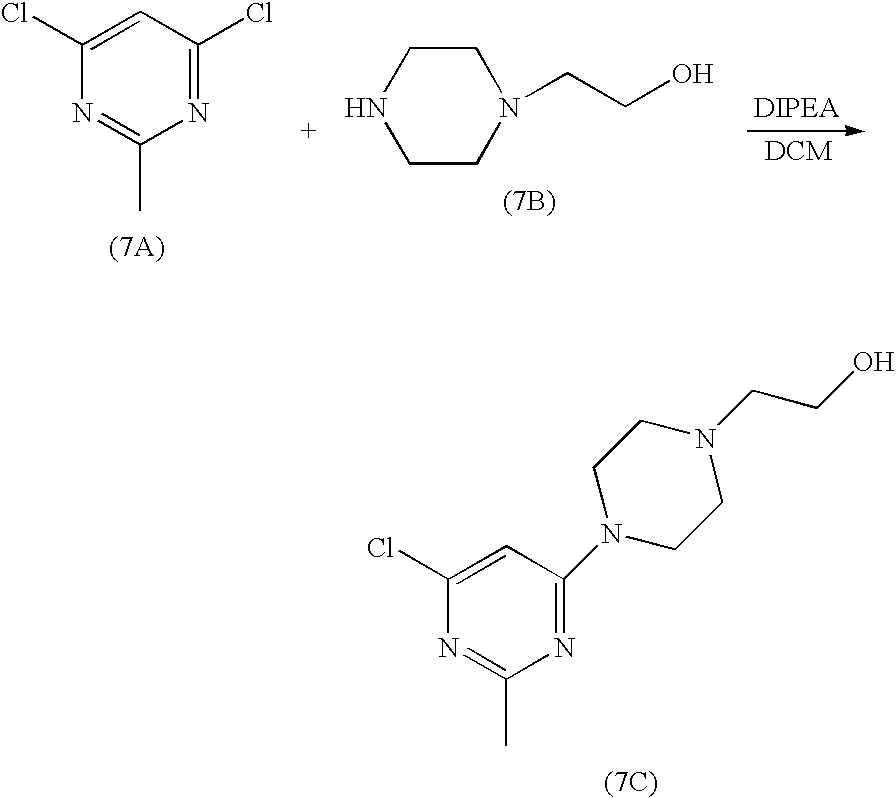
2-piperazin-1-yl-ethanol (8.2 g, 63.1 mmol) was added to a solution of 4,6-dichloro-2-methylpyrimidine (5.2 g, 31.9 mmol) in dichloromethane (80 ml) at rt. The mixture was stirred for two hours and triethylamine (0.9 ml) was added. The mixture was stirred at rt for 20h. The resultant solid was filtered. The cake was washed with dichloromethane (20 ml). The filtrate was concentrated to give an oil. This oil was dried under high vacuum for 20h to give a solid. This solid was stirred with heptane (50 ml) at rt for 5h. Filtration gave 7C (8.13 g) as a white solid
7B. Example 7

To a 250 ml of round bottom flask were charged compound 5C (1.9 g, 7.1 mmol), compound 7C (1.5 g, 5.9 mmol), K2CO3 (16 g, 115.7 mmol), Pd (OAc)2 (52 mg, 0.23 mmol) and BINAP (291 mg, 0.46 mmol). The flask was placed under vacuum and flushed with nitrogen. Toluene was added (60 ml). The suspension was heated to 100-110° C. and stirred at this temperature for 20h. After cooling to room temperature, the mixture was applied to a silica gel column. The column was first eluted with EtOAC, and then with 10% of MeOH in EtOAC. Finally, the column was washed with 10% 2M ammonia solution in MeOH/90% EtOAC. The fractions which contained the desired product were collected and concentrated to give compound IV as a yellow solid (2.3 g).
Analytical MethodsSolid State Nuclear Magnetic Resonance (SSNMR)All solid-state C-13 NMR measurements were made with a Bruker DSX-400, 400 MHz NMR spectrometer. High resolution spectra were obtained using high-power proton decoupling and the TPPM pulse sequence and ramp amplitude cross-polarization (RAMP-CP) with magic-angle spinning (MAS) at approximately 12 kHz (A. E. Bennett et al, J. Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu and S. O. Smith, J. Magn. Reson. A., 1994, 110, 219-227). Approximately 70 mg of sample, packed into a canister-design zirconia rotor was used for each experiment. Chemical shifts (δ) were referenced to external adamantane with the high frequency resonance being set to 38.56 ppm (W. L. Earl and D. L. VanderHart, J. Magn. Reson., 1982, 48, 35-54).X-Ray Powder DiffractionOne of ordinary skill in the art will appreciate that an X-ray diffraction pattern may be obtained with a measurement error that is dependent upon the measurement conditions employed. In particular, it is generally known that intensities in a X-ray diffraction pattern may fluctuate depending upon measurement conditions employed. It should be further understood that relative intensities may also vary depending upon experimental conditions and, accordingly, the exact order of intensity should not be taken into account. Additionally, a measurement error of diffraction angle for a conventional X-ray diffraction pattern is typically about 5% or less, and such degree of measurement error should be taken into account as pertaining to the aforementioned diffraction angles. Consequently, it is to be understood that the crystal forms of the instant invention are not limited to the crystal forms that provide X-ray diffraction patterns completely identical to the X-ray diffraction patterns depicted in the accompanying Figures disclosed herein. Any crystal forms that provide X-ray diffraction patterns substantially identical to those disclosed in the accompanying Figures fall within the scope of the present invention. The ability to ascertain substantial identities of X-ray diffraction patterns is within the purview of one of ordinary skill in the art.X-Ray powder diffraction data for the crystalline forms of Compound (IV) were obtained using a Bruker GADDS (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) (General Area Detector Diffraction System) manual chi platform goniometer. Powder samples were placed in thin walled glass capillaries of 1 mm or less in diameter; the capillary was rotated during data collection. The sample-detector distance was 17 cm. The radiation was Cu Kα (45 kV 111 mA, λ=1.5418 Å). Data were collected for 3<2θ<35° with a sample exposure time of at least 300 seconds.Single Crystal X-RayAll single crystal data were collected on a Bruker-Nonius (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) Kappa CCD 2000 system using Cu Kα radiation (λ=1.5418 Å) and were corrected only for the Lorentz-polarization factors. Indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W. C. Jr & Sweet, R. M. (Academic, NY), Vol. 276, pp. 307-326) in the Collect program suite (Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B. V., 1998).The structures were solved by direct methods and refined on the basis of observed reflections using either the SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia NY 11716 Scattering factors, including f′ and f″, in the SDP software were taken from the “International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1) software package with minor local modifications or the crystallographic package, MAXUS (maXus solution and refinement software suite: S. Mackay, C. J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus: a computer program for the solution and refinement of crystal structures from diffraction data).The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were varied.The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were variedDifferential Scanning CalorimetryThe DSC instrument used to test the crystalline forms was a TA Instruments® model Q1000. The DSC cell/sample chamber was purged with 100 ml/min of ultra-high purity nitrogen gas. The instrument was calibrated with high purity indium. The accuracy of the measured sample temperature with this method is within about +/−1° C., and the heat of fusion can be measured within a relative error of about +/−5%. The sample was placed into an open aluminum DSC pan and measured against an empty reference pan. At least 2 mg of sample powder was placed into the bottom of the pan and lightly tapped down to ensure good contact with the pan. The weight of the sample was measured accurately and recorded to a hundredth of a milligram. The instrument was programmed to heat at 10° C. per minute in the temperature range between 25 and 350° C.The heat flow, which was normalized by a sample weight, was plotted versus the measured sample temperature. The data were reported in units of watts/gram (“W/g”). The plot was made with the endothermic peaks pointing down. The endothermic melt peak was evaluated for extrapolated onset temperature, peak temperature, and heat of fusion in this analysis.Thermogravimetric Analysis (TGA)The TGA instrument used to test the crystalline forms was a TAInstruments® model Q500. Samples of at least 10 milligrams were analyzed at a heating rate of 10° C. per minute in the temperature range between 25° C. and about 350° C.
Example 8Preparation of:
crystalline monohydrate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)An example of the crystallization procedure to obtain the crystalline monohydrate form is shown here:
- Charge 48 g of the compound of formula (IV).
- Charge approximately 1056 mL (22 mL/g) of ethyl alcohol, or other suitable alcohol.
- Charge approximately 144 mL of water.
- Dissolve the suspension by heating to approximately 75° C.
- Optional: Polish filter by transfer the compound of formula (IV) solution at 75° C. through the preheated filter and into the receiver.
- Rinse the dissolution reactor and transfer lines with a mixture of 43 mL of ethanol and 5 mL of water.
Heat the contents in the receiver to 75-80° C. and maintain 75-80° C. to achieve complete dissolution.Charge approximately 384 mL of water at a rate such that the batch temperature is maintained between 75-80° C.Cool to 75° C., and, optionally, charge monohydrate seed crystals. Seed crystals are not essential to obtaining monohydrate, but provide better control of the crystallization.
- Cool to 70° C. and maintain 70° C. for ca. 1 h.
- Cool from 70 to 5 C over 2 h, and maintain the temperature between 0 at 5° C. for at least 2 h.
- Filter the crystal slurry.
- Wash the filter cake with a mixture of 96 mL of ethanol and 96 mL of water.
- Dry the material at ≦50° C. under reduced pressure until the water content is 3.4 to 4.1% by KF to afford 41 g (85 M %).
Alternately, the monohydrate can be obtained by:- 1) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate and heated at 80° C. to give bulk monohydrate.
- 2) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate. On standing several days at room temperature, bulk monohydrate had formed.
- 3) An aqueous suspension of compound IV was seeded with monohydrate and heated at 70° C. for 4 hours to give bulk monohydrate. In the absence of seeding, an aqueous slurry of compound IV was unchanged after 82 days at room temperature.
- 4) A solution of compound IV in a solvent such as NMP or DMA was treated with water until the solution became cloudy and was held at 75-85° C. for several hours. Monohydrate was isolated after cooling and filtering.
- 5) A solution of compound IV in ethanol, butanol, and water was heated. Seeds of monohydrate were added to the hot solution and then cooled. Monohydrate was isolated upon cooling and filtration.
One of ordinary skill in the art will appreciate that the monohydrate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 1 or by a representative sampling of peaks as shown in Table 1.Representative peaks taken from the XRPD of the monohydrate of the compound of formula (IV) are shown in Table 1.TABLE 1 2-Theta d(Å) Height 17.994 4.9257 915 18.440 4.8075 338 19.153 4.6301 644 19.599 4.5258 361 21.252 4.1774 148 24.462 3.6359 250 25.901 3.4371 133 28.052 3.1782 153The XRPD is also characterized by the following list comprising 2θ values selected from the group consisting of: 4.6±0.2, 11.2±0.2, 13.8±0.2, 15.2±0.2, 17.9±0.2, 19.1±0.2, 19.6±0.2, 23.2±0.2, 23.6±0.2. The XRPD is also characterized by the list of 2θ values selected from the group consisting of: 18.0±0.2, 18.4±0.2, 19.2±0.2, 19.6±0.2, 21.2±0.2, 24.5±0.2, 25.9±0.2, and 28.0±0.2.Single crystal x-ray data was obtained at room temperature (+25° C.). The molecular structure was confirmed as a monohydrate form of the compound of Formula (IV).The following unit cell parameters were obtained for the monohydrate of the compound of formula (IV) from the x-ray analysis at 25° C.:a(Å)=13.8632(7); b(Å)=9.3307(3); c(Å)=38.390(2);V(Å3) 4965.9(4); Z′=1; Vm=621Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.354Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).Single crystal x-ray data was also obtained at −50° C. The monohydrate form of the compound of Formula (IV) is characterized by unit cell parameters approximately equal to the following:Cell dimensions:
- a(Å)=13.862(1);
- b(Å)=9.286(1);
- c(Å)=38.143(2);
Volume=4910(1) Å3Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.369wherein the compound is at a temperature of about −50° C.The simulated XRPD was calculated from the refined atomic parameters at room temperature.The monohydrate of the compound of formula (IV) is represented by the DSC as shown in FIG. 2. The DSC is characterized by a broad peak between approximately 95° C. and 130° C. This peak is broad and variable and corresponds to the loss of one water of hydration as seen in the TGA graph. The DSC also has a characteristic peak at approximately 287° C. which corresponds to the melt of the dehydrated form of the compound of formula (IV).The TGA for the monohydrate of the compound of Formula (IV) is shown in FIG. 2 along with the DSC. The TGA shows a 3.48% weight loss from 50° C. to 175° C. The weight loss corresponds to a loss of one water of hydration from the compound of Formula (IV).The monohydrate may also be prepared by crystallizing from alcoholic solvents, such as methanol, ethanol, propanol, i-propanol, butanol, pentanol, and water.
Example 9Preparation of:
crystalline n-butanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)The crystalline butanol solvate of the compound of formula (IV) is prepared by dissolving compound (IV) in 1-butanol at reflux (116-118° C.) at a concentration of approximately 1 g/25 mL of solvent. Upon cooling, the butanol solvate crystallizes out of solution. Filter, wash with butanol, and dry.The following unit cell parameters were obtained from the x-ray analysis for the crystalline butanol solvate, obtained at room temperature:a(Å)=22.8102(6); b(Å)=8.4691(3); c(Å)=15.1436(5); β=95.794(2);V(Å3) 2910.5(2); Z′=1; Vm=728Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.283Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the butanol solvate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 3 or by a representative sampling of peaks. Representative peaks for the crystalline butanol solvate are 2θ values of: 5.9±0.2, 12.0±0.2, 13.0±0.2, 17.7±0.2, 24.1±0.2, and 24.6±0.2.
Example 10Preparation of:
crystalline ethanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)

To a 100-mL round bottom flask was charged 4.00 g (10.1 mmol) of 5D (contained 2.3 Area % 5C) 6.60 g (50.7 mmol) of 7B, 80 mL of n-butanol and 2.61 g (20.2 mmol) of DIPEA. The resulting slurry was heated to 120° C. and maintained at 120° C. for 4.5 h whereby HPLC analysis showed 0.19 relative Area % of residual 5D to compound IV. The homogeneous mixture was cooled to 20° C. and left stirring overnight. The resulting crystals were filtered. The wet cake was washed twice with 10-mL portions of n-butanol to afford a white crystalline product. HPLC analysis showed this material to contain 99.7 Area % compound IV and 0.3 Area % 5C.The resulting wet cake was returned to the 100-mL reactor, and charged with 56 mL (12 mL/g) of 200 proof ethanol. At 80° C. an additional 25 mL of ethanol was added. To this mixture was added 10 mL of water resulting in rapid dissolution. Heat was removed and crystallization was observed at 75-77° C. The crystal slurry was further cooled to 20° C. and filtered. The wet cake was washed once with 10 mL of 1:1 ethanol: water and once with 10 mL of n-heptane. The wet cake contained 1.0% water by KF and 8.10% volatiles by LOD. The material was dried at 60° C./30 in Hg for 17 h to afford 3.55 g (70 M %) of material containing only 0.19% water by KF, 99.87 Area % by HPLC. The 1H NMR spectrum, however revealed that the ethanol solvate had been formed.The following unit cell parameters were obtained from the x-ray analysis for the crystalline ethanol solvate (di-ethanolate, E2-1), obtained at −40° C.:a(Å)=22.076(1); b(Å)=8.9612(2); c(Å)=16.8764(3); β=114.783(1);V(Å3) 3031.1(1); Z′=1; Vm=758Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.271Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 4 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 5.8±0.2, 11.3±0.2, 15.8±0.2, 17.2±0.2, 19.5±0.2, 24.1±0.2, 25.3±0.2, and 26.2±0.2.In addition, during the process to form the ethanolate (diethanolate) the formation of another ethanol solvate (½ ethanolate, T1E2-1) has been observed. To date this additional ethaonol solvate is known strictly as a partial desolvation product of the original diethanolate form E2-1, and has only been observed on occasion during crystallization of E2-1The following unit cell parameters were obtained from the x-ray analysis for the crystalline ½ ethanol solvate T1E2-1, obtained at −10° C.:a(Å)=22.03(2); b(Å)=9.20(1); c(Å)=12.31(1);β=93.49(6)V(Å3) 2491(4)); Z′=1; Vm=623;Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.363Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (T1E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 7 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 7.20±0.2, 12.01±0.2, 12.81±0.2, 18.06±0.2, 19.30±0.2, and 25.24±0.2.
Example 11Preparation of:
crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (Neat form N-6)To a mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL) was added DIPEA (155 mL, 0.89 mol). The suspension was heated at 110° C. (solution obtained) for 25 min., then cooled to about 90° C. The resulting hot solution was added dropwise into hot (80° C.) water (8010) mL, keeping the temperature at about 80° C. The resulting suspension was stirred 15 min at 80° C. then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with water (2×1600 mL) and dried in vacuo at 55-60° C. affording 192.45 g (88.7% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H), 2.41 (s, 3H), 2.43 (t, 2H, J=6), 2.49 (t, 4H, J=6.3), 3.51 (m, 4H), 3.54 (q, 2H, J=6), 4.46 (t, 1H, J=5.3), 6.05 (s, 1H), 7.26 (t, 1H, J=7.6), 7.28 (dd, 1H, J=7.6, 1.7), 7.41 (dd, 1H, J=7.6, 1.7), 8.23 (s, 1H), 9.89 (s, 1H), 11.48. KF0.84; DSC: 285.25° C. (onset), 286.28° C. (max).The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline compound IV, obtained at 23° C.:a(Å)=22.957(1); b(Å)=8.5830(5); c(Å)=13.803(3); β=112.039(6);V(Å3)=2521.0(5); Z′=1; Vm=630Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.286Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the crystalline form of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 5 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (N-6) are 2θ values of: 6.8±0.2, 11.1±0.2, 12.3±0.2, 13.2±0.2, 13.7±0.2, 16.7±0.2, 21.0±0.2, 24.3±0.2, and 24.8±0.2.
Example 12Preparation of:
crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (neatform T1H1-7)The title neat form may be prepared by heating the monohydrate form of the compound of formula (IV) above the dehydration temperature.The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline (T1H1-7) compound IV, obtained at 25° C.:a(Å)=13.4916; b(Å)=9.3992(2); c(Å)=38.817(1);V(Å3)=4922.4(3); Z′=1; Vm=615Space group PbcaDensity (calculated) (g/cm3) 1.317Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the neat crystalline form (T1H1-7) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 6 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (T1H1-7)) are 2θ values of: 8.0±0.2, 9.7±0.2, 11.2±0.2, 13.3±0.2, 17.5±0.2, 18.9±0.2, 21.0±0.2, 22.0±0.2.Obviously, numerous modifications and variations of the present invention are possible in light of the above teachings. It is therefore to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein.PATENThttps://patents.google.com/patent/US8680103B2/enAminothiazole-aromatic amides of formula I

wherein Ar is aryl or heteroaryl, L is an optional alkylene linker, and R2, R3, R4, and R5, are as defined in the specification herein, are useful as kinase inhibitors, in particular, inhibitors of protein tyrosine kinase and p38 kinase. They are expected to be useful in the treatment of protein tyrosine kinase-associated disorders such as immunologic and oncological disorders [see, U.S. Pat. No. 6,596,746 (the ‘746 patent), assigned to the present assignee and incorporated herein by reference], and p38 kinase-associated conditions such as inflammatory and immune conditions, as described in U.S. patent application Ser. No. 10/773,790, filed Feb. 6, 2004, claiming priority to U.S. Provisional application Ser. No. 60/445,410, filed Feb. 6, 2003 (hereinafter the ‘410 application), both of which are also assigned to the present assignee and incorporated herein by reference.The compound of formula (IV), ′N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, is an inhibitor of SRC/ABL and is useful in the treatment of oncological diseases.

Other approaches to preparing 2-aminothiazole-5-carboxamides are described in the ‘746 patent and in the ‘410 application. The ‘746 patent describes a process involving treatment of chlorothiazole with n-BuLi followed by reaction with phenyl isocyanates to give chlorothiazole-benzamides, which are further elaborated to aminothiazole-benzamide final products after protection, chloro-to-amino substitution, and deprotection, e.g.,

The ‘410 application describes a multi-step process involving first, converting N-unsubstituted aminothiazole carboxylic acid methyl or ethyl esters to bromothiazole carboxylic acid esters via diazotization with tert-butyl nitrite and subsequent CuBr2 treatment, e.g.,

then, hydrolyzing the resulting bromothiazole esters to the corresponding carboxylic acids and converting the acids to the corresponding acyl chlorides, e.g.,

then finally, coupling the acyl chlorides with anilines to afford bromothiazole-benzamide intermediates which were further elaborated to aminothiazole-benzamide final products, e.g.,

Other approaches for making 2-aminothiazole-5-carboxamides include coupling of 2-aminothiazole-5-carboxylic acids with amines using various coupling conditions such as DCC [Roberts et al, J. Med. Chem. (1972), 15, at p. 1310], and DPPA [Marsham et al., J. Med. Chem. (1991), 34, at p. 1594)].The above methods present drawbacks with respect to the production of side products, the use of expensive coupling reagents, less than desirable yields, and the need for multiple reaction steps to achieve the 2-aminothiazole-5-carboxamide compounds.Reaction of N,N-dimethyl-N′-(aminothiocarbonyl)-formamidines with α-haloketones and esters to give 5-carbonyl-2-aminothiazoles has been reported. See Lin, Y. et al, J. Heterocycl. Chem. (1979), 16, at 1377; Hartmann, H. et al, J. Chem. Soc. Perkin Trans. (2000), 1, at 4316; Noack, A. et al; Tetrahedron (2002), 58, at 2137; Noack, A.; et al. Angew. Chem. (2001), 113, at 3097; and Kantlehner, W. et al., J. Prakt. Chem./Chem.-Ztg. (1996), 338, at 403. Reaction of β-ethoxy acrylates and thioureas to prepare 2-aminothiazole-5-carboxylates also has been reported. See Zhao, R., et al., Tetrahedron Lett. (2001), 42, at 2101. However, electrophilic bromination of acrylanilide and crotonanilide has been known to undergo both aromatic bromination and addition to the α,β-unsaturated carbon-carbon double bonds. See Autenrieth, Chem. Ber. (1905), 38, at 2550; Eremeev et al., Chem. Heterocycl. Compd. Engl. Transl. (1984), 20, at 1102.New and efficient processes for preparing 2-aminothiazole-5-carboxamides are desired.
SUMMARY OF THE INVENTION
This invention is related to processes for the preparation of 2-aminothiazole-5-aromatic amides having the formula (I),

wherein L, Ar, R2, R3, R4, R5, and m are as defined below, comprising reacting a compound having the formula (II),

wherein Q is the group —O—P*, wherein P* is selected so that, when considered together with the oxygen atom to which P* is attached, Q is a leaving group, and Ar, L, R2, R3, and m are as defined below,
with a halogenating reagent in the presence of water followed by a thiourea compound having the formula (III),

wherein, R4 and R5 are as defined below,
to provide the compound of formula (I),

wherein,Ar is the same in formulae (I) and (II) and is aryl or heteroaryl;L is the same in formulae (I) and (II) and is optionally-substituted alkylene;R2 is the same in formulae (I) and (II), and is selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo;R3 is the same in formulae (I) and (II), and is selected from hydrogen, halogen, cyano, haloalkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, aryl, heteroaryl, cycloalkyl, and heterocyclo;R4 is (i) the same in each of formulae (I) and (III), and (ii) is independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo, or alternatively, R4 is taken together with R5, to form heteroaryl or heterocyclo;R5 is (i) the same in each of formulae (I) and (III), and (ii) is independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo, or alternatively, R5 is taken together with R4, to form heteroaryl or heterocyclo; andm is 0 or 1.Applicants have surprisingly discovered said process for converting β-(P*)oxy acryl aromatic amides and thioureas to 2-aminothiazole derivatives, wherein the aromatic amides are not subject to further halogenation producing other side products. Aminothiazole-aromatic amides, particularly, 2-aminothiazole-5-benzamides, can thus be efficiently prepared with this process in high yield.In another aspect, the present invention is directed to crystalline forms of the compound of formula (IV).
EXAMPLESExample 1Preparation of Intermediate:
(S)-1-sec-Butylthiourea

To a solution of S-sec-butyl-amine (7.31 g, 0.1 mol) in chloroform (80 mL) at 0° C. was slowly added benzoyl isothiocyanate (13.44 mL, 0.1 mol). The mixture was allowed to warm to 10° C. and stirred for 10 min. The solvent was then removed under reduced pressure, and the residue was dissolved in MeOH (80 mL). An aqueous solution (10 mL) of NaOH (4 g, 0.1 mol) was added to this solution, and the mixture was stirred at 60° C. for another 2 h. The MeOH was then removed under reduced pressure, and the residue was stirred in water (50 mL). The precipitate was collected by vacuum filtration and dried to provide S-1-sec-butyl-thiourea (12.2 g, 92% yield). mp 133-134° C.; 1H NMR (500 MHz, DMSO-D6) δ 7.40 (s, 1H), 7.20 (br s, 1H), 6.76 (s, 1H), 4.04 (s, 1H), 1.41 (m, 2H), 1.03 (d, J=6.1 Hz, 3H), 0.81 (d, J=7.7 Hz, 3H); 13C NMR (125 MHz, DMSO-D6) δ 182.5, 50.8, 28.8, 19.9, 10.3; LRMS m/z 133.2 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.49; H, 8.88; N, 21.32; S, 24.27.
Example 2Preparation of Intermediate:
(R)-1-sec-Butylthiourea

(R)-1-sec-Butylthiourea was prepared in 92% yield according to the general method outlined for Example 1. mp 133-134° C.; 1H NMR (500 MHz, DMSO) δ 0.80 (m, 3H, J=7.7), 1.02 (d, 3H, J=6.1), 1.41 (m, 2H), (3.40, 4.04) (s, 1H), 6.76 (s, 1H), 7.20 (s, br, 1H), 7.39 (d, 1H, J=7.2); 13C NMR (500 MHz, DMSO) δ: 10.00, 19.56, 28.50, 50.20, 182.00; m/z 133.23 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.32; H, 9.15; N, 21.14; S, 24.38.
Example 3Preparation of:

To a solution of 3-amino-N-methyl-4-methylbenzamide hydrochloride (1.0 g, 5 mmol) in acetone (10 mL) at 0° C. was added pyridine (1.2 mL, 15 mmol) dropwise via syringe. 3-Methoxyacryloyl chloride (0.72 mL 6.5 mmol) was added and the reaction stirred at room temperature for 1 h. The solution was cooled again to 0° C. and 1N HCl (1.5 mL) was added dropwise via pipette. The reaction mixture was stirred for 5 min, then water (8.5 mL) was added via an addition funnel. The acetone was removed in vacuo and the resulting solution stirred for 4 h. Crystallization began within 15 min. After stirring for 4 h, the vessel was cooled in an ice bath for 30 min, filtered, and rinsed with ice cold water (2×3 mL) to give compound 3A (0.99 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.12 (br s, 1H), 7.76 (s, 1H), 7.29 (m, 2H), 7.05 (d, J=7.9 Hz, 1H), 5.47 (d, J=12.3 Hz, 1H), 3.48 (s, 3H), 2.54 (d, J=4.7 Hz, 3H), 2.03 (s, 3H); HPLC rt 2.28 min (Condition A).
3B. Example 3To a 50 mL RBF containing the above compound 3A (0.5 g, 2.0 mmol) was added THF (2.5 mL) and water (2 mL), followed by NBS (0.40 g, 2.22 mmol), and the solution was stirred for 90 min. R-sec-butylthiourea (Ex. 2) (267 mg), was added, and the solution was heated to 75° C. for 8 h. Conc. NH4OH was added to adjust the pH to 10 followed by the addition of EtOH (15 mL). Water (15 mL) was added and the slurry stirred for 16 h, filtered, and washed with water to give Example 3 as a light brown solid (0.48 g, 69% yield, 98% purity). MS 347.1; HPLC 2.59.
Example 4Preparation of:

Example 4 is prepared following the methods of Example 3 but using the appropriate acryl benzamide and Example 1.
Example 5Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (The compound of Formula (IV))

5A. 1-(6-Chloro-2-methylpyrimidin-4-yl)thiourea

To a stirring slurry of 4-amino-5-chloro-2-methylpyrimidine (6.13 g, 42.7 mmol) in THF (24 mL) was added ethyl isothiocyanatoformate (7.5 mL, 63.6 mmol), and the mixture heated to reflux. After 5 h, another portion of ethyl isothiocyanato formate (1.0 mL, 8.5 mmol) was added and after 10 h, a final portion (1.5 mL, 12.7 mmol) was added and the mixture stirred 6 h more. The slurry was evaporated under vacuum to remove most of the solvent and heptane (6 mL) added to the residue. The solid was collected by vacuum filtration and washed with heptane (2×5 mL) giving 8.01 g (68% yield) of the intermediate ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate.A solution of ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate (275 mg, 1.0 mmol) and 1N sodium hydroxide (3.5 eq) was heated and stirred at 50° C. for 2 h. The resulting slurry was cooled to 20-22° C. The solid was collected by vacuum filtration, washed with water, and dried to give 185 mg of 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea (91% yield). 1H NMR (400 MHz, DMSO-d6): δ2.51 (S, 3H), 7.05 (s, 1H), 9.35 (s, 1H), 10.07 (s, 1H), 10.91 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 25.25, 104.56, 159.19, 159.33, 167.36, 180.91.
5B. (E)-N-(2-Chloro-6-methylphenyl)-3-ethoxyacrylamide

To a cold stirring solution of 2-chloro-6-methylaniline (59.5 g 0.42 mol) and pyridine (68 ml, 0.63 mol) in THF (600 mL) was added 3-ethoxyacryloyl chloride (84.7 g, 0.63 mol) slowly keeping the temp at 0-5° C. The mixture was then warmed and stirred for 2 h. at 20° C. Hydrochloric acid (1N, 115 mL) was added at 0-10° C. The mixture was diluted with water (310 mL) and the resulting solution was concentrated under vacuum to a thick slurry. The slurry was diluted with toluene (275 mL) and stirred for 15 min. at 20-22° C. then 1 h. at 0° C. The solid was collected by vacuum filtration, washed with water (2×75 mL) and dried to give 74.1 g (73.6% yield) of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide). 1H NMR (400 Hz, DMSO-d6) δ 1.26 (t, 3H, J=7 Hz), 2.15 (s, 3H), 3.94 (q, 2H, J=7 Hz), 5.58 (d, 1H, J=12.4 Hz), 7.10-7.27 (m, 2H, J=7.5 Hz), 7.27-7.37 (d, 1H, J=7.5 Hz), 7.45 (d, 1H, J=12.4 Hz), 9.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 14.57, 18.96, 67.17, 97.99, 126.80, 127.44, 129.07, 131.32, 132.89, 138.25, 161.09, 165.36.
5C. 2-Amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide

To a mixture of compound 5B (5.00 g, 20.86 mmol) in 1,4-dioxane (27 mL) and water (27 mL) was added NBS (4.08 g, 22.9 mmol) at −10 to 0° C. The slurry was warmed and stirred at 20-22° C. for 3 h. Thiourea (1.60 g, 21 mmol) was added and the mixture heated to 80° C. After 2 h, the resulting solution was cooled to 20-22° and conc. ammonium hydroxide (4.2 mL) was added dropwise. The resulting slurry was concentrated under vacuum to about half volume and cooled to 0-5° C. The solid was collected by vacuum filtration, washed with cold water (10 mL), and dried to give 5.3 g (94.9% yield) of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6) δ δ 2.19 (s, 3H), 7.09-7.29 (m, 2H, J=7.5), 7.29-7.43 (d, 1H, J=7.5), 7.61 (s, 2H), 7.85 (s, 1H), 9.63 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 18.18, 120.63, 126.84, 127.90, 128.86, 132.41, 133.63, 138.76, 142.88, 159.45, 172.02.
5D. 2-(6-Chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide

To a stirring solution of compound 5C (5.00 g, 18.67 mmol) and 4,6-dichloro-2-methylpyrimidine (3.65 g 22.4/mmol) in THF (65 mL) was added a 30% wt. solution of sodium t-butoxide in THF (21.1 g, 65.36 mmol) slowly with cooling to keep the temperature at 10-20° C. The mixture was stirred at room temperature for 1.5 h and cooled to 0-5° C. Hydrochloric acid, 2N (21.5 mL) was added slowly and the mixture stirred 1.75 h at 0-5° C. The solid was collected by vacuum filtration, washed with water (15 mL) and dried to give 6.63 g (86.4% yield) of compound 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).
5E. Example 5To a mixture of compound 5D (4.00 g, 10.14 mmol) and hydroxyethylpiperazine (6.60 g, 50.69 mmol) in n-butanol (40 mL) was added DIPEA (3.53 mL, 20.26 mmol). The slurry was heated at 118° C. for 4.5 h, then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with n-butanol (5 mL), and dried. The product (5.11 g) was dissolved in hot 80% EtOH—H2O (80 mL), and the solution was clarified by filtration. The hot solution was slowly diluted with water (15 mL) and cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with 50% ethanol-water (5 mL) and dried affording 4.27 g (83.2% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide as monohydrate. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.40 (s, 3H), 2.42 (t, 2H, J=6), 2.48 (t, 4H, J=6.3), 3.50 (m, 4H), 3.53 (q, 2H, J=6), 4.45 (t, 1H, J=5.3), 6.04 (s, 1H), 7.25 (t, 1H, J=7.6), 7.27 (dd, 1H, J=7.6, 1.7), 7.40 (dd, 1H, J=7.6, 1.7), 8.21 (s, 1H), 9.87 (s, 1H), 11.47.
Example 6Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide

To a slurry of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide 5B (120 mg, 0.50 mmol) in THF (0.75 ml) and water (0.5 mL) was added NBS (98 mg, 0.55 mmol) at 0° C. The mixture was warmed and stirred at 20-22° C. for 3 h. To this was added 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea 5A (100 mg, 0.49 mmol), and the slurry heated and stirred at reflux for 2 h. The slurry was cooled to 20-22° C. and the solid collected by vacuum filtration giving 140 mg (71% yield) of 2-(6-chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).Compound 5D was elaborated to N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide, following Step 5E.
Example 7Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide7A. 2-[4-(6-Chloro-2-methyl-pyrimidin-4-yl)-piperazin-1-yl]-ethanol

2-Piperazin-1-yl-ethanol (8.2 g, 63.1 mmol) was added to a solution of 4,6-dichloro-2-methylpyrimidine (5.2 g, 31.9 mmol) in dichloromethane (80 ml) at rt. The mixture was stirred for two hours and triethylamine (0.9 ml) was added. The mixture was stirred at rt for 20 h. The resultant solid was filtered. The cake was washed with dichloromethane (20 ml). The filtrate was concentrated to give an oil. This oil was dried under high vacuum for 20 h to give a solid. This solid was stirred with heptane (50 ml) at rt for 5 h. Filtration gave 7C (8.13 g) as a white solid
7B. Example 7

To a 250 ml of round bottom flask were charged compound 5C (1.9 g, 7.1 mmol), compound 7C (1.5 g, 5.9 mmol), K2CO3 (16 g, 115.7 mmol), Pd (OAc)2 (52 mg, 0.23 mmol) and BINAP (291 mg, 0.46 mmol). The flask was placed under vacuum and flushed with nitrogen. Toluene was added (60 ml). The suspension was heated to 100-110° C. and stirred at this temperature for 20 h. After cooling to room temperature, the mixture was applied to a silica gel column. The column was first eluted with EtOAC, and then with 10% of MeOH in EtOAC. Finally, the column was washed with 10% 2M ammonia solution in MeOH/90% EtOAC. The fractions which contained the desired product were collected and concentrated to give compound IV as a yellow solid (2.3 g).
Analytical MethodsSolid State Nuclear Magnetic Resonance (SSNMR)All solid-state C-13 NMR measurements were made with a Bruker DSX-400, 400 MHz NMR spectrometer. High resolution spectra were obtained using high-power proton decoupling and the TPPM pulse sequence and ramp amplitude cross-polarization (RAMP-CP) with magic-angle spinning (MAS) at approximately 12 kHz (A. E. Bennett et al, J. Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu and S. O, Smith, J. Magn. Reson. A, 1994, 110, 219-227). Approximately 70 mg of sample, packed into a canister-design zirconia rotor was used for each experiment. Chemical shifts (6) were referenced to external adamantane with the high frequency resonance being set to 38.56 ppm (W. L. Earl and D. L. VanderHart, J. Magn. Reson., 1982, 48, 35-54).X-Ray Powder DiffractionOne of ordinary skill in the art will appreciate that an X-ray diffraction pattern may be obtained with a measurement error that is dependent upon the measurement conditions employed. In particular, it is generally known that intensities in a X-ray diffraction pattern may fluctuate depending upon measurement conditions employed. It should be further understood that relative intensities may also vary depending upon experimental conditions and, accordingly, the exact order of intensity should not be taken into account. Additionally, a measurement error of diffraction angle for a conventional X-ray diffraction pattern is typically about 5% or less, and such degree of measurement error should be taken into account as pertaining to the aforementioned diffraction angles. Consequently, it is to be understood that the crystal forms of the instant invention are not limited to the crystal forms that provide X-ray diffraction patterns completely identical to the X-ray diffraction patterns depicted in the accompanying Figures disclosed herein. Any crystal forms that provide X-ray diffraction patterns substantially identical to those disclosed in the accompanying Figures fall within the scope of the present invention. The ability to ascertain substantial identities of X-ray diffraction patterns is within the purview of one of ordinary skill in the art.X-Ray powder diffraction data for the crystalline forms of Compound (IV) were obtained using a Bruker GADDS (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) (General Area Detector Diffraction System) manual chi platform goniometer. Powder samples were placed in thin walled glass capillaries of 1 mm or less in diameter; the capillary was rotated during data collection. The sample-detector distance was 17 cm. The radiation was Cu Kα (45 kV 111 mA, λ=1.5418 Å). Data were collected for 3<2θ<35° with a sample exposure time of at least 300 seconds.Single Crystal X-RayAll single crystal data were collected on a Bruker-Nonius (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) Kappa CCD 2000 system using Cu Kα radiation (λ=1.5418 Å) and were corrected only for the Lorentz-polarization factors. Indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W. C. Jr. & Sweet, R. M. (Academic, NY), Vol. 276, pp. 307-326) in the Collect program suite (Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B. V., 1998).The structures were solved by direct methods and refined on the basis of observed reflections using either the SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia N.Y. 11716 Scattering factors, including f′ and f″, in the SDP software were taken from the “International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1) software package with minor local modifications or the crystallographic package, MAXUS (maXus solution and refinement software suite: S. Mackay, C. J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus: a computer program for the solution and refinement of crystal structures from diffraction data).The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were varied.The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were variedDifferential Scanning CalorimetryThe DSC instrument used to test the crystalline forms was a TA INSTRUMENTS° model Q1000. The DSC cell/sample chamber was purged with 100 ml/min of ultra-high purity nitrogen gas. The instrument was calibrated with high purity indium. The accuracy of the measured sample temperature with this method is within about +/−1° C., and the heat of fusion can be measured within a relative error of about +/−5%. The sample was placed into an open aluminum DSC pan and measured against an empty reference pan. At least 2 mg of sample powder was placed into the bottom of the pan and lightly tapped down to ensure good contact with the pan. The weight of the sample was measured accurately and recorded to a hundredth of a milligram. The instrument was programmed to heat at 10° C. per minute in the temperature range between 25 and 350° C.The heat flow, which was normalized by a sample weight, was plotted versus the measured sample temperature. The data were reported in units of watts/gram (“W/g”). The plot was made with the endothermic peaks pointing down. The endothermic melt peak was evaluated for extrapolated onset temperature, peak temperature, and heat of fusion in this analysis.Thermogravimetric Analysis (TGA)The TGA instrument used to test the crystalline forms was a TA INSTRUMENTS® model Q500. Samples of at least 10 milligrams were analyzed at a heating rate of 10° C. per minute in the temperature range between 25° C. and about 350° C.
Example 8Preparation of:
Crystalline monohydrate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)An example of the crystallization procedure to obtain the crystalline monohydrate form is shown here:Charge 48 g of the compound of formula (IV).Charge approximately 1056 mL (22 mL/g) of ethyl alcohol, or other suitable alcohol.Charge approximately 144 mL of water.Dissolve the suspension by heating to approximately 75° C.Optional: Polish filter by transfer the compound of formula (IV) solution at 75° C. through the preheated filter and into the receiver.Rinse the dissolution reactor and transfer lines with a mixture of 43 mL of ethanol and 5 mL of water.Heat the contents in the receiver to 75-80° C. and maintain 75-80° C. to achieve complete dissolution.Charge approximately 384 mL of water at a rate such that the batch temperature is maintained between 75-80° C.Cool to 75° C., and, optionally, charge monohydrate seed crystals. Seed crystals are not essential to obtaining monohydrate, but provide better control of the crystallization.Cool to 70° C. and maintain 70° C. for ca. 1 h.Cool from 70 to 5 C over 2 h, and maintain the temperature between 0 at 5° C. for at least 2 h.Filter the crystal slurry.Wash the filter cake with a mixture of 96 mL of ethanol and 96 mL of water.Dry the material at ≦50° C. under reduced pressure until the water content is 3.4 to 4.1% by KF to afford 41 g (85 M %).Alternately, the monohydrate can be obtained by:1) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate and heated at 80° C. to give bulk monohydrate.2) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate. On standing several days at room temperature, bulk monohydrate had formed.3) An aqueous suspension of compound IV was seeded with monohydrate and heated at 70° C. for 4 hours to give bulk monohydrate. In the absence of seeding, an aqueous slurry of compound IV was unchanged after 82 days at room temperature.4) A solution of compound IV in a solvent such as NMP or DMA was treated with water until the solution became cloudy and was held at 75-85° C. for several hours. Monohydrate was isolated after cooling and filtering.5) A solution of compound IV in ethanol, butanol, and water was heated. Seeds of monohydrate were added to the hot solution and then cooled. Monohydrate was isolated upon cooling and filtration.One of ordinary skill in the art will appreciate that the monohydrate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 1 or by a representative sampling of peaks as shown in Table 1.Representative peaks taken from the XRPD of the monohydrate of the compound of formula (IV) are shown in Table 1.TABLE 1 2-Theta d(Å) Height 17.994 4.9257 915 18.440 4.8075 338 19.153 4.6301 644 19.599 4.5258 361 21.252 4.1774 148 24.462 3.6359 250 25.901 3.4371 133 28.052 3.1782 153The XRPD is also characterized by the following list comprising 2θ values selected from the group consisting of: 4.6±0.2, 11.2±0.2, 13.8±0.2, 15.2±0.2, 17.9±0.2, 19.1±0.2, 19.6±0.2, 23.2±0.2, 23.6±0.2. The XRPD is also characterized by the list of 2θ values selected from the group consisting of: 18.0±0.2, 18.4±0.2, 19.2±0.2, 19.6±0.2, 21.2±0.2, 24.5±0.2, 25.9±0.2, and 28.0±0.2.Single crystal x-ray data was obtained at room temperature (+25° C.). The molecular structure was confirmed as a monohydrate form of the compound of Formula (IV).The following unit cell parameters were obtained for the monohydrate of the compound of formula (IV) from the x-ray analysis at 25° C.:a(Å)=13.8632(7); b(Å)=9.3307(3); c(Å)=38.390(2);V(Å3) 4965.9(4); Z′=1; Vm=621Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.354wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).Single crystal x-ray data was also obtained at −50° C. The monohydrate form of the compound of Formula (IV) is characterized by unit cell parameters approximately equal to the following:Cell dimensions: a(Å)=13.862(1);
- b(Å)=9.286(1);
- c(Å)=38.143(2);
Volume=4910(1) Å3Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.369wherein the compound is at a temperature of about −50° C.The simulated XRPD was calculated from the refined atomic parameters at room temperature.The monohydrate of the compound of formula (IV) is represented by the DSC as shown in FIG. 2. The DSC is characterized by a broad peak between approximately 95° C. and 130° C. This peak is broad and variable and corresponds to the loss of one water of hydration as seen in the TGA graph. The DSC also has a characteristic peak at approximately 287° C. which corresponds to the melt of the dehydrated form of the compound of formula (IV).The TGA for the monohydrate of the compound of Formula (IV) is shown in FIG. 2 along with the DSC. The TGA shows a 3.48% weight loss from 50° C. to 175° C. The weight loss corresponds to a loss of one water of hydration from the compound of Formula (IV).The monohydrate may also be prepared by crystallizing from alcoholic solvents, such as methanol, ethanol, propanol, i-propanol, butanol, pentanol, and water.
Example 9Preparation of:
Crystalline n-butanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)The crystalline butanol solvate of the compound of formula (IV) is prepared by dissolving compound (IV) in 1-butanol at reflux (116-118° C.) at a concentration of approximately 1 g/25 mL of solvent. Upon cooling, the butanol solvate crystallizes out of solution. Filter, wash with butanol, and dry.The following unit cell parameters were obtained from the x-ray analysis for the crystalline butanol solvate, obtained at room temperature:a(Å)=22.8102(6); b(Å)=8.4691(3); c(Å)=15.1436(5); β=95.794(2);V(Å3) 2910.5(2); Z′=1; Vm=728Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.283wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the butanol solvate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 3 or by a representative sampling of peaks. Representative peaks for the crystalline butanol solvate are 2θ values of: 5.9±0.2, 12.0±0.2, 13.0±0.2, 17.7±0.2, 24.1±0.2, and 24.6±0.2.
Example 10Preparation of:
Crystalline ethanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)

To a 100-mL round bottom flask was charged 4.00 g (10.1 mmol) of 5D (contained 2.3 Area % 5C) 6.60 g (50.7 mmol) of 7B, 80 mL of n-butanol and 2.61 g (20.2 mmol) of DIPEA. The resulting slurry was heated to 120° C. and maintained at 120° C. for 4.5 h whereby HPLC analysis showed 0.19 relative Area % of residual 5D to compound IV. The homogeneous mixture was cooled to 20° C. and left stirring overnight. The resulting crystals were filtered. The wet cake was washed twice with 10-mL portions of n-butanol to afford a white crystalline product. HPLC analysis showed this material to contain 99.7 Area % compound IV and 0.3 Area % 5C.The resulting wet cake was returned to the 100-mL reactor, and charged with 56 mL (12 mL/g) of 200 proof ethanol. At 80° C. an additional 25 mL of ethanol was added. To this mixture was added 10 mL of water resulting in rapid dissolution. Heat was removed and crystallization was observed at 75-77° C. The crystal slurry was further cooled to 20° C. and filtered. The wet cake was washed once with 10 mL of 1:1 ethanol:water and once with 10 mL of n-heptane. The wet cake contained 1.0% water by KF and 8.10% volatiles by LOD. The material was dried at 60° C./30 in Hg for 17 h to afford 3.55 g (70 M %) of material containing only 0.19% water by KF, 99.87 Area % by HPLC. The 1H NMR spectrum, however revealed that the ethanol solvate had been formed.The following unit cell parameters were obtained from the x-ray analysis for the crystalline ethanol solvate (di-ethanolate, E2-1), obtained at −40° C.:a(Å)=22.076(1); b(Å)=8.9612(2); c(Å)=16.8764(3); β=114.783(1);V(Å3) 3031.1(1); Z′=1; Vm=758Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.271wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 4 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 5.8±0.2, 11.3±0.2, 15.8±0.2, 17.2±0.2, 19.5±0.2, 24.1±0.2, 25.3±0.2, and 26.2±0.2.In addition, during the process to form the ethanolate (diethanolate) the formation of another ethanol solvate (½ ethanolate, T1E2-1) has been observed. To date this additional ethanol solvate is known strictly as a partial desolvation product of the original diethanolate form E2-1, and has only been observed on occasion during crystallization of E2-1The following unit cell parameters were obtained from the x-ray analysis for the crystalline ½ ethanol solvate T1E2-1, obtained at −10° C.:a(Å)=22.03(2); b(Å)=9.20(1); c(Å)=12.31(1);β=93.49(6)V(Å3) 2491(4)); Z′=1; Vm=623;Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.363wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (T1E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 7 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 7.20±0.2, 12.01±0.2, 12.81±0.2, 18.06±0.2, 19.30±0.2, and 25.24±0.2.
Example 11Preparation of:
Crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (Neat form N-6)To a mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL) was added DIPEA (155 mL, 0.89 mol). The suspension was heated at 110° C. (solution obtained) for 25 min., then cooled to about 90° C. The resulting hot solution was added dropwise into hot (80° C.) water (8010) mL, keeping the temperature at about 80° C. The resulting suspension was stirred 15 min at 80° C. then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with water (2×1600 mL) and dried in vacuo at 55-60° C. affording 192.45 g (88.7% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H), 2.41 (s, 3H), 2.43 (t, 2H, J=6), 2.49 (t, 4H, J=6.3), 3.51 (m, 4H), 3.54 (q, 2H, J=6), 4.46 (t, 1H, J=5.3), 6.05 (s, 1H), 7.26 (t, 1H, J=7.6), 7.28 (dd, 1H, J=7.6, 1.7), 7.41 (dd, 1H, J=7.6, 1.7), 8.23 (s, 1H), 9.89 (s, 1H), 11.48. KF0.84; DSC: 285.25° C. (onset), 286.28° C. (max).The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline compound IV, obtained at 23° C.:a(Å)=22.957(1); b(Å)=8.5830(5); c(Å)=13.803(3); β=112.039(6);V(Å3)=2521.0(5); Z′=1; Vm=630Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.286wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the crystalline form of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 5 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (N-6) are 2θ values of: 6.8±0.2, 11.1±0.2, 12.3±0.2, 13.2±0.2, 13.7±0.2, 16.7±0.2, 21.0±0.2, 24.3±0.2, and 24.8±0.2.
Example 12Preparation of:
Crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (neat form T1H1-7)The title neat form may be prepared by heating the monohydrate form of the compound of formula (IV) above the dehydration temperature.The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline (T1H1-7) compound IV, obtained at 25° C.:a(Å)=13.4916; b(Å)=9.3992(2); c(Å)=38.817(1);V(Å3)=4922.4(3); Z′=1; Vm=615Space group PbcaDensity (calculated) (g/cm3) 1.317wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the neat crystalline form (T1H1-7) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 6 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (T1H1-7)) are 2θ values of: 8.0±0.2, 9.7±0.2, 11.2±0.2, 13.3±0.2, 17.5±0.2, 18.9±0.2, 21.0±0.2, 22.0±0.2.Obviously, numerous modifications and variations of the present invention are possible in light of the above teachings. It is therefore to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein.
PAPERhttps://pubs.acs.org/doi/abs/10.1021/jm060727j
2-Aminothiazole (1) was discovered as a novel Src family kinase inhibitor template through screening of our internal compound collection. Optimization through successive structure−activity relationship iterations identified analogs 2 (Dasatinib, BMS-354825) and 12m as pan-Src inhibitors with nanomolar to subnanomolar potencies in biochemical and cellular assays. Molecular modeling was used to construct a putative binding model for Lck inhibition by this class of compounds. The framework of key hydrogen-bond interactions proposed by this model was in agreement with the subsequent, published crystal structure of 2 bound to structurally similar Abl kinase. The oral efficacy of this class of inhibitors was demonstrated with 12m in inhibiting the proinflammatory cytokine IL-2 ex vivo in mice (ED50 ∼ 5 mg/kg) and in reducing TNF levels in an acute murine model of inflammation (90% inhibition in LPS-induced TNFα production when dosed orally at 60 mg/kg, 2 h prior to LPS administration). The oral efficacy of 12m was further demonstrated in a chronic model of adjuvant arthritis in rats with established disease when administered orally at 0.3 and 3 mg/kg twice daily. Dasatinib (2) is currently in clinical trials for the treatment of chronic myelogenous leukemia.

PATENT
https://patents.google.com/patent/WO2019209908A1/enDasatinib (DAS), having the chemical designation N-(2-chloro-6-methylphenyl)-2- [[6-[4-(2-hydroxyethyl)-l-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide, monohydrate, is an orally bioavailable inhibitor of the receptor tyrosine kinase (RTK) epidermal growth factor receptor (ErbB; EGFR) family, with antineoplastic activity. Dasatinib has the following structure:

Dasatinib is commercially marketed under the name SPRY CEL® and is indicated for the treatment of patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase, for the treatment of patients chronic, accelerated, or myeloid or lymphoid blast phase Philadelphia chromosome-positive chronic myeloid leukemia with resistance or intolerance to prior therapy and for the treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia with resistance or intolerance to prior therapy.Solid forms of dasatinib are described in U.S. Patent Nos. 7491725 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 8680103 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 7973045 (anhydrous), 8067423 (isopropyl alcohol solvate), 8242270 (butanol solvate, monohydrate, diethanolate, hemi- ethanolate, anhydrous), 8884013 (monohydrates), 9249134 (amorphous), 9456992 (solid dispersion nanoparticles), 9556164 (saccharin salt crystal) and 9884857 (saccharinate, glutarate, nicotinate); in U.S. Publication Nos. 20160250153 (solid dispersion nanoparticles), 20160264565 (Form-SDI), 20160361313 (solid dispersion nanoparticles), 20170183334 (salts) and 20140031352 (anti-oxidative acid); in International Publication Nos.W02010067374 (solvated forms and Form I), W02010139980, W02010139981,W02013065063 (anhydrous), W02017103057, W02017108605 (solid dispersion),WO2017134617 (amorphous), WO2014086326 (NMP, isoamyl-OH, 1, 3-propanediol process), WO2015107545, WO2015181573, WO2017134615 (PG solvate), W02010062715 (isosorbide dimethyl ether, N,N’-dimethylethylene urea, N,N’-dimethyl-N,N’-propylene urea), WO2010139979 (DCM, DMSP, monohydrate), WO2011095588 (anhydrate, hydrochloride, hemi-ethanol), W02012014149 (N-methylformamide) and W02017002131 (propandiol, monohydrate); and in Chinese Patent Nos. CN102643275, CN103059013, CN103819469, CN104341410. None of the references describe an ethyl formate solvate of dasatinib.Dasatinib co-crystals are described in U.S. Patent No. 9,340,536 (co-crystals selected from methyl-4-hydroxybenzoate, nicotinamide, ethyl gallate, methyl gallate, propyl gallate, ethyl maltol, vanillin, menthol, and (lR,2S,5R)-(-)-menthol) and International Publication No. W02016001025 (co-crystal selected from menthol or vanillin). None of the references describe dasatinib co-crystal comprising dasatinib and a second compound, as a co-crystal former, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin.Dasatinib (DAS), having the chemical designation N-(2-chloro-6-methylphenyl)-2- [[6-[4-(2-hydroxyethyl)-l-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide, monohydrate, is an orally bioavailable inhibitor of the receptor tyrosine kinase (RTK) epidermal growth factor receptor (ErbB; EGFR) family, with antineoplastic activity. Dasatinib has the following structure:
Dasatinib is commercially marketed under the name SPRY CEL® and is indicated for the treatment of patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase, for the treatment of patients chronic, accelerated, or myeloid or lymphoid blast phase Philadelphia chromosome-positive chronic myeloid leukemia with resistance or intolerance to prior therapy and for the treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia with resistance or intolerance to prior therapy.Solid forms of dasatinib are described in U.S. Patent Nos. 7491725 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 8680103 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 7973045 (anhydrous), 8067423 (isopropyl alcohol solvate), 8242270 (butanol solvate, monohydrate, diethanolate, hemi- ethanolate, anhydrous), 8884013 (monohydrates), 9249134 (amorphous), 9456992 (solid dispersion nanoparticles), 9556164 (saccharin salt crystal) and 9884857 (saccharinate, glutarate, nicotinate); in U.S. Publication Nos. 20160250153 (solid dispersion nanoparticles), 20160264565 (Form-SDI), 20160361313 (solid dispersion nanoparticles), 20170183334 (salts) and 20140031352 (anti-oxidative acid); in International Publication Nos.W02010067374 (solvated forms and Form I), W02010139980, W02010139981,W02013065063 (anhydrous), W02017103057, W02017108605 (solid dispersion),WO2017134617 (amorphous), WO2014086326 (NMP, isoamyl-OH, 1, 3-propanediol process), WO2015107545, WO2015181573, WO2017134615 (PG solvate), W02010062715 (isosorbide dimethyl ether, N,N’-dimethylethylene urea, N,N’-dimethyl-N,N’-propylene urea), WO2010139979 (DCM, DMSP, monohydrate), WO2011095588 (anhydrate, hydrochloride, hemi-ethanol), W02012014149 (N-methylformamide) and W02017002131 (propandiol, monohydrate); and in Chinese Patent Nos. CN102643275, CN103059013, CN103819469, CN104341410. None of the references describe an ethyl formate solvate of dasatinib.Dasatinib co-crystals are described in U.S. Patent No. 9,340,536 (co-crystals selected from methyl-4-hydroxybenzoate, nicotinamide, ethyl gallate, methyl gallate, propyl gallate, ethyl maltol, vanillin, menthol, and (lR,2S,5R)-(-)-menthol) and International Publication No. W02016001025 (co-crystal selected from menthol or vanillin). None of the references describe dasatinib co-crystal comprising dasatinib and a second compound, as a co-crystal former, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin. hereafter. ClaimsHide Dependent What is claimed is:1. A dasatinib co-crystal comprising dasatinib and a second compound, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin.2. The dasatinib co-crystal according to claim 1, wherein a molar ratio of the dasatinib to the second compound is about 1: 1.3. The dasatinib co-crystal according to claim 1, wherein the second compound is butyl paraben.4. The dasatinib co-crystal according to claim 3, wherein a molar ratio of the dasatinib to the butyl paraben is about 1 : 1.5. The dasatinib co-crystal according to claim 1, which is Form I co-crystal of dasatinib and butyl paraben.6. The dasatinib co-crystal according to claim 5, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 4.9, 9.8, 11.3, 14.9, 17.5, 20.8, 21.6, 22.6 and 25.4° 2Q degrees.7. The dasatinib co-crystal according to claim 5, characterized by a thermal event at about 287.3 °C, as measured by differential scanning calorimetry.8. The dasatinib co-crystal according to claim 5, characterized by a weight loss of 8.1% from about 70 °C through about 165 °C, as measured by thermal gravimetric analysis.9. The dasatinib co-crystal of claim 5 monoclinic, P2i/C.10. The dasatinib co-crystal d of claim 5 which has single crystal parametersa = 18.630 (2) Ab = 8.725 (1) Ac = 22.331 (2) Aa = g = 90°, b = 104.575 (8)°.11. The dasatinib co-crystal of claim 5 which has a cell volume is about 3512.9 A3.12. The dasatinib co-crystal according to claim 1, wherein the second compound is ethyl vanillin.13. The dasatinib co-crystal according to claim 9, wherein a molar ratio of the dasatinib to the ethyl vanillin is about 1 : 1.14. The dasatinib co-crystal according to claim 1, which is Form II co-crystal of dasatinib and ethyl vanillin.15. The dasatinib co-crystal according to claim 14, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 5.7, 10.9, 13.5, 17.1, 18.4, 19.4, 23.7 and 26.3° 2Q degrees.16. The dasatinib co-crystal according to claim 14, characterized by one or more thermal events selected from about 140 °C, about 181 °C, and about 293 °C, as measured by differential scanning calorimetry.17. The dasatinib co-crystal according to claim 14, characterized by a weight loss of 24.3% from about 120 through 250 °C, as measured by thermal gravimetric analysis.18. The dasatinib co-crystal of claim 14 monoclinic, P2i/n.19. The dasatinib co-crystal d of claim 14 which has single crystal parametersa = 18.452 (1) Ab = 9.441 (6) Ac = 19.377 (1) Aa = g = 90°, b = 108.78 (1)°.20. The dasatinib co-crystal of claim 5 which has a cell volume is about 3195.71 A3.21. The dasatinib co-crystal according to claim 1, wherein the second compound is propyl paraben.22. The dasatinib co-crystal according to claim 21, wherein a molar ratio of the dasatinib to the propyl paraben is about 1 : 1.23. The dasatinib co-crystal according to claim 1, which is Form III co-crystal ofdasatinib and propyl paraben.24. The dasatinib co-crystal according to claim 23, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 4.8, 9.6, 11.9, 14.8, 18.4, 22.2, 23.9 and 26.1° 2Q degrees.25. The dasatinib co-crystal of claim 23 monoclinic, P2i/n.26. The dasatinib co-crystal of claim 23 which has single crystal parametersa = 18.859 (9) Ab = 8.131 (6) Ac = 22.473 (1) Aa = g = 90°, b = 103.87(1)°.27. The dasatinib co-crystal of claim 23 which has a cell volume is about 3345.51 A3.28. An ethyl formate solvate of dasatinib.29. The ethyl formate solvate of dasatinib according to claim 28, wherein a molar ratio of the dasatinib to the ethyl formate is about 1 : 1.30. The ethyl formate solvate of dasatinib according to claim 1, which is Form I of ethyl formate solvate of dasatinib.31. The ethyl formate solvate of dasatinib according to claim 30, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 6.0, 12.1, 15.1, 18.0, 23.8 and 24.8° 2Q degrees.32. The ethyl formate solvate of dasatinib according to claim 30, characterized by athermal event at about 287.3 °C, as measured by differential scanning calorimetry.33. The ethyl formate solvate of dasatinib according to claim 30, characterized by aweight loss of 8.1% from about 70 °C through about 165 °C, as measured by thermal gravimetric analysis.34. The ethyl formate solvate of dasatinib of claim 23 orthorhombic, P2i/c.35. The ethyl formate solvate of dasatinib of claim 23 which has single crystal parameters a = 14.8928 (5) Ab = 8.3299 (3) Ac = 22.18990 (6) Aa = g =b = 90°.36. The ethyl formate solvate of dasatinib of claim 23 which has a cell volume is about 2731.9 A3.37. A pharmaceutical composition comprising a pharmaceutically effective amount of the dasatinib co-crystal according to claim 1 and pharmaceutically acceptable excipient.38. A method of treating disease in a patient comprising administering a pharmaceutical formulation according to claim 37 to the patient in need thereof.39. A method of treating disease according to claim 38, wherein the disease ismyelogenous leukemia.40. A method of treating disease according to claim 38, wherein the disease isPhiladelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase.41. A method of treating disease according to claim 38, wherein the disease Ph+ acute lymphoblastic leukemia (Ph+ ALL).42. A method of making the dasatinib co-crystal according to claim 1, comprisingdissolving dasatinib and a second compound, wherein the second compound is selected from the group consisting of butyl paraben, propyl paraben and ethyl vanillin, in heated methanol (-10: 1 – wt(mg)DAs:v(mL)MeOH and molD,\s:mohnci compound is 1 : 1.1) to form a clear solution, heating the solution under vacuum for about l8-20h to yield the dasatinib co-crystal.43. A process for the preparation Form II co-crystal of dasatinib and ethyl vanillin,according to claim 14, comprising: (g) dissolving Form I of ethyl formate solvate of dasatinib and ethyl vanillin in N-methyl-2-pyrrolidone to form a solution;(h) adding water to the solution;(i) stirring the solution for about 12-24 hours to form a slurry;(j) filtering the slurry to yield a precipitate;(k) washing the precipitate with water; and(l) drying the precipitate under vacuum with warming to yield Form II co crystal of dasatinib and ethyl vanillin.44. A process for the preparation of Form I of ethyl formate solvate of dasatinib,according to claim 30, comprising:(d) dissolving dasatinib in ethyl formate to form a solution;(e) stirring the solution for about 12-24 hours form a slurry;(f) filtering the slurry to yield Form I of ethyl formate solvate of dasatinib.45. A process for the preparation of Form I of ethyl formate solvate of dasatinib,according to claim 30, comprising:(g) dissolving dasatinib in N-Methyl-2-pyrrolidone to form a solution;(h) adding ethyl formate to the solution to form a slurry;(i) adding additional ethyl formate to the slurry;(j) stirring the slurry for about 2 hours;(k) filtering the slurry to yield a precipitate; and(l) washing the precipitate with ethyl formate to yield Form I of ethyl formate solvate of dasatinib.
ATENThttps://patents.google.com/patent/WO2013065063A1/en
Dasatinib, N-(2-chloro-6-methylphenyl)-2- [(6-[4-(2-hydroxyl)- 1 -piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide compound having the following chemical structure of Formula (I)

Formula IAlso known as BMS-354825, it is a drug produced by Bristol Myers Squibb and sold under the trade name Sprycel. Dasatinib is an oral dual BCR/ABL and SRC family tyrosine kinase inhibitor approved for use in patients with chronic myelogenous leukemia (CML) after Imatinib treatment has failed and Philadelphia chromosome- positive acute lymphoblastic leukemia (Ph + ALL). It is also being assessed for use in metastatic melanoma.A preparation of Dasatinib is described in US patent No. 6596746 (B l ), where the process is done by reacting compound of the following formula III with N-(2- hydroxyethyl) piperazine at 80° C.

Formula IIIThe compound of Formula (I) and its preparation is described in US Patent No. 6596746, US patent application No. 2005/0176965 Al , and US patent application No. 2006/0004067 Al .l Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, 1R etc. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR). Solvent medium and mode of crystallization play very important role in obtaining a crystalline form.The discovery of new polymorphic forms is a continuing goal of formulators. The new polymorphs may be advantageous for dosage form development and enhancing bioavailability owing to the altered physiochemical properties. Some form may turn out to be more efficacious. Discovering novel processes to prepare known polymorphic forms is also a primary goal of the pharmaceutical development scientists. New processes can provide novel intermediates or synthetic pathways that result in product with increased chemical and polymorphic purity in addition to providing cost and other advantages. There is thus a need to provide novel synthetic routes and intermediates that can realize these goals.Several crystalline forms of Dasatinib are described in the literature; these are designated as HI -7, BU-2, E2-1 , N-6, T1 H1 -7 and TIE2-1. Crystalline Dasatinib monohydrate (H I -7) and butanol solvate (BU-2) along with the processes for their preparation are described in WO 2005077945. In addition US 2006/0004067, which is continuation of US 2005215795 also describe two ethanol solvates (E2-1 ; TIE2-1) and two anhydrous forms (N-6 and T1 H1 -7).WO 2009053854 discloses various Dasatinib solvates including their crystalline form, amorphous form and anhydrous form.US patent No. 7973045 discloses the anhydrous form of Dasatinib and process for preparation thereof. The anhydrous form disclosed therein have typical characteristic XRD peaks at about 7.2, 1 1.9, 14.4, 16.5, 17.3, 19.1 , 20.8, 22.4, 23.8, 25.3 and 29.1 on the 2- theta value. WO 2010062715 discloses isosorbide dimethyl ether solvate, Ν,Ν’- dimethylethylene urea solvate and N,N’-dimethyl-N,N’-propylene urea solvate of Dasatinib.WO 2010067374 discloses novel crystalline form I, solvates of DMF, DMSO, toluene, isopropyl acetate and processes for their preparation.WO 2010139979 discloses MDC solvate and process of preparation, for use in the manufacture of pure Dasatinib.WO 2010139980 discloses a process for the preparation of crystalline Dasatinib monohydrate.The present invention is a step forward in this direction and provides a novel anhydrous form and process for its preparation, which can be used for the preparation of pure Dasatinib, in particularly Dasatinib monohydrate.The process for preparing Dasatinib monohydrate is described in US 2006/0004067. Further studies by the inventors have shown that the preparation of Dasatinib by using the method, which is disclosed in US 2006/0004067 yields the monohydrate with ~ 90% purity. Therefore the present invention provides a novel anhydrous form which can be used to get Dasatinib monohydrate with high yield and purity.Preparing API with increased purity is always an aim of the pharmaceutical development team. The inventors of the present invention have found that preparingDasatinib monohydrate using the novel anhydrous form of the present invention resulted in a highly pure product with a good yield.Scheme 1 shows a general process for the preparation of Dasatinib as disclosed in US 2006/0004067. Intermediate 3 and N-(2-hydroxyethyl) piperazine are heated together in a solvent system comprising n-butanol as a solvent and diisopropyl ethylamine (DIPEA) as a base. On cooling of the reaction mixture, Dasatinib precipitates out which is isolated by filtration.



DasatinibScheme 1Example – 1In a reaction vessel, N-(2-chloro-6-methylphenyl)-2-[(6-chloro-2-methyl-4- pyrimidinyl) amino] -5-thiazolecarboxamide (1 gm, 2.54 mmol) and N-(2- hydroxyethyl) piperazine (5.3 gm, 40.70 mmol) was added under stirring. The reaction mixture was heated at 80 °C for 2H. Acetonitrile was added into reaction mixture at 80 °C and stirred for 30 min. Cooled the suspension to room temperature and stirred for 30 min. Filtered, washed with acetonitrile and dried at 60 °C under vacuum to get 950 mg anhydrous N-(2-chloro-6-methylphenyl)-2-[(6-[4-(2-hydroxy 1)- 1 -piperaziny l]-2- methyl-4-pyrimidinyl]amino]-5-thiazole carboxamide (76.73 % Yield).HPLC Purity 99.90 %M/C by KF 0.12 %DSC 278.17 °CTGA 2.05 %XRD as provided in Fig. 2
Patent
Publication numberPriority datePublication dateAssigneeTitleUS7491725B22004-02-062009-02-17Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-aromatic carboxamides as kinase inhibitorsWO2009147238A12008-06-062009-12-10Boehringer Ingelheim International GmbhSolid pharmaceutical formulations comprising bibw 2992WO2010062715A22008-11-032010-06-03Teva Pharmaceutical Industries Ltd.Polymorphs of dasatinib and process for preparation thereofWO2010067374A22008-12-082010-06-17Hetero Research FoundationPolymorphs of dasatinibWO2010139980A12009-06-032010-12-09Generics [Uk] LimitedProcess for preparing crystalline dasatinib monohydrateWO2010139979A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2010139981A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2011003853A22009-07-062011-01-13Boehringer Ingelheim International GmbhProcess for drying of bibw2992, of its salts and of solid pharmaceutical formulations comprising this active ingredientUS7973045B22007-10-232011-07-05Teva Pharmaceutical Industries Ltd.Anhydrous form of dasatinib and process for preparation thereofWO2011095588A12010-02-042011-08-11Ratiopharm GmbhPharmaceutical composition comprising n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamidWO2012014149A12010-07-302012-02-02Ranbaxy Laboratories LimitedN-methylformamide solvate of dasatinibCN102643275A2011-02-212012-08-22江苏先声药物研究有限公司A new preparation method for Dasatinib N-6 crystal formCN103059013A2011-10-182013-04-24北京本草天源药物研究院New crystal of Dasatinib monohydrate and preparation method thereofWO2013065063A12011-11-032013-05-10Cadila Healthcare LimitedAnhydrous form of dasatinib, process for its preparation and its useUS20140031352A12012-07-242014-01-30Laurus Labs Private LimitedSolid forms of tyrosine kinase inhibitors, process for the preparation and their pharmaceutical composition thereofCN103819469A2012-11-162014-05-28重庆医药工业研究院有限责任公司Crystal form of dasatinib and preparation method for crystal form of dasatinibWO2014086326A12012-12-062014-06-12Zentiva, K.S.A method for the preparation and purification of new and known polymorphs and solvates of dasatinibUS8884013B22010-02-082014-11-11Nan Jing Cavendish Bio-Engineering Technology Co., Ltd.Polymorphs of Dasatinib, preparation methods and pharmaceutical compositions thereofCN104341410A2013-08-092015-02-11上海科胜药物研发有限公司New Dasatinib crystal form and preparation method thereofWO2015107545A12013-12-182015-07-23Dharmesh Mahendrabhai ShahWater soluble salts of dasatinib hydrateWO2015181573A12014-05-262015-12-03Egis Gyógyszergyár Zrt.Dasatinib saltsWO2016001025A12014-06-302016-01-07Basf SeMulticomponent crystals of dasatinib with menthol or vanillinUS9249134B22013-03-262016-02-02Cadila Healthcare LimitedProcess for preparation of amorphous form of dasatinibUS9340536B22012-06-152016-05-17Basf SeMulticomponent crystals comprising dasatinib and selected co-crystal formersUS20160250153A12012-01-132016-09-01Xspray Microparticles AbNovel methodsUS20160264565A12013-11-082016-09-15Shilpa Medicare LimitedCrystalline dasatinib processWO2017002131A12015-06-292017-01-05Msn Laboratories Private LimitedCrystalline forms of n-(2-chloro-6-methy]phenvn-2-[f6-[4-(2-hvdroxvethvl)-l- piperazinvil-2-methvl-4-pvrimidinvllaminol-5-thiazolecarboxamide and their process thereofUS9556164B22013-07-252017-01-31Basf SeSalts of Dasatinib in crystalline formWO2017103057A12015-12-162017-06-22Synthon B.V.Pharmaceutical composition comprising anhydrous dasatinibWO2017108605A12015-12-222017-06-29Synthon B.V.Pharmaceutical composition comprising amorphous dasatinibWO2017134615A12016-02-032017-08-10Dr. Reddy’s Laboratories LimitedSolid state forms of dasatinib and processes for their preparationWO2017134617A12016-02-032017-08-10Dr. Reddy’s Laboratories LimitedProcess for the preparation of amorphous dasatinibUS9884857B22013-07-252018-02-06Basf SeSalts of dasatinib in amorphous form
PATENTSPublication numberPriority datePublication dateAssigneeTitleUS20060079563A1 *1999-04-152006-04-13Jagabandhu DasCyclic protein tyrosine kinase inhibitorsUS20070219370A1 *2006-03-152007-09-20Bristol-Myers Squibb CompanyProcess for preparing n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino] -5-thiazolecarboxamide and related metabolites thereofUS20080275009A1 *2005-09-212008-11-06Bristol-Myers Squibb CompanyOral administration of n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-1,3-thiazole-5-carboxamide and salts thereofUS20090030203A1 *2005-08-052009-01-29Bristol-Myers Squibb CompanyPreparation of 2-amino-thiazole-5-carboxylic-acid derivativesUS20090118297A1 *2007-10-232009-05-07Ondrej SimoPolymorphs of dasatinib and process for preparation thereofWO2012014149A12010-07-302012-02-02Ranbaxy Laboratories LimitedN-methylformamide solvate of dasatinibUS20120309968A1 *2010-02-082012-12-06Nan Jing Cavendish Bio-Engineering Technology Co., Ltd.Polymorphs of dasatinib, preparation methods and pharmaceutical compositions thereofUS8530492B22009-04-172013-09-10Nektar TherapeuticsOligomer-protein tyrosine kinase inhibitor conjugatesUS8680103B22004-02-062014-03-25Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-aromatic carboxamides as kinase inhibitorsWO2014102759A22012-12-312014-07-03Ranbaxy Laboratories LimitedProcess for the preparation of dasatinib and its intermediatesUS8816077B22009-04-172014-08-26Nektar TherapeuticsOligomer-protein tyrosine kinase inhibitor conjugatesUS20150057446A1 *2012-04-202015-02-26Shilpa Medicare LimitedProcess for preparing dasatinib monohydrateWO2016001025A12014-06-302016-01-07Basf SeMulticomponent crystals of dasatinib with menthol or vanillinUS9340536B22012-06-152016-05-17Basf SeMulticomponent crystals comprising dasatinib and selected co-crystal formersUS9556164B22013-07-252017-01-31Basf SeSalts of Dasatinib in crystalline formUS9884857B22013-07-252018-02-06Basf SeSalts of dasatinib in amorphous formWO2018078392A12016-10-292018-05-03Cipla LimitedPolymorphs of dasatinibWO2018100585A12016-12-012018-06-07Natco Pharma LimitedAn improved process for the preparation of dasatinib polymorphWO2018134189A12017-01-202018-07-26Cerbios-Pharma SaCo-crystal of an antitumoral compoundWO2018134190A12017-01-202018-07-26Cerbios-Pharma SaCo-crystals of an antitumoral compoundUS10174018B22016-12-132019-01-08Princeton Drug Discovery IncProtein kinase inhibitorsWO2019209908A12018-04-252019-10-31Johnson Matthey Public Limited CompanyCrystalline forms of dasatinibUS10722484B22016-03-092020-07-28K-Gen, Inc.Methods of cancer treatmentUS10799459B12019-05-172020-10-13Xspray Microparticles AbRapidly disintegrating solid oral dosage forms containing dasatinibFamily To Family CitationsUS7396935B22003-05-012008-07-08Bristol-Myers Squibb CompanyAryl-substituted pyrazole-amide compounds useful as kinase inhibitorsUS7652146B2 *2004-02-062010-01-26Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-carboxamides useful as kinase inhibitorsTW200600513A *2004-06-302006-01-01Squibb Bristol Myers CoA method for preparing pyrrolotriazine compoundsPE20061394A1 *2005-03-152006-12-15Squibb Bristol Myers CoMetabolites of n- (2-chloro-6-methylphenyl) -2 – [[6- [4- (2-hydroxyethyl) -1-piperazinyl] -2-methyl-4-pyrimidinyl] amino] -5-thiazolecarboxamidesUS20060235006A1 *2005-04-132006-10-19Lee Francis YCombinations, methods and compositions for treating cancerPL1885339T32005-05-052015-12-31Bristol Myers Squibb Holdings IrelandFormulations of a src/abl inhibitorWO2008076883A22006-12-152008-06-26Abraxis Bioscience, Inc.Triazine derivatives and their therapeutical applicationsWO2010139979A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2010139980A1 *2009-06-032010-12-09Generics [Uk] LimitedProcess for preparing crystalline dasatinib monohydrateEP2359813A12010-02-042011-08-24Ratiopharm GmbHPharmaceutical composition comprising N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamidCN102250084A *2010-02-082011-11-23南京卡文迪许生物工程技术有限公司Dasatinib polymorphic substance as well as preparation method and pharmaceutical composition thereofCN102643275B *2011-02-212016-04-20江苏先声药物研究有限公司The preparation method that a kind of Dasatinib N-6 crystal formation is newWO2013065063A12011-11-032013-05-10Cadila Healthcare LimitedAnhydrous form of dasatinib, process for its preparation and its useUS20150087687A12012-03-232015-03-26Dennis BrownCompositions and methods to improve the therapeutic benefit of indirubin and analogs thereof, including meisoindigoSG10201610869TA2012-06-262017-02-27Del Mar PharmaceuticalsMethods for treating tyrosine-kinase-inhibitor-resistant malignancies in patients with genetic polymorphisms or ahi1 dysregulations or mutations employing dianhydrogalactitol, diacetyldianhydrogalactiCN103664929B *2012-08-302016-08-03石药集团中奇制药技术(石家庄)有限公司Dasatinib polycrystalline form medicament and preparation methodCN102838595B *2012-09-132014-09-24江苏奥赛康药业股份有限公司Preparation method of high-purity dasatinib and by-product of dasatinibCN103819469A *2012-11-162014-05-28重庆医药工业研究院有限责任公司Crystal form of dasatinib and preparation method for crystal form of dasatinibCZ306598B62012-12-062017-03-22Zentiva, K.S.A method of preparation and purification of new and known polymorphs and dasatinib solvatesCN105764502A2013-07-262016-07-13现代化制药公司Combinatorial methods to improve the therapeutic benefit of bisantrene and analogs and derivatives thereofCN103408542B *2013-08-132016-06-29南京优科生物医药研究有限公司A kind of preparation method of highly purified Dasatinib anhydrideWO2015049645A2 *2013-10-042015-04-09Alembic Pharmaceuticals LimitedAn improved process for the preparation of dasatinibCZ306732B62013-12-192017-05-31Zentiva, K.S.A method of preparation of the anhydrous polymorphic form of N-6 DasatinibCN104788445B *2015-04-102017-06-23山东新时代药业有限公司A kind of synthetic method of Dasatinib intermediateCN106668022B *2015-11-052020-09-15武汉应内药业有限公司Application of aminothiazole MyD88 specific inhibitor TJM2010-5* Cited by examiner, † Cited by third party, ‡ Family to family citation
References[edit]
- ^ “Sprycel (Dasatinib)” (PDF). Therapeutic Goods Administration(TGA). Retrieved 18 July 2020.
- ^ Jump up to:a b c d e f g h i j “Sprycel EPAR”. European Medicines Agency(EMA). Retrieved 28 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h “Dasatinib”. The American Society of Health-System Pharmacists. Retrieved 8 December 2017.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Keating GM (January 2017). “Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia”. Drugs. 77 (1): 85–96. doi:10.1007/s40265-016-0677-x. PMID 28032244. S2CID 207489056.
- ^ Olivieri, A.; Manzione, L. (2007). “Dasatinib: a new step in molecular target therapy”. Annals of Oncology. 18 Suppl 6: vi42–vi46. doi:10.1093/annonc/mdm223. PMID 17591830.
- ^ “NHS – Healthcare News”. nelm.nhs.uk. Archived from the original on 5 May 2013. Retrieved 27 September 2011.
- ^ Yurttaş NO, Eşkazan AE (2018). “Dasatinib-induced pulmonary arterial hypertension”. British Journal of Clinical Pharmacology. 84 (5): 835–845. doi:10.1111/bcp.13508. PMC 5903230. PMID 29334406.
- ^ Jump up to:a b c d “Sprycel (dasatinib) and risk of pulmonary arterial hypertension”. U.S. Food and Drug Administration (FDA). 23 September 2011. Retrieved 28 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M, Kiefer SE, et al. (June 2006). “The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants”. Cancer Research. 66 (11): 5790–7. doi:10.1158/0008-5472.CAN-05-4187. PMID 16740718.
- ^ Jump up to:a b Piscitani L, Sirolli V, Morroni M, Bonomini M (2020). “Nephrotoxicity Associated with Novel Anticancer Agents (Aflibercept, Dasatinib, Nivolumab): Case Series and Nephrological Considerations”. International Journal of Molecular Sciences. 21(14): e4878. doi:10.3390/ijms21144878. PMC 7402330. PMID 32664269.
- ^ Jump up to:a b Braun TP, Eide CA, Druker BJ (2020). “Response and Resistance to BCR-ABL1-Targeted Therapies”. Cancer Cell. 37(4): 530–542. doi:10.1016/j.ccell.2020.03.006. PMC 7722523. PMID 32289275.
- ^ “Otsuka and Bristol-Myers Squibb Announce a Change in Contract Regarding Collaboration in Japan in the Oncology Therapy Area”.
- ^ “FDA Approves U.S. Product Labeling Update for Sprycel (dasatinib) to Include Three-Year First-Line and Five-Year Second-Line Efficacy and Safety Data in Chronic Myeloid Leukemia in Chronic Phase”. Bristol-Myers Squibb (Press release).
- ^ “Bristol-Myers Squibb Announces Extension of U.S. Agreement for ABILIFY and Establishment of an Oncology Collaboration with Otsuka”. Bristol-Myers Squibb (Press release).
- ^ Drahl C (16 January 2012). “How Jagabandhu Das made dasatinib possible”. The Safety Zone blog. Chemical & Engineering News. Retrieved 29 August 2016.
- ^ “Drug Approval Package: Sprycel (Dasatinib) NDA #021986 & 022072”. U.S. Food and Drug Administration (FDA). 6 September 2006. Retrieved 28 April 2020.
- ^ “2010 Notifications”. U.S. Food and Drug Administration (FDA). 18 November 2010. Retrieved 28 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e “FDA approves dasatinib for pediatric patients with CML”. U.S. Food and Drug Administration (FDA). 9 November 2017. Retrieved 28 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c Cohen D (November 2014). “US trade rep is pressing Indian government to forbid production of generic cancer drug, consortium says”. BMJ. 349: g6593. doi:10.1136/bmj.g6593. PMID 25370846. S2CID 206903723.
- ^ Jump up to:a b c Kirkland JL, Tchkonia T (2020). “Senolytic drugs: from discovery to translation”. Journal of Internal Medicine. 288 (5): 518–536. doi:10.1111/joim.13141. PMC 7405395. PMID 32686219.
- ^ Jump up to:a b Paez-Ribes M, González-Gualda E, Doherty GJ, Muñoz-Espín D (2019). “Targeting senescent cells in translational medicine”. EMBO Molecular Medicine. 11 (12): e10234. doi:10.15252/emmm.201810234. PMC 6895604. PMID 31746100.
- ^ Wyld L, Bellantuono I, Tchkonia T, Danson S, Kirkland JL (2020). “Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies”. Cancers. 12 (8): e2134. doi:10.3390/cancers12082134. PMC 7464619. PMID 32752135.
Further reading[edit]
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. (December 2004). “Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays”. Journal of Medicinal Chemistry. 47 (27): 6658–61. doi:10.1021/jm049486a. PMID 15615512.
External links[edit]
- “Dasatinib”. Drug Information Portal. U.S. National Library of Medicine.
/////////////DASATINIB, BMS 35482503, KIN 001-5, NSC 759877, Sprycel, BMS, APOTEX, ダサチニブ水和物 , X78UG0A0RN, дазатиниб , دازاتينيب , 达沙替尼 ,
#DASATINIB, #BMS 35482503, #KIN 001-5, #NSC 759877, #Sprycel, #BMS, #APOTEX, #ダサチニブ水和物 , #X78UG0A0RN, #дазатиниб , #دازاتينيب , #达沙替尼 ,
O.Cc1nc(Nc2ncc(s2)C(=O)Nc3c(C)cccc3Cl)cc(n1)N4CCN(CCO)CC4
PATENT
https://patents.google.com/patent/US8884013B2/enDasatinib, with the trade name SPRYCEL™, is a oral tyrosine kinase inhibitor and developed by BMS Company. It is used to cure adult chronic myelogenous leukemia (CML), acute lymphatic leukemia (ALL) with positive Philadelphia chromosome, etc. Its chemical name is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidyl]amino]-5-thiazolformamide and its chemical structure is as following:

Five polymorphs of Dasatinib and the preparation methods thereof were described by Bristol-Myers Squibb in the Chinese Patent Application No. CN200580011916.6 (publication date is 13 Jun. 2007). The preparation methods instructed in this document are:Monohydrate: Dasatinib (48 g) was added into ethanol (1056 mL 22 ml/g) and water (144 mL), and dissolved by heating to 75° C.; the mixture was purified, filtrated and transferred to the receiver. The solution reactor and transferring pipes were washed with the mixture of ethanol (43 mL) and water (5 mL). The solution was heated to 75˜80° C. to be soluble completely and water (384 mL) was heated and the temperature of the solution was kept between 75° C. and 80° C. The seed crystal of monohydrate (preferable) was added when cooling to 75° C., and keep the temperature at 70° C. for 1 h; cooling to 5° C. within 2 h and keeping the temperature at 0˜5° C. for 2 h. The slurry was filtrated and the filter cake was washed by the mixture of ethanol (96 mL) and water (96 mL); after being dried under vacuum≦50° C. 41 g of solid was obtained.Butanol solvate: under refluxing (116° C.˜118° C.), Dasatinib was dissolved in 1-butanol (about 1 g/25 mL) to yield crystalline butanol solvate of Dasatinib. When cooling, this butanol solvate was recrystallized from solution. The mixture was filtrated and the filter cake was dried after being washed with butanol.Ethanol solvate: 5D (4 g, 10.1 mmol), 7B (6.6 g, 50.7 mmol), n-bubanol (80 mL) and DIPEA (2.61 g, 20.2 mmol)) were added into a 100 ml round flask. The obtained slurry was heated to 120° C. and kept the temperature for 4.5 h, and then cooled to 20° C. and stirred over night. The mixture was filtrate, and the wet filter cake was washed with n-butanol (2×10 mL) to yield white crystal product. The obtained wet filter cake was put back to the 100 ml reactor and 56 mL (12 mL/g) of 200 proof ethanol was added. Then additional ethanol (25 mL) was added at 80° C., and water (10 mL) was added into the mixture to make it dissolved rapidly. Heat was removed and crystallization was observed at 75° C.˜77° C. The crystal slurry was further cooled to 20° C. and filtrated. The wet filter cake was washed with ethanol:water (1:1, 10 mL) once and then washed with n-heptane (10 mL) once. After that it was dried under the condition of 60° C./30 in Hg for 17 h to yield 3.55 g of substance only containing 0.19% water.Neat form of N-6: DIPEA (155 mL, 0.89 mmol) was added into the mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL). The suspension was heated at 110° C. for 25 min to be solution, which was then cooled down to about 90° C. The obtained solution was added dropwise into hot water (80° C., 8010 mL), and the mixture was stirred at 80° C. with heat preservation for 15 min and cooled to room temperature slowly. The solid was filtrated under vacuum and collected, washed by water (2×1600 mL) and dried under vacuum at 55° C.˜60° C. to give 192.45 of compound.Neat form of T1H1-7 (neat form and pharmaceutically acceptable carrier): monohydrate of Dasatinib was heated over dehydrate temperature to yield.Because Dasatinib is practically insoluble in water or organic solvent (e.g. methanol, ethanol, propanol, isopropanol, butanol, pentanol, etc.), even in the condition of heating, a large amount (over 100 times) of solvent is needed, which is disadvantageous in industrial production; in addition, with the method described in the Patent document of CN200580011916.6, the related substances in products can not be lowed effectively during the process of crystal preparation to improve the products quality.In terms of polymorphs of drug, each polymorph has different chemical and physical characteristics, including melting point, chemical stability, apparent solubility, rate of dissolution, optical and mechanical properties, vapor pressure as well as density. Such characteristics can directly influence the work-up or manufacture of bulk drug and formulation, and also affect the stability, solubility and bioavailability of formulation. Consequently, polymorph of drug is of great importance to quality, safety and efficacy of pharmaceutical preparation. When it comes to Dasatinib, there are still needs in the art for new polymorphs suitable for industrial production and with excellent physical and chemical properties as well.Example 1Preparation of the Polymorph IA. Dasatinib (10 g) and DMSO (40 ml) were added into a flask and heated up to 60˜70° C. by stirring, after dissolving, the mixture (120 mL) of water and acetone (1:1) was added under heat preservation. When crystal was precipitated, cooled it down to 0° C. to grow the grains for 10 minutes. Filtrate it and the cake was washed by water and then by the mixture of water and acetone (1:1). After that it was dried under −0.095 MPa at about 50° C. using phosphorus pentoxide as drying aid to give 7.7 g of white solid. Yield was 77%.Contrasts Index of raw material Items before transformation Index of Polymorph I Appearance off-white powder White crystal powder Related substance 0.85% 0.07% KF moisture 0.67% 3.59% 70~150 0.72% 3.63% TGA weight loss
The following items of products prepared by Method A were detected: microscope-crystal form (See. FIG. 1); XRPD Test (See. FIG. 2), IR Test (See. FIG. 3), DSC-TGA Test (See. FIG. 4-1, 4–2), 13C Solid-state NMR Test (See. FIG. 5).B. Dasatinib (10 g) and DMSO (40 ml) were added into a flask and heated slowly up to 60˜70° C. by stirring, after dissolving, the mixture (160 mL) of ethanol and water (1:1) was added under heat preservation. When crystal was precipitated, cooled it down to 0° C. to grow the grains for 10 minutes. Filtrate it and the cake was washed by the mixture of ethanol and water (1:1) and dried under −0.095 MPa at about 50° C. using phosphorus pentoxide as drying aid to give 7.7 g of white solid. Yield was 87%.Contrasts Index of raw material Items before transformation Index of Polymorph I Appearance off-white powder White crystal powder Related substance 0.85% 0.08% KF moisture 0.67% 3.58% 70~150 0.72% 3.67% TGA weight lossHPLC.Related Substances DeterminationHPLC conditions and system applicability: octadecylsilane bonded silica as the filler; 0.05 mol/L of potassium dihydrogen phosphate (adjusted to pH 2.5 by phosphoric acid, 0.2% triethylamine)-methanol (45:55) as the mobile phase; detection wavelength was 230 nm; the number of theoretical plates should be not less than 2000, calculated according to the peak of Dasatinib. The resolution of the peak of Dasatinib from the peaks of adjacent impurities should meet requirements.Determination method: sample was dissolved in mobile phase to be the solution containing 0.5 mg per milliliter. 20 μL of such solution was injected into liquid chromatograph, and chromatogram was recorded until the sixfold retention time of major component peak. If there were impurities peaks in the chromatogram of sample solution, total impurities and any single impurity were calculated by normalization method on the basis of peak area.Stability of Polymorph in the FormulationsThe XRPD patterns of capsules and tablets respectively prepared in the Example 3 and Example 4 have been tested, and compared with XRPD characteristic peaks of Polymorph I of Dasatinib prepared by the Method A in the Example 1 in the present invention, as listed in the following table:Bulk Drug Capsules 1 Capsules 2 Tablets 2 (Polymorph (Polymorph (Polymorph Tablets 1 (Polymorph I) I) I) (Polymorph I) I) 2θ 2θ 2θ 2θ 2θ 9.060 9.080 9.070 9.060 9.070 11.100 11.120 11.110 11.100 11.110 13.640 13.670 13.650 13.640 13.650 15.100 15.120 15.110 15.100 15.110 17.820 17.840 17.830 17.820 17.820 19.380 19.400 19.390 19.380 19.390 22.940 22.970 22.950 22.950 22.950The results in the above-mentioned comparative table have shown that the crystal form had substantially no change after Polymorph I of Dasatinib in the invention were prepared into capsules or tablets by the formulation process.In addition, The relative substances of capsules and tablets respectively prepared in the Example 3 and Example 4 have been tested, and compared with those of Polymorph I of Dasatinib prepared by the Method A in the Example 1 in the present invention, as listed in the following table:Bulk Drug (Polymorph I) Capsules 1 Capsules 2 Tablets 1 Tablets 2 0.07% 0.08% 0.08% 0.07% 0.08%The results in the above-mentioned comparative table have shown that the Polymorph I of Dasatinib was stable, and there were no significantly changes in respect to the relative substances, after Polymorph I of Dasatinib in the invention were prepared into capsules or tablets by the formulation process.INDUSTRIAL APPLICATIONThe present invention provides novel polymorphs of Dasatinib, preparing methods, and pharmaceutical composition comprising them. These polymorphs have better physicochemical properties, are more stable and are more suitable for industrial scale production, furthermore, are suitable for long-term storage, and are advantageous to meet the requirements of formulation process and long-term storage of formulations. The preparation technique of this invention was simple, quite easy for operation and convenient for industrial production, and the quality of the products was controllable with paralleled yields. In addition, by the methods of polymorph preparation in this invention, the amount of organic solvent used in crystal transformation could be reduced greatly, which led to reduced cost of products; organic solvents in Class III with low toxicity could be used selectively to prepare the polymorphs of this invention, reducing the toxic effects of the organic solvents potentially on human body to some extent.PATENThttps://patents.google.com/patent/WO2010067374A2/enDasatinib are antineoplastic agents, which were disclosed in WO Patent Publication No. 00/62778 and U.S. Patent No. 6,596,746. Dasatinib, chemically N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4- pyrimidinyl]amino]-5-thiazolecarboxamide, is represented by the following structure:

Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, etc. Although those differences disappear once the compound is dissolved, they can appreciably influence pharmaceutically relevant properties of the solid form, such as handling properties, dissolution rate and stability. Such properties can significantly influence the processing, shelf life, and commercial acceptance of a polymorph. It is therefore important to investigate all solid forms of a drug, including all polymorphic forms, and to determine the stability, dissolution and flow properties of each polymorphic form. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR).Solvent medium and mode of crystallization play very important role in obtaining a crystalline form over the other. Dasatinib can exist in different polymorphic forms, which differ from each other in terms of stability, physical properties, spectral data and methods of preparation.U.S. Patent Application No. 2005/0215795 A1 (herein after referred to as the 795 patent application) described five crystalline forms of dasatinib (monohydrate, butanol solvate, ethanol solvate, neat form (N-6) and neat form (T1H1-7)), characterized by powder X-ray diffraction (P-XRD) pattern.According to the ‘795 patent application, dasatinib monohydrate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 18.0, 18.4, 19.2, 19.6, 21.2, 24.5, 25.9 and 28.0 ± 0.2 degrees. As per the process exemplified in the ‘795 patent application, dasatinb monohydrate can be obtained in dasatinib, by heating and dissolving the dasatinib in an ethanol and water mixture. Crystallizing the monohydrate from the ethanol and water mixture and cooled to get dasatinib monohydrate.According to the ‘795 patent application, dasatinib crystalline butanol solvate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 5.9, 12.0, 13.0, 17.7, 24.1 and 24.6 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline ethanol solvate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 5.8, 11.3, 15.8, 17.2, 19.5, 24.1, 25.3 and 26.2 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline neat form (N-6) is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 6.8, 11.1, 12.3, 13.2, 13.7, 16.7, 21.0, 24.3 and 24.8 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline neat form (T1H1-7) is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 8.0, 9.7, 11.2, 13.3, 17.5, 18.9, 21.0 and 22.0 ± 0.2 degrees.U.S. Patent application No. 2006/0094728 disclosed ethanolate form (T1E2-1) of dasatinib, characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 7.2, 12.0, 12.8, 18.0, 19.3 and 25.2 ± 0.2 degrees. We have discovered novel crystalline form of dasatinib, dasatinib dimethylformamide solvate, dasatinib dimethyl sulfoxide solvate, dasatinib toluene solvate and dasatinib isopropyl acetate solvate.Another object of the present invention is to provide process for preparing the novel crystalline form of dasatinib, dasatinib dimethylformamide solvate, dasatinib dimethyl sulfoxide solvate, dasatinib toluene solvate, dasatinib isopropyl acetate solvate and known crystalline dasatinib monohydrate.Still another object of the present invention is to provide pharmaceutical compositions containing the novel crystalline form of dasatinib.Reference Example2-(6-Cholro-2-methylpyrimidin-4-yl-amino)-N-(2-chloro-6-methylphenyl) thiazole-5-carboxamide (15 gm) was added to 1-(2-hydroxyethyl)piperazine at 250C and heated to 850C, stirred for 2 hours 30 minutes at 850C. To the solution was added water (500 ml) at 800C and slowly cooled to 250C, stirred for 1 hour at 250C. The solid was collected by filtration and the solid was washed with water (50 ml), and then dried the solid at 550C under vacuum to obtain 15 gm of dasatinib.Example 1Dasatinib (5 gm) obtained according to reference example was dissolved in ethyl acetate (300 ml) at 250C and heated to reflux temperature. To the solution was added methanol (100 ml) and stirred for 30 minutes at reflux temperature to form clear solution. The solution was slowly cooled to room temperature and then cooled to O0C, stirred for 1 hour at O0C. The solid was collected by filtration and the solid was washed with mixture of ethyl acetate and methanol (20 ml, 3:1), and then dried the solid at 500C under vacuum to obtain 3.5 gm of crystalline dasatinib form I.Example 2Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in acetone (100 ml) and methanol (250 ml) and heated to reflux temperature, stirred for 30 minutes at reflux temperature to form clear solution. The solution was cooled to room temperature and then cooled to 200C, stirred for 1 hour at 200C. The solid was collected by filtration and the solid was washed with mixture of acetone (10 ml) and methanol (25 ml), and then dried the solid at 500C under vacuum to obtain 4 gm of crystalline dasatinib form I (HPLC purity: 99.85%).Example 3Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) at 250C and heated to 650C to form clear solution. To the solution was slowly added acetone (50 ml) at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C and stirred for 1 hour at 250C. The contents are filtered and the solid obtained was washed with mixture of dimethylformamide and acetone (15 ml, 1:2), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.94%).Example 4Dasatinib (5 gm) was dissolved in dimethylformamide (25 ml) at 250C and heated to 650C to form clear solution. Ethyl acetate (50 ml) was added slowly to the solution at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C, stirred for 1 hour at 250C and filtered. The solid obtained was washed with mixture of dimethylformamide and ethyl acetate (30 ml, 1:2), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate.Example 5Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) and heated to 650C to form a clear solution. The solution was cooled to 250C and then cooled to 50C, stirred for 4 hour at 50C. The solid was collected by filtration and the solid was washed with chilled dimethylformamide (10 ml), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.9%).Example 6Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) and heated to 650C to form a clear solution. Water (50 ml) was added slowly to the solution at 650C and stirred for 1 hour at 650C. The solution was cooled to 250C and stirred for 30 minutes at 250C. The solid was collected by filtration and the solid was washed with mixture of dimethylformamide and water (15 ml, 1 :2), and then dried the solid at 500C under vacuum to obtain 4.7 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.93%).Example 7Dasatinib dimethylformamide solvate (4.7 gm) obtained as in example 6 was dissolved in water (50 ml) and heated to 750C, stirred for 4 hours at 750C. The solution was cooled to 250C, stirred for 30 minutes at 250C and filtered. The solid obtained was washed with water (15 ml), and then dried at 500C under vacuum to obtain 4.7 gm of dasatinib monohydrate.Example 8Dasatinib (20 gm) was dissolved in dimethyl sulfoxide (100 ml) at 250C and heated to 650C to form clear solution. To the solution was slowly added water (200 ml) at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C and stirred for 30 minutes at 250C. The solid was collected by filtration and the solid was washed with mixture of dimethyl sulfoxide and water (30 ml, 1 :2), and then dried the solid at 500C under vacuum to obtain 19.5 gm of dasatinib monohydrate.Example 9Dasatinib (5 gm) was dissolved in isopropyl acetate (65 ml) and heated to 800C, stirred for 1 hour at 800C to form a clear solution. The solution was cooled to 250C, stirred for 1 hour at 250C and filtered. The solid obtained was washed with isopropyl acetate (15 ml) to obtain 5 gm of dasatinib isopropyl acetate solvate.Example 10Dasatinib (6 gm) was dissolved in toluene (100 ml) and heated to reflux temperature, stirred for 2 hours at reflux temperature to form a clear solution. The solution was slowly cooled to 250C. The contents are filtered and the solid obtained was washed with toluene (20 ml) to obtain 5.5 gm of dasatinib toluene solvate.Example 11Dasatinib (5 gm) was dissolved in dimethyl sulfoxide (20 ml) at 250C and heated to 650C. To the solution was slowly added ethyl acetate (200 ml) at 650C and the solution was slowly cooled to O0C, stirred for 2 hours at O0C. The solid was collected by filtration and the solid was washed with mixture of dimethyl sulfoxide and ethyl acetate (55 ml, 1 :10), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethyl sulfoxide solvate.
PATENThttps://patents.google.com/patent/WO2014086326A1/enDasatinib, N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)- 1 -piperazinyl]-2- methyl-4-pyrimidmyl]amino]-5-thiazole carboxamide of formula I, also known as BMS- 354825, is a cancer treatment drug developed by Bristol-Myers Squibb and sold under the trade name Sprycel®. Dasatinib is a multi- BCR/ABL and Src family tyrosine kinase inhibitor and it is used for treatment of chronic myelogenous leukaemia (CML) as a secondary drug after primary treatment with imatinib (Gleevec®). It is also used for treatment of acute lymphoblastic leukaemia caused by mutation/translocation of chromosomes and development of the so-called Philadelphia chromosome (Ph+ ALL). However, its potential is so wide that the possibility of using it for treatment of other types of cancer, including advanced stages of prostate cancer, is still being investigated.

(I)In accordance with the basic patent WO2000062778A1, dasatinib is prepared by reaction of the key intermediate of formula II with l-(2-hydroxyethyl)piperazine in the presence of a base and a suitable solvent (Scheme 1). A similar preparation method was later used in a number of other process patents, only varying the corresponding base or solvent. Through the selection of a suitable solvent or procedure a great number of solvates or polymorphs can be prepared. Polymorphs have been one of the most frequently studied physical characteristics of active pharmaceutical substances (API) recently. Thus, different polymorphs of one API may have entirely different physical-chemical properties such as solubility, melting point, mechanical resistance of crystals but they may also influence the chemical and physical stability. Then, these properties may have an impact on further processes such as handling of the particular API, grinding or formulation method. These various physical-chemical characteristics of polymorphs influence the resulting bioavailability of the solid dosage form. Therefore, looking for new polymorphs and solvates is becoming an important tool for obtaining a polymorph form with the desired physical-chemical characteristics.

The process patent WO2005077945A2 describes preparation of the following solvates of dasatinib: monohydrate, butanol solvate, as well as two anhydrous forms (N-6 and T1H1- 7). A related patent also mentions two ethanol solvates, the hemi-ethanol and diethanol solvates (US 8 242 270 B2). Salts, various combinations of salts and their solvates have been described in detail in the patent application WO2007035874A1.Another process patent, WO2009053854A2, dealt with the preparation of a number of solvates or mixed solvates out of which especially the isopropanol and mixed isopropanol/dimethyl sulfoxide solvates, as well as a new solid form B, another anhydrous polymorph of dasatinib, are worth mentioning. Other patent applications have also dealt with the preparation of other solvates/mixed solvates (WO2010067374A2), or processes for the preparation and purification of the monohydrate/anhydrous form (WO2010139981A2) and its polymorphs (WO2011095059 Al).API solvates or salts are used in drug formulations in many cases. In the case of solvates the limits for individual solvents, their contents or maximum daily doses have to be strictly observed. Then, these limits can dramatically restrict their effective use. Thus, the clearly most convenient option is the use of sufficiently stable polymorphs of API that do not contain any solvents bound in the crystalline structure.Some of the above mentioned patent documents describe preparation of a stable anhydrous form of dasatinib (N-6). In accordance with individual patent documents the main disadvantages of the preparation of N-6 is the necessity of desolvation of the solvated form of the API at high temperatures (WO2009053854A2), or application of an increased temperature (50°C and more) and vacuum for a relatively long time (8-12h; WO2010139981A2 and WO2005077945A2). These procedures are very demanding from the point of view of general technology, energy and time, to say nothing of the necessity to work under an inert atmosphere to prevent possible oxidation-degradation reactions of the API. This is because dasatinib may be oxidized by atmospheric oxygen to the corresponding N-oxide (oxidation occurs in the piperazine ring), which may undergo the Cope elimination at increased temperatures. This secondary reaction may subsequently impair the purity of the prepared API.With a view to the above mentioned facts it is obvious that completely new methods and processes have to be developed even for polymorphs or solvates that are already well- known. Generally, the development of technologically and economically more efficient procedures is the main decisive parameter in their industrial utilization for the preparation of the API.Dasatinib of formula I is prepared by a reaction of the intermediate of formula II with l-(2- hydroxyethyl)piperazine in the presence of diisopropylethylamine (DIPEA) in an organic solvent from the group of dipolar aprotic solvents, higher alcohols or diols.If a dipolar aprotic solvent from the group of N-methyl-2-pyrrolidone (NMP), N^iV-dimethyl formamide (DMF), AyV-dimethyl acetamide (DMA), dimethyl sulfoxide (DMSO), formamide (FA), N,N -dimethyl propylene urea (DMPU) and l,3-dimethyl-2-imidazolidinone (DMI) is used, the reaction is carried out at 50-110°C under an inert atmosphere for 1/2-6 hours. In a preferable embodiment, NMP, DMSO, DMPU or DMI is used and the reaction is carried out at 90°C for 1-3 hours. The result of the reaction is crude dasatinib in the form of a solution in the corresponding solvent.If an alcohol from the group of isoamyl alcohol or 1,3-propanediol is used as a solvent for preparation of the crude dasatinib, the reaction mixture is heated at 120-160°C for 2-12 hours, in a preferable embodiment at 135°C for 3-6 hours.If dipolar aprotic solvents (NMP, DMF, DMA, DMSO, FA, DMPU and DMI) are used, in step a) a precipitant is added to the hot solution (90°C) under continuous stirring in an inert atmosphere in a 2- 15 fold, most preferably 4-10fold (by volume) amount with respect to the dipolar aprotic solvent. Suitable precipitants comprise especially acetonitrile, propionitrile, most preferably acetonitrile.After addition of the precipitant the obtained solution is withdrawn from the heating bath and is slowly left to cool down to 22°C under continuous stirring in an inert atmosphere. Crystallization occurs within 1-120 minutes (depending on the volume, until complete cooling). After having cooled down to 22°C (laboratory temperature), the suspension is stirred for another hour. The corresponding solvate of dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35 °C, most preferably at 22°C, and washed with the respective co-solvent.The solvate of dasatinib obtained this way can be directly used in the next step – recrystallization, without the necessity of drying. If necessary, the product may be dried at 10- 35°C, most preferably at 25°C, and at the pressure of 10-200 kPa, most preferably 50 kPa, for 6-24 hours, most preferably 12 hours.If NMP is used as the solvent in step a), the corresponding NMP solvate is isolated. The obtained dried crystalline NMP solvate (NM) of dasatinib has a characteristic XRPD pattern, which is presented in Figure no. 1. The NMP solvate (NM) has the following characteristic peaks: 5.88; 6.73; 10.73; 11.92; 13.39; 14.97; 16.72; 18.95; 20.17; 21.46; 22.81; 24.65; 25.18; 26.02 and 28.06 ± 0.2° 2-theta.If isoamyl alcohol or 1,3-propanediol are used as the solvents in step a), the reaction mixture is left to cool down to 22°C after expiration of the reaction time (3-6 h). Crystallization generally begins when the inner temperature of the reaction mixture drops to 100°C. After cooling down to 22°C (laboratory temperature), the suspension is further stirred for another 1 hour. Crystalline dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35°C, most preferably at 22°C, and washed with the corresponding solvent.The obtained product is dried at 10-35°C, most preferably at 25°C, and at the pressure of 10-200 kPa, most preferably 50 kPa, for 6-24 hours, most preferably 12 hours.The obtained crystalline isoamyl alcohol solvate (SI) of dasatinib has a characteristic XRPD pattern, which is shown in Figure no. 2. The solvate (SI) has the following characteristic peaks: 5.72; 10.35; 11.42; 12.61; 13.14; 14.27; 15.33; 17.18; 17.44; 17.97; 19.12; 19.95; 20.38; 22.05; 22.42; 23.01; 23.46; 23.68; 25.26; 26.20; 26.45; 26.62 and 27.78 ± 0.2° 2-theta.The obtained crystalline 1,3-propanediol solvate (SP) of dasatinib has a characteristic XRPD pattern, which is shown in Figure no. 3. The solvate (SP) has the following characteristic peaks: 6.04; 12.01; 15.10; 17.95; 18.35; 18.77; 21.25; 21.51; 22.96; 24.08; 24.62; 25.80; 26.16; 28.16 and 33.6578 ± 0.2° 2-theta.These solvates (or polymorph forms) are then easily converted to the desired anhydrous polymorph N-6 or another solvate in steps b) and c). All the forms prepared this way are sufficiently stable and can easily be isolated in the chemical purities of 99% and higher (in accordance with HPLC).The anhydrous polymorph form N-6 is prepared in the following way: any solvate or another polymorph is dissolved under an inert atmosphere at 90°C (reflux) in a 10-30 times, most preferably 20 times, the (weight) amount of the crystallization solvent. Suitable crystallization solvents include especially methanol, ethanol, isopropanol, most preferably methanol.A co-solvent is added in 0.1-10 times, most preferably ½-l times, the volume of the crystallization solvent used in an inert atmosphere at 90°C. The co-solvent can be, e.g., acetonitrile, propionitrile and their mixtures, most preferably acetonitrile. After addition of the co-solvent the obtained solution is withdrawn from the heating bath and is slowly left to cool down to 22°C under continuous stirring in an inert atmosphere. Crystallization occurs during 1-120 minutes (depending on the volume, until complete cooling). After having cooled down to 22°C (laboratory temperature), the suspension is stirred for another hour. Crystalline dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35°C, most preferably at 22°C, and washed with the corresponding co-solvent. The chemical purity of the obtained product is 99% (in accordance with HPLC); it is the polymorph form N-6 and its XRPD pattern is shown in Figure no. 4. The polymorph form N-6 has the following characteristic peaks: 6.77; 12.31; 13.16; 13.75; 16.70; 17.20; 18.54; 19.34; 20.25; 20.95; 21.94; 24.28; 24.82; and 27.80 ± 0.2° 2-theta.Brief Description of Drawings:Figure 1: shows an X-ray powder diffraction pattern of the crystalline solvate NM. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Figure 2: shows an X-ray powder diffraction pattern of the isoamyl alcohol crystalline solvate SI. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation. Figure 3: shows an X-ray powder diffraction pattern of the 1,3 propanediol crystalline solvate SP. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Figure 4: shows an X-ray powder diffraction pattern of the crystalline anhydrous form N-6. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Examples: The following working examples illustrate methods for the preparation of dasatinib of formula I, its polymorph form N-6 and its solvates NM, SI, SP.The polymorph forms and solvates of dasatinib were characterized with X-ray powder diffraction using the following methods:The diffraction patterns were measured using an X’PERT PRO MPD PANalytical diffractometer with a graphite monochromator, radiation used CuKa (λ=1.542 A), excitation voltage: 45 kV, anode current: 40 mA, measured range: 2 – 40° 2Θ, increment: 0.01° 2Θ. The measurement was carried out using a flat powder sample that was placed on a Si plate. For the primary optic setting programmable divergence diaphragms with the irradiated sample area of 10 mm, Soller diaphragms 0.02 rad and an anti-dispersion diaphragm ¼ were used. For the secondary optic setting an X’Celerator detector with the maximum opening of the detection slot, Soller diaphragms 0.02 rad and an anti-dispersion diaphragm 5.0 mm were used. HPLC method:Stock solution of samples: dissolve 5.0 mg of the sample in 10.0 ml of 50% acetonitrile R with water.Dimensions of the chromatographic HPLC column: / = 0.10 m, d= 3 mm- stationary phase: Zorbax Eclipse Plus Phenyl-Hexyl RRHD 1.8 μιη; temperature: 35 °C. Mobile phase: A: phosphate buffer (0.01 M sodium dihydrogen phosphate, pH treated by addition of sodium hydroxide to 7.00 ± 0.05); B: acetonitrile R.Gradient (A/B; flow 0.6 ml/min): 0 min 80/20; 10 min 50/50; 11 min 50/50; 12 min 80/20. Detection at the wavelength of 220 nm.Feed: 2 μΐ of the sample stock solution Example 1.Preparation of the NMP solvate (NM) of dasatinib:The intermediate of formula II (1.00 g; 2.54 mmol) and l-(2-hydroxyethyl)piperazine (1.66 g; 12.77 mmol) were dissolved in N-methylpyrrolidone (5 ml) under an inert atmosphere and diisopropylethylamine (0.9 ml, 5.18 mmol) was added to the reaction mixture. The reaction mixture was stirred and heated up to 90°C for 70 minutes and then acetonitrile (30 ml) was added to the reaction. The mixture was withdrawn from the heating bath and stirred intensively. Crystallization started after 5 minutes, the suspension was left to cool down under continuous stirring. After achieving the laboratory temperature it was stirred for another 2 hours. The crystalline substance was aspirated on frit S3, washed with acetonitrile (5 ml) and dried by suctioning under an inert nitrogen atmosphere for 15 minutes. The XRPD pattern of the sample obtained this way corresponds to the NMP solvate (NM) and can be used in the subsequent steps without the necessity of drying. Drying after 6 hours in an exsiccator at the laboratory temperature in vacuo (50 kPa) provided 1.2 g of crystalline dasatinib; 80% of the theoretical yield. HPLC purity 99.12%. The 1H NMR and 13C NMR spectra correspond to the data known from the literature. The XRPD pattern of the dried product corresponds to the NMP solvate (NM). The NM solvate is characterized by the reflections presented in Table 1 :Table 1 – NM forminterplanarpos. distance[°2Th.] [nm] rel. int. [%]5.88 1.5024 81.86.73 1.3131 100.010.73 0.8236 10.611.92 0.7420 59.213.39 0.6606 19.614.97 0.5915 38.416.72 0.5298 45.018.95 0.4679 10.920.17 0.4399 13.921.46 0.4138 13.422.81 0.3895 21.024.65 0.3608 13.325.18 0.3534 14.426.02 0.3422 11.928.06 0.3177 5.8


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}