
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Anamorelin249921-19-5[RN]
3-{(2R)-3-{(3R)-3-Benzyl-3-[(trimethylhydrazino)carbonyl]-1-piperidinyl}-2-[(2-methylalanyl)amino]-3-oxopropyl}-1H-indole
3-Piperidinecarboxylic acid, 1-[(2R)-2-[(2-amino-2-methyl-1-oxopropyl)amino]-3-(1H-indol-3-yl)-1-oxopropyl]-3-(phenylmethyl)-, 1,2,2-trimethylhydrazide, (3R)-8846анаморелинأناموريلين阿那瑞林
| Formula | C31H42N6O3 |
|---|---|
| Molar mass | 546.716 g·mol−1 |
.HCL
Anamorelin hydrochloride
3-Piperidinecarboxylic acid, 1-[(2R)-2-[(2-amino-2-methyl-1-oxopropyl)amino]-3-(1H-indol-3-yl)-1-oxopropyl]-3-(phenylmethyl)-, 1,2,2- trimethylhydrazide, hydrochloride (1:1), (3R)-
| Formula | C31H42N6O3. HCl |
|---|---|
| CAS | 861998-00-7 |
| Mol weight | 583.1645 |
APPROVED JAPAN PMDA Adlumiz, 22/1/2021
アナモレリン塩酸塩
ONO-7643, RC-1291, ST-1291
Antineoplastic, Growth hormone secretagogue receptor (GHSR) agonist
Anamorelin is a non-peptidic ghrelin mimetic
Treatment of cancer anorexia and cancer cachexia
Anamorelin hydrochloride has been submitted New Drug Application (NDA) for the treatment of cachexia in non-small cell lung cancer (NSCLC) patients.
It was originally developed by Novo Nordisk, then it was licensed to Ono and Helsinn Therapeutics for the treatment of cachexia and anorexia in cancer patients.
Anamorelin hydrochloride has been submitted New Drug Application (NDA) for the treatment of cachexia in non-small cell lung cancer (NSCLC) patients.
It was originally developed by Novo Nordisk, then it was licensed to Ono and Helsinn Therapeutics for the treatment of cachexia and anorexia in cancer patients.
Company:Novo Nordisk (Originator) , Helsinn,Ono
Anamorelin (INN) (developmental code names ONO-7643, RC-1291, ST-1291), also known as anamorelin hydrochloride (USAN, JAN), is a non-peptide, orally-active, centrally-penetrant, selective agonist of the ghrelin/growth hormone secretagogue receptor (GHSR) with appetite-enhancing and anabolic effects which is under development by Helsinn Healthcare SA for the treatment of cancer cachexia and anorexia.[2][3][4]
Anamorelin significantly increases plasma levels of growth hormone (GH), insulin-like growth factor 1 (IGF-1), and insulin-like growth factor-binding protein 3 (IGFBP-3) in humans, without affecting plasma levels of prolactin, cortisol, insulin, glucose, adrenocorticotropic hormone (ACTH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), or thyroid-stimulating hormone (TSH).[3][5] In addition, anamorelin significantly increases appetite, overall body weight, lean body mass, and muscle strength,[4][5] with increases in body weight correlating directly with increases in plasma IGF-1 levels.[3]
As of February 2016, anamorelin has completed phase III clinical trials for the treatment of cancer cachexia and anorexia associated with non-small-cell lung carcinoma.[6][7]
On 18 May 2017, the European Medicines Agency recommended the refusal of the marketing authorisation for the medicinal product, intended for the treatment of anorexia, cachexia or unintended weight loss in patients with non-small cell lung cancer. Helsinn requested a re-examination of the initial opinion. After considering the grounds for this request, the European Medicines Agency re-examined the opinion, and confirmed the refusal of the marketing authorisation on 14 September 2017.[8] The European Medicines Agency concluded that the studies show a marginal effect of anamorelin on lean body mass and no proven effect on hand grip strength or patients’ quality of life. In addition, following an inspection at clinical study sites, the agency considered that the safety data on the medicine had not been recorded adequately. Therefore, the agency was of the opinion that the benefits of anamorelin did not outweigh its risks.[9]
EMA
The chemical name of anamorelin hydrochloride is 2-Amino-N-((R)-1-((R)-3-benzyl-3-(1,2,2-trimethylhydrazine-1-carbonyl)piperidin-1-yl)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-methylpropanamide hydrochloride corresponding to the molecular formula C31H42N6O3•HCl and has a relative molecular mass 583.16 g/mol and has the following structure:
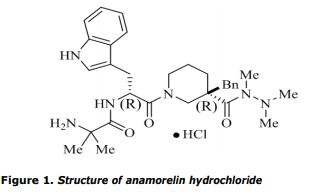
The structure of the active substance was elucidated by a combination of 1 H-NMR, 13C-NMR, elemental analysis, FT-IR, UV and and mass spectrometry. Anamorelin HCl appears as a white to off-white hygroscopic solid, freely soluble in water, methanol and ethanol, sparingly soluble in acetonitrile and practically insoluble in ethyl acetate, isopropyl acetate and n-heptane. Its pka was found to be 7.79 and the partition coefficient 2.98. It has two chiral centres with the R,R absolute configuration, which is controlled in the active substance specification by chiral HPLC. Based on the presented data, neither anamorelin hydrochloride, nor any of its salts have been previously authorised in medicinal products in the European Union. Anamorelin is therefore considered as a new active substance.
SYN
OPRD

PATENT
WO 9958501
PATENT
WO 2001034593
https://patents.google.com/patent/WO2001034593A1/enExample 1A procedure for the preparation of the compound which is either 2-Amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide

or2-Amino-N-[(1R)-2-[(3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide

Step aPiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester 3-ethyl ester

A one-necked round-bottom flask (1 I) equipped with a magnetic stirrer and addition funnel was charged with NaOH-pellets (15,6 g), tetrahydrofuran (400 ml) and ethylnipecotate (50 ml, 324 mmol). To the stirred mixture at room temperature was added dropwise a solution of Boc2O (84,9 g, 389 mmol) dissolved in tetrahydrofuran (150 ml) (1 hour, precipitation of white solid, NaOH-pellets dissolved, exoterm). The mixture was stirred overnight at room temperature. The mixture was added to EtOAc (500 ml) and H2O (2000 ml), and the aqueous layer was re-extracted with EtOAc (2 X 500 ml) and the combined organic layers were washed with brine (100 ml), dried over MgSO4, filtered and concentrated in vacuo to afford piperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester 3-ethyl ester (82,5 g) as a thin yellow oil.1H-NMR (300 MHz, CDCI3): δ 1,25 (t, 3H, CH3); 1 ,45 (s, 9H, 3 X CH3); 2,05 (m, 1H); 2,45 (m, 1H); 2,85 (m, 1 H); 3,95 (d (broad), 1 H); 4,15 (q, 2H, CH2)Step b3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester 3-ethyl ester (racemic mixture)

A three-necked round-bottom flask (2 I) equipped with a magnetic stirrer, thermometer, nitrogen bubbler and addition funnel was evacuated, flushed with nitrogen, charged with anhydrous tetrahydrofuran (500 ml) and cooled to -70 °C. Then lithium diisopropylamine (164 ml of a 2,0 M solution in tetrahydrofuran, 327 mmol) was added. To the stirred solution at -70 °C was added dropwise over 45 min. a solution of piperidine-1 ,3-dicarboxylic acid 1- tert-butyl ester 3-ethyl ester (80 g, 311 mmol) in anhydrous tetrahydrofuran (50 ml) (temperature between -70 °C and -60 °C, clear red solution). The mixture was stirred for 20 min. and followed by dropwise addition over 40 min. of a solution of benzylbromide (37 ml, 311 mmol) in anhydrous tetrahydrofuran (250 ml) (temperature between -70 °C and -60 °C). The mixture was stirred for 1 hour at -70 °C, and then left overnight at room temperature (pale orange).The reaction mixture was concentrated in vacuo to approx. 300 ml, transferred to a separating funnel, diluted with CH2CI2 (900 ml) and washed with H2O (900 ml). Due to poor separation the aqueous layer was re-extracted with CH2CI2 (200 ml), the combined organic layers were washed with aqueous NaHSO4 (200 ml, 10%), aqueous NaHCO3 (200 ml, saturated), H2O (200 ml), brine (100 ml), dried over MgSO4> filtered and concentrated in vacuo to afford an oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The solids formed was removed by filtration, washed with heptane and dried in vacuo to give a racemic mixture of 3-benzylpiperidine-1 ,3-dicarboxylic acid 1-ter–butyl ester 3-ethyl ester (81 ,4 g). ■ HPLC (h8): Rt = 15,79 min.LC-MS: Rt = 7,67 min. (m+1) = 348,0Step c 3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester (racemic mixture)

3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester 3-ethyl ester (81 g, 233 mmol) was dissolved in EtOH (400 ml) and NaOH (400 ml, 16% aqueous solution) in a one neck round- bottom flask (1 L) equipped with a condenser and a magnetic stirrer. The mixture was refluxed for 10 h under nitrogen, and cooled to room temperature, concentrated in vacuo to approx. 600 ml (precipitation of a solid), diluted with H2O (400 ml), cooled in an icebath, and under vigorous stirring acidified with 4 M H2SO4 until pH = 3 (final temperature: 28 °C). The mixture was extracted with EtOAc (2 X 700 ml), and the combined organic layers were washed with brine (200 ml), dried over MgSO4, filtered and concentrated in vacuo to afford an oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The crystals formed were removed by filtration, washed with heptane and dried in vacuo to give a racemic mixture of 3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester (66,0 g)HPLC (h8): Rt = 12,85 min.LC-MS: Rt = 5,97 min. (m+1) = 320,0Chirale HPLC (Chiracel OJ, heptane(92):iPrOH(8):TFA(0,1)): Rt = 8,29 min. 46,5 % Rt = 13,69 min. 53,5 %Step d(3R)-3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-Benzylpiperidine-1,3-dicarboxylic acid 1-tert-butyl ester
(Resolution of 3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester)

3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester (76 g, 238 mmol) was dissolved in EtOAc (3,0 L) in a one neck flask (5L) equipped with magnetic stirring. Then H2O (30 ml), R(+)-1-phenethylamine (18,2 ml, 143 mmol) and Et3N (13,2 ml, 95 mmol) were added and the mixture was stirred overnight at room temperature resulting in precipitation of white crystals (41 ,9 g), which were removed by filtration, washed with EtOAc and dried in vacuo. The precipitate was dissolved in a mixture of aqueous NaHSO4 (300 ml, 10%) and EtOAc (600 ml), layers were separated and the aqueous layer re-extracted with EtOAc (100 ml). The combined organic layers were washed with brine (100 ml), dried over MgSO4 and filtered. The solvent was removed in vacuo to afford a colourless oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The crystals that had been formed were removed by filtration, washed with heptane and dried in vacuo to give one compound which is either (3R)-3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-benzylpiperidine- 1,3-dicarboxylic acid 1-tert-butyl ester (27,8 g).Chirale HPLC (Chiracel OJ, heptane(92):iPrOH(8):TFA(0,1)):Rt = 7,96 min. 95,8 % eeStep e(3R)-3-Benzyl-3-(N,N’1N’-trimethylhvdrazinocarbonyl)piperidine-1-carboxylic acid tert-butyl ester or (3S)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidine-1-carboxylic acid tert-butyl ester

Trimethylhydrazine dihydrochloride (15,3 g, 104 mmol) was suspended in tetrahydrofuran (250 ml) in a one-neck round-bottom flask (1 I) equipped with a large magnetic stirrer, and an addition funnel/nitrogen bubbler. The flask was then placed in a water-bath (temp: 10- 20°C), bromo-rrts-pyrrolydino-phosphonium-hexafluorophosphate (40,4 g, 86,7 mmol) was added, and under vigorous stirring dropwise addition of diisopropylethylamine (59 ml, 347 mmol). The mixture (with heavy precipitation) was stirred for 5 min., and a solution of the product from step d which is either (3R)-3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-benzylpiperidine-1,3-dicarboxylic acid 1-tert-butyl ester (27,7 g, 86,7 mmol) in tetrahydrofuran (250 ml) was added slowly over 1 ,5 hour. The mixture was stirred overnight at room temperature. The reaction was diluted with EtOAc (1000 ml), washed with H2O (500 ml), aqueous NaHSO4, (200 ml, 10%), aqueous NaHCO3 (200 ml, saturated), brine (200 ml), dried over MgSO4, filtered and concentrated in vacuo to afford a thin orange oil. The mixture was dissolved in EtOAc (300 ml), added to SiO2 (150 g) and concentrated in vacuo to a dry powder which was applied onto a filter packed with SiO2 (150 g), washed with heptan (1 I) and the desired compound was liberated with EtOAc (2,5 I). After concentration in vacuo, the product which is either (3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)-piperidine-1- carboxylic acid tert-butyl ester or (3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)- piperidine-1-carboxylic acid tert-butyl ester (49 g) as an orange oil was obtained.HPLC (h8): Rt = 14,33 min.Ste f(3R)-3-Benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-Benzyl-piperidine-3- carboxylic acid trimethylhydrazide

The product from step e which is either (3R)-3-Benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)-piperidine-1 -carboxylic acid tert-butyl ester or (3S)-3-Benzyl-3- (N,N’,N’-trimethylhydrazinocarbonyl)-piperidine-1 -carboxylic acid tert-butyl ester (56,7 g, 100,9 mmol) was dissolved in EtOAc (500 ml) (clear colourless solution) in a one-neck roundbottom flask (2L) equipped with magnetic stirring. The flask was then placed in a waterbath (temp: 10-20 °C), and HCI-gas was passed through the solution for 5 min. (dust- like precipitation). After stirring for 1 hour (precipitation of large amount of white crystals), the solution was flushed with N2 to remove excess of HCI. The precipitate was removed by gentle filtration, washed with EtOAc (2 X 100 ml), and dried under vacuum at 40 °C overnight to give the product which is either (3R)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide (37,0 g).HPLC (h8): Rt = 7,84 min.Step q r(1 R)-2-r(3R)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-((1 H-indol-3- yl)methyl)-2-oxoethvncarbamic acid tert-butyl ester or .(1 R)-2-..3S)-3-Benzyl-3-(N,N’,N’- trimethylhvdrazinocarbonyl)piperidin-1-vn-1-((1 H-indol-3-yl)methyl)-2-oxoethyllcarbamic acid tert-butyl ester

Boc-D-Trp-OH (32,3 g, 106 mmol) was dissolved in dimethylacetamide (250 ml) in a one- neck roundbottom flask (500 ml) equipped with a magnetic stirrer and a nitrogen bubbler. The solution was cooled to 0-5 °C and 1-hydroxy-7-azabenzotriazole (14,4 g, 106 mmol), 1- ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (20,3 g, 106 mmol), N- methylmorpholine (11 ,6 ml, 106 mmol) were added. After stirring for 20 min. at 0-5 °C the product from step f which is either (3R)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide (37,0 g, 106 mmol) and N-methylmorpholine (24,4 ml, 223 mmol) were added. The reaction was stirred overnight at room temperature. The mixture was then added to EtOAc (750 ml) and washed with aqueous NaHSO4 (300 ml, 10 %). The layers were allowed to separate, and the aqueous layer was re-extracted with EtOAc (500 ml). The combined organic layers were washed with H2O (100 ml), aqueous NaHCO3 (300 ml, saturated), H2O (100 ml), brine (300 ml), dried over MgSO4, filtered and concentrated in vacuo to afford the product which is either [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-((1H- indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester or [(1 R)-2-[(3S)-3-benzyl-3- (N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-((1 H-indol-3-yl)methyl)-2- oxoethyljcarbamic acid tert-butyl ester (56,7g) as an orange oil.HPLC (h8): Rt = 14,61 min.LC-MS: Rt = 7,35 min. (m+1 ) = 562,6Step h1 -f(2R)-2-Amino-3-(1 H-indol-3-yl)propionylH3R)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide or 1-f(2R)-2-Amino-3-(1 H-indol-3-yl)propionvn-(3S)-3-benzylpiperidine-3- carboxylic acid trimethylhydrazide

The product from step g which is either [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1 -yl]-1 -((1 H-indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester or [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1- yl]-1-((1 H-indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester (56,7 g, 100,9 mmol) was dissolved in EtOAc (500 ml) (clear colourless solution) in a one-neck round-bottom flask (2L) equipped with magnetic stirring. The flask was then placed in a water-bath (temp: 10-20 °C), and HCI-gas was passed through the solution for 10 min. (heavy precipitation of oil). The mixture was flushed with N2 to remove excess of HCI and then separated into an oil and an EtOAc-layer. The EtOAc-layer was discarded. The oil was dissolved in H2O (500 ml), CH2CI2 (1000 ml), and solid Na2CO3 was added until pH > 7. The layers were separated, and the organic layer was washed with H2O (100 ml), brine (100 ml), dried over MgSO4, filtered and concentrated in vacuo to afford the product which is either 1-[(2R)-2-amino-3-(1 H-indol- 3-yl)propionyl]-(3R)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide or 1-[(2R)-2- amino-3-(1H-indol-3-yl)propionyl]-(3S)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide (27 g) as an orange foam.HPLC (h8): Rt = 10,03 min.Step i(1-r(1 R)-2-r(3R)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-(1H-indol-3- ylmethyl)-2-oxo-ethylcarbamovπ-1 -methylethyl fcarbamic acid tert-butyl ester or1-r(1 R)-2-r(3S)-3-Benzyl-3-(N,N’.N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-(1 H-indol-3- ylmethyl)-2-oxo-ethylcarbamovπ-1-methylethyl)carbamic acid tert-butyl ester

Boc-Aib-OH (11 ,9 g, 58,4 mmol) was dissolved in dimethylacetamide (125 ml) in a one-neck roundbottom flask (500 ml) equipped with a magnetic stirrer and nitrogen bubbler. To the stirred solution at room temperature were added 1-hydroxy-7-azabenzotriazole (7,95 g, 58,4 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (11 ,2 g, 58,4 mmol), and diisopropylethylamine (13,0 ml, 75,8 mmol). After 20 min. (yellow with precipitation) a solution of the product from step h which is either 1-[(2R)-2-amino-3-(1 H-indol-3- yl)propionyl]-(3R)-3-benzylpiperidine-3:carboxylic acid trimethylhydrazide or 1-[(2R)-2- amino-3-(1 H-indol-3-yl)propionyl]-(3S)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide (27,0 g, 58,4 mmol) in dimethylacetamide (125 ml) was added. The reaction was stirred at room temperature for 3 h. The mixture was added to EtOAc (750 ml) and washed with aqueous NaHSO4 (300 ml, 10 %). The layers were allowed to separate, and the aqueous layer was re-extracted with EtOAc (500 ml). The combined organic layers were washed with H2O (100 ml), aqueous NaHCO3 (300 ml, saturated), H2O (100 ml), brine (300 ml), dried over MgSO4, filtered and concentrated in vacuo to approx. 500 ml. Then SiO2 (150 g) was added and the remaining EtOAc removed in vacuo to give a dry powder which was applied onto a filter packed with SiO2 (150 g), washed with heptan (1 L), and the desired compound was liberated with EtOAc (2,5 L). After concentration in vacuo, the product which is either {1-[(1 R)-2-[(3R)-3-benzyl-3-(N, N’, N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1-methylethyl}carbamic acid tert-butyl ester or {1-[(1R)-2-[(3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1 H-indol-3- ylmethyl)-2-oxo-ethylcarbamoyl]-1-methylethyl}carbamic acid tert-butyl ester 33,9 g as an orange foam was obtained.HPLC (h8): Rt = 14,05 min.Step j2-Amino-N-r(1 R)-2-f(3R)-3-benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vπ-1- (1 H-indol-3-ylmethyl)-2-oxoethyll-2-methylpropionamide, fumarate or2-Amino-N-r(1 R)-2-r(3S)-3-benzyl-3-(N1N’1N’-trimethylhvdrazinocarbonyl)piperidin-1-yll-1- (1H-indol-3-ylmethyl)-2-oxoethvπ-2-methylpropionamide, fumarate

The product from step i which is either {1-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1- methylethyl}carbamic acid tert-butyl ester or {1-[(1 R)-2-[(3S)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1 -yl]-1 -(1 H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1 – methylethyljcarbamic acid tert-butyl ester (23,8 g, 36,8 mmol) was dissolved in of EtOAc (800 ml) (clear yellow solution) in a one neck round-bottom flask (1L) equipped with magnetic stirring. The flask was then placed in a water-bath (temp: 10-20 °C), and HCI-gas was passed through the solution for 5 min. (dust-like precipitation). After stirring for 1 hour (precipitation of large amount of yellow powder), the solution was flushed with N2 to remove excess of HCI. The precipitate was removed by gentle filtration and dried under vacuum at 40 °C overnight.The non-crystallinic precipitate was dissolved in H2O (500 ml) and washed with EtOAc (100 ml). Then CH2CI2 (1000 ml) and solid Na2CO3 was added until pH > 7. The 2 layers were separated, and the aqueous layer was e-extracted with CH2CI2 (200 ml). The combined organic layers were washed with brine (100 ml), dried over MgSO4 and filtered. The solvent was evaporated under reduced pressure and redissolved in EtOAc (500 ml) in a one neck round-bottom flask (1 L) equipped with magnetic stirring. A suspension of fumaric acid (3,67 g) in isopropanol (20 ml) and EtOAc (50 ml) was slowly added (5 min.), which resulted in precipitation of a white crystallinic salt. After 1 hour the precipitation was isolated by filtration and dried overnight in vacuum at 40 °C to give the fumarate salt of the compound which is either 2-amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1- yl]-1-(1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide or 2-amino-N-[(1 R)-2-[(3S)-3- benzyl-3-(N,N,,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1 H-indol-3-ylmethyl)-2- oxoethyl]-2-methylpropionamide (13,9 g) as a white powder.HPLC (A1): Rt = 33,61 min.HPLC (B1): Rt = 34,62 min. LC-MS: Rt = 5,09 min. (m+1) = 547,4
ClaimsHide Dependent
1. The compound obtainable by the procedure as described in example 1 , or a pharmaceutically acceptable salt thereof.2. The compound obtainable by the procedure as described in example 1 , and which compound is2-Amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide

or a pharmaceutically acceptable salt thereof.3. A pharmaceutical composition comprising, as an active ingredient, a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof together with a pharmaceutically acceptable carrier or diluent.4. A pharmaceutical composition according to claim 3 for stimulating the release of growth hormone from the pituitary.5. A pharmaceutical composition according to claim 3 or claim 4 for administration to animals to increase their rate and extent of growth, to increase their milk and wool production, or for the treatment of ailments.6. A method of stimulating the release of growth hormone from the pituitary of a mammal, the method comprising administering to said mammal an effective amount of a compound according to any one of claims 1 or 2 or a pharmaceutically acceptable salt thereof, or of a composition according to any one of claims 3 – 5.7. A method of increasing the rate and extent of growth, the milk and wool production, or for the treatment of ailments, the method comprising administering to a subject in need thereof an effective amount of a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof, or of a composition according to any one of claims 3-5.8. Use of a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof for the preparation of a medicament.9. Use according to claim 8 wherein the medicament is for stimulating the release of growth hormone from the pituitary of a mammal.
PATENT
CN 108239141
PATENT
US 20130281701
| Growth hormone is a major participant in the control of several complex physiologic processes, including growth and metabolism. Growth hormone is known to have a number of effects on metabolic processes, e.g., stimulation of protein synthesis and free fatty acid mobilization and to cause a switch in energy metabolism from carbohydrate to fatty acid metabolism. Deficiency in growth hormone can result in a number of severe medical disorders, e.g., dwarfism. |
| The release of growth hormone from the pituitary is controlled, directly or indirectly, by number of hormones and neurotransmitters. Growth hormone release can be stimulated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin. In both cases the hormones are released from the hypothalamus but their action is mediated primarily via specific receptors located in the pituitary. Other compounds which stimulate the release of growth hormone from the pituitary have also been described. For example, arginine, L-3,4-dihydroxyphenylalanine (1-Dopa), glucagon, vasopressin, PACAP (pituitary adenylyl cyclase activating peptide), muscarinic receptor agonists and a synthetic hexapeptide, GHRP (growth hormone releasing peptide) release endogenous growth hormone either by a direct effect on the pituitary or by affecting the release of GHRH and/or somatostatin from the hypothalamus. |
| The use of certain compounds for increasing the levels of growth hormone in mammals has previously been proposed. For example, U.S. Pat. Nos. 6,303,620 and 6,576,648 (the entire contents of which are incorporated herein by reference), disclose a compound: (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide, having the following chemical structure: |
(MOL) (CDX) which acts directly on the pituitary cells under normal experimental conditions in vitro to release growth hormone therefrom. This compound is also known under the generic name “anamorelin.” This growth hormone releasing compound can be utilized in vitro as a unique research tool for understanding, inter alia, how growth hormone secretion is regulated at the pituitary level. Moreover, this growth hormone releasing compound can also be administered in vivo to a mammal to increase endogenous growth hormone release.
Example 1
Crystallization of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide form A
| 0.0103 g of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide was dissolved in methanol (0.1 mL) in a glass vial. The glass vial was then covered with PARAFILM® (thermoplastic film) which was perforated with a single hole. The solvent was then allowed to evaporate under ambient conditions. An X-ray diffraction pattern showed the compound was crystalline ( FIG. 1). |
PATENT
WO 2017067438
https://patents.google.com/patent/WO2017067438A1/enAnamorelin, whose chemical name is: (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2- Trimethylformylhydrazide is a compound that increases mammalian growth hormone levels and has a compound structure as shown in Formula I:

Cancer cachexia is a state of consumption in which patients lose a lot of weight and muscle mass. It is necessary for the treatment of cachexia because it weakens the patient, affects the quality of life and interferes with the patient’s treatment plan. The drug alamorelin produces the same effect as the so-called “starved hormone” ghrelin, which stimulates hunger. Alamolin is a mimetic of ghrelin, which is secreted by the stomach and is a ligand for growth hormone receptors. . Alamolin binds to this receptor, causing the release of growth hormone, causing a metabolic cascade that affects a variety of different factors, including fat-removing body weight, as well as blood sugar metabolism. Therefore, alamorelin can also enhance the appetite of patients and help patients stay healthy. The 2014 European Society of Medical Oncology (ESMO) in Madrid, Spain, announced that Alamolin is expected to be the first drug in history to effectively improve cancer cachexia.Alamolin is a drug developed by Helsinn Therapeutics (Switzerland) from Novo Nordisk for the development of a cachexia and anorexia for patients with cancer, including non-small cell lung cancer. It can also be used to treat hip fractures and preventive diseases. The strength of the elderly and the elderly has continued to decline. In two key, 12-week Phase III clinical trials (ROMANA 1, ROMANA 2), alamorelin can significantly increase the body fat loss, and is generally tolerated; the incidence of serious adverse drug reactions is less than 3%, mainly related to hyperglycemia and diabetes. Compared with the placebo group, alamorelin continued to increase body weight and improve cancer anorexia-cachexia-related symptoms and concerns; however, there was no significant difference in the improvement of grip strength between the alamolin group and the placebo group. Therefore, this product has excellent clinical value and market value.The polymorphic form of the drug free base and its preparation are reported as follows:Synthesis of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylmethyl is disclosed in the patent ZL99806010.0 A method for synthesizing hydrazide, and using [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylmethylcarbonyl)piperidin-1-yl tert-Butyl ester of 1-((1H-indol-3-yl)methyl)-2-oxoethyl]carbamate is dissolved in dichloromethane, then trifluoroacetic acid is added to remove tert-butyl formate After the base, the mixture was concentrated to remove the solvent, and then the product was extracted with dichloromethane, and the obtained extract was concentrated to dryness to give (3R)-1-(2-methylalanyl-D-color ammonia as an amorphous powder. Acyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide.Patent ZL00815145.8 discloses the synthesis of alamorelin and its compounds as pharmaceutically acceptable salts, relating to novel diastereomeric compounds, pharmaceutically acceptable salts thereof, compositions containing them and their use in therapy Lack of use of medical conditions caused by growth hormone. Synthesis of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformyl is disclosed in this patent. The synthesis method of hydrazine, and using [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylmethylcarbonylcarbonyl)piperidin-1-yl] 1-((1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was dissolved in ethyl acetate, and then hydrogen chloride gas was passed to remove the tert-butyl formate protection group. , the solid is dissolved in water, and then the pH is adjusted to about 7 with sodium carbonate, and the product is extracted with dichloromethane; the extract phase is concentrated to obtain (3R)-1-(2-methylalanyl-D-tryptophan). -3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide.Patent WO2006016995 discloses (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylmethyl as a medicament Crystalline polymorphs of hydrazides, methods of producing and separating these polymorphs, and pharmaceutical compositions and drug therapies containing these polymorphs, the crystalline polymorphs for direct application to the pituitary Gland cells release the growth hormone. This patent discloses (4R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone 4 Crystal form: Form A, Form B, Form C and Form D. The patent also provides the preparation of 3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone. The method of crystal form, especially the preparation method of Form C, in which the method of removing the tert-butyl formate protecting group of methanesulfonic acid in methanol is utilized without exception. As a well-known cause in the art, clinical studies have found that mesylate is genotoxic, and its DNA alkylation leads to mutagenic effects, in which methyl methanesulfonate and ethyl methanesulfonate have been reported. (eg document EMEA/44714/2008). The invention adopts hydrochloric acid or hydrogen chloride gas to remove the tert-butyl formate protecting group, avoids the method of removing methanesulfonic acid, thereby avoiding the risk of the genotoxic impurities in the process, and increasing the risk. The safety of the drug.(3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-three prepared by the patent ZL99806010.0 and the patent ZL00815145.8 Methyl formyl hydrazide, no data on the purity of its compounds, we found that (3R)-1-(2-methylalanyl-D-tryptophan)-3 was prepared by this method. -Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide does not help to remove the impurities produced, and the purity of the obtained product is not high, and it is difficult to meet the medicinal requirements. And (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethyl obtained by the preparation method of the present invention. The crystal form of the formyl hydrazide has a purity of 99.8% and a single impurity of less than 0.1%, which fully meets the requirements for medicinal purity. Moreover, the crystal form is stable to conditions such as pressure, temperature, humidity and illumination, and the preparation method is simple in operation and suitable for industrial production.Example 1:300 g of [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylcarbamidocarbonyl)piperidin-1-yl]-1-(( 1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was added to the reaction flask, and then 4 L of dichloromethane was added to the reaction flask, and the raw material was completely dissolved by stirring.Then, the reaction system is cooled to 10 ° C or lower in an ice bath, hydrogen chloride gas is continuously supplied to the reaction liquid, and solids are gradually precipitated, and the reaction is further maintained at about 10 ° C for 3 to 5 hours, and the sample is detected. After the reaction of the raw materials is completed, the reaction system is completed. 1.5 L of water was added thereto, the solid was completely dissolved, and then the pH was adjusted to about 8 with a 20% aqueous sodium hydroxide solution, and the layers were separated; the aqueous phase was extracted once more with dichloromethane, and the organic phases were combined.The organic phase was dried over anhydrous sodium sulfate for 3 hrs, filtered, and then evaporated to ethylamine 3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude 246 g, yield 97.2%. HPLC content (area normalization method) was 96.1%.Example 2:300 g of [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylcarbamidocarbonyl)piperidin-1-yl]-1-(( 1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was added to the reaction flask, 36% concentrated hydrochloric acid was added to the reaction flask, and the reaction system was heated to 40 with stirring. The reaction was carried out at ° C to 50 for 3 hours.Then, the sample is detected. After the reaction of the raw material is completed, the reaction system is cooled to 10 or less, and 2.0 L of dichloromethane is added to the reaction system, and then the pH is adjusted to about 8 with a 20% aqueous sodium hydroxide solution, and the aqueous phase is further separated. It was extracted once with dichloromethane and the organic phases were combined.The organic phase was dried over anhydrous sodium sulfate for 3 hrs, filtered, and then evaporated to ethylamine 3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude 248 g, yield 98%. HPLC content (area normalization method) was 96.2%.Preparation of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone E crystal formExample 3Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10g was added to the reaction flask and 30 ml of N- was added.Methylpyrrolidone, stirred and dissolved completely. Then, 60 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 60 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 20 ° C, filtered, and the filter cake was washed with a mixture of N-methylpyrrolidone / H 2 O; the cake was vacuum dried at about 55 ° C to obtain (3R)-1-(2-methylalanyl) -D-tryptophan)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.5 g), HPLC content (area normalization) 99.72%. The XRD pattern is shown in Fig. 1, the DSC chart is shown in Fig. 2, and the TGA pattern is shown in Fig. 3, where the crystal form is defined as the E crystal form. The DSC of the crystal form has an endotherm at 120.05, the TGA is heated at 60A, and the crystal loss of 5 is about 3.1%. Combined with the Karl Fischer method, the moisture content of the product is determined. 3.1% and 3.2% indicate that the sample is present as a monohydrate.Example 4:Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 30 ml of N,N-dimethylformamide was added, stirred, and dissolved completely. Then, 30 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 50 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 h.Slowly cool to below 10 ° C, filter, filter cake washed with N, N-dimethylformamide / H 2 O mixture; vacuum cake dried at around 55 ° C to obtain (3R)-1-(2-A Alanyl-D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.5 g), HPLC content (area normalization) ) 99.87%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 5:Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 30 ml of dimethyl sulfoxide was added, stirred, and dissolved completely. Then, 40 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 60 ° C, the solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 10 ° C, filtered, and the filter cake was washed with a mixture of dimethyl sulfoxide / H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2-methylalanyl) -D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.1 g), HPLC content (area normalization) 99.61%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 6Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 40 ml of 1,4-dioxane was added, stirred, and dissolved completely. Then, 50 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 70 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 10 ° C, filtered, and the filter cake was washed with a mixture of 1,4-dioxane/H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2-methyl alanyl-D-tryptophan-3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.7 g), HPLC content (area normalization) 99.11%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 7Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 40 ml of N,N-dimethylacetamide was added, stirred, and dissolved completely. Then, 40 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 70 ° C. The solution became cloudy, and was slowly cooled to about 50 ° C. Seed crystals were added thereto, and cooling was continued to gradually precipitate a solid.The reaction system was cooled to about 10 ° C, filtered, and the filter cake was washed with a mixture of N,N-dimethylacetamide/H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2- Methylalanyl-D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.1 g), HPLC content (area normalized) Law) 99.78%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 7Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 50 ml of acetone was added, stirred, and dissolved completely. Then, 70 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 45 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cool to below 10 ° C, filter, filter cake washed with acetone / H 2 O mixture; filter cake vacuum dried at around 50 ° C to obtain (3R)-1-(2-methylalanyl-D-color Aminoacyl-3-phenylmethyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.3 g), HPLC content (area normalization) 98.9%. Upon comparison, it was confirmed that the solid was in the E crystal form.
SYN
Reference:
1. Org. Process Res. Dev. 2006, 10, 339–345.

Abstract

The rapid process development of a scaleable synthesis of the pseudotripeptide RC-1291 for preclinical and clinical evaluation is described. By employing a nontraditional N-to-C coupling strategy, the peptide chain of RC-1291 was assembled in high yield, with minimal racemization and in an economical manner by introducing the most expensive component last. A one-pot deprotection/crystallization procedure was developed for the isolation of RC-1291 free base, which afforded the target compound in excellent yield and with a purity of >99.5% without chromatographic purification.
(R,R)-2-Amino-N-[2-[3-benzyl-3-(N,N′,N′-trimethyl-hydrazinocarbonyl)piperidin-1-yl]-1-(1H-indol-3-ylmethyl)- 2-oxo-ethyl]-2-methyl-propionamide (1). Crude 7 (911 g; 1.28 mol theoretical)10 was dissolved in methanol (4.12 L) in a 22-L round-bottom flask equipped with a mechanical stirrer, a temperature probe, a reflux condenser, a gas (N2) inlet, and an addition funnel. The solution was heated to 55 °C; then methanesulfonic acid (269.5 g, 2.805 mol) was added over a period of 15 min. (Caution: gas evolution!) The solution was then heated to 60 °C for a period of 1 h, after which HPLC analysis showed that no 7 remained. The temperature of the reaction mixture was increased to reflux (68-72 °C) over a period of 35 min, while simultaneously adding a solution of KOH (85%, 210.4 g, 3.187 mol) in water (4.12 L). The clear, slightly yellow solution was then allowed to cool to 20 °C at a rate of 5 °C/h. The free base of RC1291 (1) crystallized as a pale-yellow solid, which was isolated by filtration. The filter cake was washed with two portions of 50% aqueous methanol (500 mL each) and then dried under high vacuum at 20 ( 5 °C to afford 1 as an off-white, crystalline solid (595 g, 85% yield for two steps, >99.5% AUC by HPLC).
HRMS (ESI) calcd for C31H43N6O3 [M + H]+ 547.3397, found 547.3432.
1H NMR (DMSO-d6; 413 K) δ 10.30 (s, 1H), 7.85 (bs, 1H), 7.50 (d, J ) 7.8 Hz, 1H), 7.27 (d, J ) 8.1 Hz, 1H), 7.1-7.2 (m, 3H), 6.95-7.0 (m, 5H), 5.07 (t, J ) 6.3 Hz, 1H), 3.54 (d, J ) 12.3 Hz, 1H), 3.36 (bs, 1H), 3.15-3.30 (m, 1H), 3.06 (dd, J ) 7.2, 14.4 Hz, 1H), 2.96 (dd, J ) 6.0, 14.3 Hz, 2H), 2.7-2.8 (m, 6H), 2.43 (m, 6H), 2.09 (bs, 1H), 1.73 (bs, 1H), 1.45-1.55 (m, 2H), 1.3-1.40 (m, 1H), 1.18 (s, 3H), 1.15 (s, 3H).
13C NMR (DMSO-d6; 413 K) δ 175.8, 173.4, 170.3, 137.0, 135.7, 129.0, 127.2, 127.1, 125.3, 122.9, 120.1, 117.6, 110.7, 109.4, 53.6, 49.0, 47.0, 42.7, 38.5, 30.7, 28.2, 28.0, 23.2, 21.1.

PAPER
https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.8b00322
Cachexia and muscle wasting are very common among patients suffering from cancer, chronic obstructive pulmonary disease, and other chronic diseases. Ghrelin stimulates growth hormone secretion via the ghrelin receptor, which subsequently leads to increase of IGF-1 plasma levels. The activation of the GH/IGF-1 axis leads to an increase of muscle mass and functional capacity. Ghrelin further acts on inflammation, appetite, and adipogenesis and for this reason was considered an important target to address catabolic conditions. We report the synthesis and properties of an indane based series of ghrelin receptor full agonists; they have been shown to generate a sustained increase of IGF-1 levels in dog and have been thoroughly investigated with respect to their functional activity.

Patent
https://patents.google.com/patent/EP2838892A1/enGrowth hormone is a major participant in the control of several complex physiologic processes including growth and metabolism. Growth hormone is known to have a number of effects on metabolic processes such as stimulating protein synthesis and mobilizing free fatty acids, and causing a switch in energy metabolism from carbohydrate to fatty acid metabolism. Deficiencies in growth hormone can result in dwarfism and other severe medical disorders.The release of growth hormone from the pituitary gland is controlled directly and indirectly by a number of hormones and neurotransmitters. Growth hormone release can be stimulated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin.The use of certain compounds to increase levels of growth hormone in mammals has previously been proposed. Anamorelin is one such compound. Anamorelin is a synthetic orally active compound originally synthesized in the 1990s as a growth hormone secretogogue for the treatment of cancer related cachexia. The free base of anamorelin is chemically defined as:® (3R) 1 -(2-methylaIanyl~D ryptophyl)~3-(phenylraethyl)~3~piperidineearboxylie acid 1 ,2,2trimethyihydrazide,* 3-{(2R)-3-{(3R)-3-benzyi-3-| (trimethylhydrazino)carbonyi]piperidin-l»yl}-2-[(2»met hylaianyl)amino]-3-ox.opropyi}-IH-indole, or• 2-Amino-N-[(lR)-2-[(3R)-3~benzyWcarbony piperidin- 1 -yl] – 1-( 1 H-indol-3 -yl^^and has the below chemical structure;
U.S. Patent No. 6,576,648 to Artkerson reports a process of preparing anamorelin as the fumarate salt, with the hydrochloride salt produced as an intermediate in Step (j) of Example 1 . U.S. Patent No. 7,825, 138 to Lorimer describes a process for preparing crystal forms of the free base of anamorelin.There is a need to develop anamorelin monohydrochloride as an active pharmaceutical ingredient with reduced impurities and improved stability over prior art forms of anamorelin hydrochloride, such as those described in U.S. Patent No, 6,576,648, having good solubility, bioavailability and processabi!ity. There is also a need to develop methods of producing pharmaceutically acceptable forms of anamorelin monohydrochloride thai have improved yield over prior art processes, reduced residual solvents, and controlled distribution of chloride content,it has unexpectedly been discovered that the process of making the hydrochloride salt of anamorelin described in Step (j) of U.S. Patent No. 6.576,648 can result in excessive levels of chloride in the final product, and that this excess chloride leads to the long-term instability of the final product due at least, partially to an increase in the amount of the less stable dihydrochloride salt of anamorelin. Conversely, because anamorelin free base is less soluble in water than the hydrochloride salt, deficient chloride content in the final product can lead to decreased solubility of the molecule. The process described in U.S. Patent No, 6,576,648 also yields a final product that contains more than 5000 ppm (0.5%) of residual solvents, which renders the product less desirable from a pharmaceutical standpoint, as described in CH Harmonized Tripartite Guideline. See Impurities; Guideline for residual solvents Q3C(R3). in order to overcome these problems, methods have been developed which, for the first time, allow for the efficient and precise control of the reaction between anarnorehn tree base and hydrochloric acid in situ, thereby increasing the yield of anarnorehn monohydrochioride from the reaction and reducing the incidence of unwanted anamorelin dihydroeh ride. According to the method, the free base of anamorelin is dissolved in an organic solvent and combined with water and hydrochloric acid, with the molar ratio of anarnorehn and chloride tightly controlled to prevent an excess of chloride in the final product. The water and hydrochloric acid can be added either sequentially or at the same time as long as two separate phases are formed. Without wishing to be bound by any theory, it is believed thai as the anamorelin free base in the organic phase is protonated by the hydrochloric acid it migrates into the aqueous phase. The controlled ratio of anamorelin free base and hydrochloric acid and homogenous distribution in the aqueous phase allows for the controlled formation of the monohydrochioride salt over the dihydrochloride, and the controlled distribution of the resulting chloride levels within individual batches and among multiple batches of anamorelin monohydrochioride.Thus, in a fust embodiment the invention provides methods for preparing anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride comprising: (a) dissolving anamorelin free base in an organic solvent to form a solution; (b) mixing said solution with water and hydrochloric acid for a time sufficient to: (i) react said anamorelin free base with said hydrochloric acid, and (ii) form an organic phase and an aqueous phase; (c) separating the aqueous phase from the organic phase; and (d) isolating anamorelin monohydrochioride from the aqueous phase.In a particularly preferred embodiment, the molar ratio of anamorelin to hydrochloric acid used in the process is less than or equal to 1 : 1 , so as to reduce the production of anamorelin dihydrochloride and other unwanted chemical species. Thus, for example, hydrochloric acid can be added at a molar ratio of from 0,90 to 1 ,0 relative to said anamorelin, from 0.90 to 0.99, or from 0.93 to 0.97.n another particularly preferred embodiment, the anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride is isolated from the aqueous phase via spray drying, preferably preceded by distillation. This technique has proven especially useful in the manufacture of anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride because of the excellent reduction in solvent levels observed, and the production of a stable amorphous form of anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride. In other embodiments, the invention relates to the various forms of anamorelin monohvdrochloride and compositions comprising anamorelin monohvdrochloride produced by the methods of the present invention. In a first embodiment, which derives from the controlled chloride content among batches accomplished by the present methods, the invention provides anamorelin monohvdrochloride or a composition comprising anamorelin monohydrochloride having an inter-batch chloride content of from 5.8 to 6.2%, preferably from 5.8 to less than 6.2%. Alternatively, the invention provides anamorelin monohydrochloride or a composition comprising anamorelin monohydrochloride having a molar ratio of chloride to anamorelin less than or equal to 1 : 1 , such as from 0.9 to 1.0 or 0.99, in yet another embodiment the invention provides an amorphous form of anamorelin monohydrochloride or a composition comprising anamorelin monohydrochloride. Further descriptions of the anamorelin monohydrochloride and compositions comprising the anamorelin monohydrochloride are given in the detailed description which follows.EXAMPLE 1 . PREPARATION OF ANAMOREUN HYDROCHLORIDEVarious methods have been developed to prepare the hydrochloric acid salt of anarnorelin, with differing results.In a first method, which is the preferred method of the present invention, anarnorelin free base was carefully measured and dissolved in isopropyl acetate. Anarnorelin free base was prepared according to known method (e.g., U.S. Patent No, 6,576,648). A fixed volume of HCl in water containing various molar ratios (0.80, 0,95, 1.00 or 1.05) of HCl relative to the anarnorelin free base was then combined with the anamorelin/isopropyl acetate solution, to form a mixture having an organic and an aqueous phase, The aqueous phase of the mixture was separated from the organic phase and the resulting aqueous phase was concentrated by spray drying to obtain the batches of anarnorelin monohydrochloride (or a composition comprising anarnorelin monohydrochloride ) shown in Table 1 A.Approximately 150mg of the resulting spray dried sample of anarnorelin monohydrochloride (or composition comprising anarnorelin monohydrochloride) was accurately weighed out and dissolved in methanol (50mL). Acetic acid (5mL) and distilled water (5mL) were added to the mixture. The resulting mixture was potentiometricaJ ly titrated using 0,0 IN silver nitrate and the e dpoint was determined. A blank determination was also performed and correction was made, if necessary. The chloride content in the sample was calculated by the following formula. This measurement method of chloride content was performed without any cations other than proton (! ! ‘ ).Chloride content (%) = VxNx35.453x l 00x l 00/{Wx[1 00-(water content (%))-(residual solvent (%))]}V: volume at the endpoint (ml.)N; actual normality of 0.01 mol/L silver nitrate35.453 : atomic weight of ChlorineW: weight of sample (mg)TABLE 1 AHCl Chloride ContentThis data showed that anamorelin monohydrochlonde produced by a fixed volume of HCl in water containing 0.80 or 1 .05 molar equivalents of HC1 relative to anamorelin free base had levels of chloride thai were undesirable, and associated with product instability as shown in Example 3.Alternatively, a fixed volume of HCl in water containing 0.95 moles of HCl relative to anamorelin free base was used to prepare anamorelin monohydrochlonde (or composition comprising anamorelin monohydrochloride) as follows. Anamorelin free base (18.8g, 34.4mmoi) and isopropyl acetate (341.8g) were mixed in a 1000 mL flask. The mixture was heated at 40±5°C to confirm dissolution of the crystals and then cooled at 25±5°C. Distilled water (22.3g) and 3.6% diluted hydrochloric acid (33. Ig, 32.7mmoL 0.95 equivalents) were added into the flask and washed with distilled water. After 30 minutes stirring, the reaction was static for more than 15 minutes and the lower layer (aqueous layer) was transferred into a separate 250mL flask. Distilled water was added to the flask and concentrated under pressure at 50i5cC. The resulting aqueous solution was then filtered and product isolated by spray drying to afford anamorelin monohydrochlonde A (the present invention).The physical properties of anamorelin monohydrochloride A were compared to anamorelin monohydrochloride produced by a traditional comparative method (“anamorelin monohydrochloride B”) (comparative example). Anamorelin mono hydrochloride B in the comparative example was produced by bubbling HCl gas into isopropyl acetate to produce a 2M solution of HCl, and reacting 0.95 molar equivalents of the 2M HCl in isopropyl acetate with anamorelin free base. The physical properties of anamorelin monohydrochloride B are reported in Table IB. This data shows that when 0.95 equivalents of HCl is added to anamorelin free base, the chloride content (or amount of anamorelin dihydrochloride) is increased, even when a stoichiometric ratio of hydrochloride to anamorelin of less than 1 ,0 is used, possibly due to uncontrolled precipitation. In addition, this data shows that the concentration of residual solvents in anamorelin monohydrochloride B was greater than the concentration in anamorelin monohydrochloride A, TABLE I B
A similar decrease in residual solvent concentration was observed when 2-methyltetrahydrofuran was used as the dissolving solvent for anamorelin free base instead of isopropvi acetate in the process for preparing spray dried anamorelin monohydrochloride A (data not reported).The residual solvent (organic volatile impurities) concentration (specifically isopropyl acetate) of anamorelin monohydrochloride in TABLE IB was measured using gas chromatography (GC-2010, Shimadzu Corporation) according to the conditions shown in TABLE 1 C,
References
- ^ Leese PT, Trang JM, Blum RA, de Groot E (March 2015). “An open-label clinical trial of the effects of age and gender on the pharmacodynamics, pharmacokinetics and safety of the ghrelin receptor agonist anamorelin”. Clinical Pharmacology in Drug Development. 4 (2): 112–120. doi:10.1002/cpdd.175. PMC 4657463. PMID 26640742.
- ^ Currow DC, Abernethy AP (April 2014). “Anamorelin hydrochloride in the treatment of cancer anorexia-cachexia syndrome”. Future Oncology. 10 (5): 789–802. doi:10.2217/fon.14.14. PMID 24472001.
- ^ Jump up to:a b c Garcia JM, Polvino WJ (June 2009). “Pharmacodynamic hormonal effects of anamorelin, a novel oral ghrelin mimetic and growth hormone secretagogue in healthy volunteers”. Growth Hormone & IGF Research. 19 (3): 267–73. doi:10.1016/j.ghir.2008.12.003. PMID 19196529.
- ^ Jump up to:a b Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, Friend J (January 2015). “Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials”. The Lancet. Oncology. 16 (1): 108–16. doi:10.1016/S1470-2045(14)71154-4. PMID 25524795.
- ^ Jump up to:a b Garcia JM, Friend J, Allen S (January 2013). “Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study”. Supportive Care in Cancer. 21 (1): 129–37. doi:10.1007/s00520-012-1500-1. PMID 22699302. S2CID 22853697.
- ^ Zhang H, Garcia JM (June 2015). “Anamorelin hydrochloride for the treatment of cancer-anorexia-cachexia in NSCLC”. Expert Opinion on Pharmacotherapy. 16 (8): 1245–53. doi:10.1517/14656566.2015.1041500. PMC 4677053. PMID 25945893.
- ^ Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, Fearon KC (April 2016). “Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials”. The Lancet. Oncology. 17 (4): 519–531. doi:10.1016/S1470-2045(15)00558-6. PMID 26906526.
- ^ “Adlumiz”. European Medicines Agency.
- ^ “Refusal of the marketing authorisation for Adlumiz (anamorelin hydrochloride): Outcome of re-examination” (PDF). European Medicines Agency. 15 September 2017.
External links
| Clinical data | |
|---|---|
| Routes of administration | Oral |
| ATC code | None |
| Pharmacokinetic data | |
| Elimination half-life | 6–7 hours[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 249921-19-5 |
| PubChem CID | 9828911 |
| ChemSpider | 8004650 |
| UNII | DD5RBA1NKF |
| CompTox Dashboard (EPA) | DTXSID20179702 |
| Chemical and physical data | |
| Formula | C31H42N6O3 |
| Molar mass | 546.716 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCC(C)(C(=O)NC(CC1=CNC2=CC=CC=C21)C(=O)N3CCCC(C3)(CC4=CC=CC=C4)C(=O)N(C)N(C)C)N | |
| hideInChIInChI=1S/C31H42N6O3/c1-30(2,32)28(39)34-26(18-23-20-33-25-15-10-9-14-24(23)25)27(38)37-17-11-16-31(21-37,29(40)36(5)35(3)4)19-22-12-7-6-8-13-22/h6-10,12-15,20,26,33H,11,16-19,21,32H2,1-5H3,(H,34,39)/t26-,31-/m1/s1Key:VQPFSIRUEPQQPP-MXBOTTGLSA-N |
///////Anamorelin hydrochloride, Anamorelin, APPROVALS 2021, JAPAN 2021, PMDA, Adlumiz, 22/1/2021, アナモレリン塩酸塩, анаморелин , أناموريلين ,阿那瑞林 , ONO 7643, RC 1291, ST 1291,
#Anamorelin hydrochloride, #Anamorelin, #APPROVALS 2021, #JAPAN 2021, #PMDA, #Adlumiz, 22/1/2021, #アナモレリン塩酸塩, #анаморелин , #أناموريلين ,阿那瑞林 , #ONO 7643, #RC 1291, #ST 1291,


{kind=link}
#Anamorelin hydrochloride, #Anamorelin, #APPROVALS 2021, #JAPAN 2021, #PMDA, #Adlumiz, 22/1/2021, #アナモレリン塩酸塩, #анаморелин , #أناموريلين ,阿那瑞林 , #ONO 7643, #RC 1291, #ST 1291,
LikeLike