
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
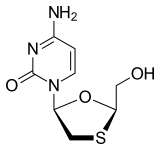
Lamivudine (2′,3′-dideoxy-3′-thiacytidine, commonly called 3TC) is an antiretroviral medication used to prevent and treat HIV/AIDS and used to treat chronic hepatitis B.[1]
It is of the nucleoside analog reverse transcriptase inhibitor (NRTI) class. It is marketed in the United States under the tradenames Epivir and Epivir-HBV.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[2] As of 2015 the cost for a typical month of medication in the United States is more than 200 USD.[3]
Medical uses
Lamivudine has been used for treatment of chronic hepatitis B at a lower dose than for treatment of HIV/AIDS. It improves the seroconversion of e-antigen positive hepatitis B and also improves histology staging of the liver. Long term use of lamivudine leads to emergence of a resistant hepatitis B virus (YMDD) mutant. Despite this, lamivudine is still used widely as it is well tolerated.
Resistance
In HIV, high level resistance is associated with the M184V/I mutation in the reverse transcriptase gene as reported by Raymond Schinazi’s group at Emory University. GlaxoSmithKline claimed that the M184V mutation reduces “viral fitness”, because of the finding that continued lamivudine treatment causes the HIV viral load to rebound but at a much lower level, and that withdrawal of lamivudine results in a higher viral load rebound with rapid loss of the M184V mutation; GSK therefore argued that there may be benefit in continuing lamivudine treatment even in the presence of high level resistance, because the resistant virus is “less fit”. The COLATE study has suggested that there is no benefit to continuing lamivudine treatment in patients with lamivudine resistance.[4] A better explanation of the data is that lamivudine continues to have a partial anti-viral effect even in the presence of the M184V mutation.
In hepatitis B, lamivudine resistance was first described in the YMDD (tyrosine–methionine–aspartate-aspartate) locus of the HBV reverse transcriptase gene. The HBV reverse transcriptase gene is 344 amino acids long and occupies codons 349 to 692 on the viral genome. The most commonly encountered resistance mutations are M204V/I/S.[5] The change in amino acid sequence from YMDD to YIDD results in a 3.2 fold reduction in the error rate of the reverse transcriptase, which correlates with a significant growth disadvantage of the virus. Other resistance mutations are L80V/I, V173L and L180M.[6]
Mechanism of action
Lamivudine is an analogue of cytidine. It can inhibit both types (1 and 2) of HIV reverse transcriptase and also the reverse transcriptase of hepatitis B virus. It is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The lack of a 3′-OH group in the incorporated nucleoside analogue prevents the formation of the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation, and therefore, the viral DNA growth is terminated.
Lamivudine is administered orally, and it is rapidly absorbed with a bio-availability of over 80%. Some research suggests that lamivudine can cross the blood–brain barrier. Lamivudine is often given in combination with zidovudine, with which it is highly synergistic. Lamivudine treatment has been shown to restore zidovudine sensitivity of previously resistant HIV. Lamivudine showed no evidence of carcinogenicity or mutagenicity in in vivo studies in mice and rats at doses from 10 to 58 times those used in humans.[7]
History
Racemic BCH-189 (the minus form is known as Lamivudine) was invented by Dr. Bernard Belleau while at work at McGill University and Dr Paul Nguyen-Ba at the Montreal-based IAF BioChem International, Inc. laboratories in 1988 and the minus enantiomer isolated in 1989. Samples were first sent to Dr. Yung-Chi Cheng of Yale University for study of its toxicity. When used in combination with AZT, he discovered that Lamivudine’s negative form reduced side effects and increased the drug’s efficiency at inhibiting reverse transcriptase.[8] The combination of Lamivudine and AZT increased the efficiency at inhibiting an enzyme HIV uses to reproduce its genetic material. As a result, Lamivudine was identified as a less toxic agent to mitochondria DNA than other retroviral drugs.[9]
Lamivudine was approved by the Food and Drug Administration (FDA) on November 17, 1995 for use with zidovudine (AZT) and again in 2002 as a once-a-day dosed medication. The fifth antiretroviral drug on the market, it was the last NRTI for three years while the approval process switched to protease inhibitors. According to the manufacturer’s 2004 annual report, its patent will expire in the United States in 2010 and in Europe in 2011.
On September 2014, Dr. Gorbee Logan, a Liberian physician, reported positive results while treating Ebola virus disease with Lamivudine. Out of 15 patients treated with the antiviral, 13 (those treated within the third to fifth day of symptoms being manifested) survived the disease and were declared virus-free; the remaining cases (treated from the fifth day or later) died.[10][11]
Presentation
- Epivir 150 mg or 300 mg tablets (GlaxoSmithKline; US and UK) for the treatment of HIV;
- Epivir-HBV 100 mg tablets (GlaxoSmithKline; US only) for the treatment of hepatitis B;
- Zeffix 100 mg tablets (GlaxoSmithKline; UK only) for the treatment of hepatitis B.
- 3TC 150 mg tablets (GlaxoSmithKline; South Africa) for the treatment of HIV;
Lamivudine is also available in fixed combinations with other HIV drugs:
- Combivir (with zidovudine);
- Epzicom/Kivexa (with abacavir);
- Trizivir (with zidovudine and abacavir)
Lamivudine (I) (CAS No. 134678-17-4) is chemically known as (-)-[2R,5S]-4T amino- 1 – [2-(hydroxymethyl)- 1 ,3 -oxathiolan-5-yl] -2( 1 H)-pyrimidin-2-one.

Formula (I)
Lamivudine is a reverse transcriptase inhibitor used alone or in combination with other classes of Anti-HIV drugs in the treatment of HIV infection. It is available commercially as a pharmaceutical composition under the brand name EPIVIR®, marketed by GlaxoSmithKline, and is covered under US 5,047,407.
This molecule has two stereo-centres, thus giving rise to four stereoisomers: (±)- Cis Lamivudine and (±)-Trans Lamivudine. The pharmaceutically active isomer however is the (-)-Cis isomer which has the absolute configuration [2R,5S] as show in Formula (I).
US 5,047,407 discloses the 1,3-oxathiolane derivatives; their geometric (cis/trans) and optical isomers. This patent describes the preparation of Lamivudine as a mixture of cis and trans isomers (shown in scheme I). The diastereomers obtained are converted into N-acetyl derivatives before separation by column chromatography using ethylacetate and methanol (99:1); however, this patent remains silent about further resolution of the cis isomer to the desired (-)- [2R,5S]-Cis-Lamivudine. Secondly, as the ethoxy group is a poor leaving group, the condensation of cytosine with compound VI gives a poor yield, i.e. 30 – 40%, of compound VII. Thirdly, chromatographic separation that has been achieved only after acetylation requires a further step of de-acetylation of the cis-(±)- isomer. Also, separation of large volumes of a compound by column chromatography makes the process undesirable on a commercial scale.

(+/-) Cis (+/-) Cis Lamivudine (VIII)
Scheme – 1 Efforts have been made in the past to overcome the shortcomings of low yield and enantiomeric enrichment, hi general, there have been two approaches to synthesize (— )-[2R,5S]-Cis-Lamivudine. One approach involves stereoselective synthesis, some examples of which are discussed below.
US 5,248,776 describes an asymmetric process for the synthesis of enantiomerically pure β-L-(-)-l,3-oxathiolone-nucleosides starting from optically pure 1,6-thioanhydro-L-gulose, which in turn can be easily prepared from L- Gulose. The condensation of the 1,3-oxathiolane derivative with the heterocyclic base is carried out in the presence of a Lewis acid, most preferably SnCl4, to give the [2R,5R] and [2R,5S] diastereomers that are then separated chromatographically.
US 5,756,706 relates a process where compound A is esterified and reduced to compound B. The hydroxy group is then converted to a leaving group (like acetyl) and the cis- and trans-2R-tetrahydrofuran derivatives are treated with a pyrimidine base, like N-acetylcytosine, in the presence trimethylsilyl triflate to give compound C in the diastereomeric ratio 4: 1 of cis and trans isomers.

A B C
Z = S5 CH
Dissolving compound C in a mixture of 3:7 ethyl acetate-hexane separates the cis isomer. The product containing predominantly the cis-2R,5S isomer and some trans-2R,5R compound is reduced with NaBH4 and subjected to column chromatography (30% MeOH-EtOAc) to yield the below compound.

US 6,175,008 describes the preparation of Lamivudine by reacting mercaptoacetaldehyde dimer with glyoxalate and further with silylated pyrimidine base to give mainly the cis-isomer by using an appropriate Lewis acid, like TMS-
I5 TMS-Tf, TiCl4 et cetera. However the stereoselectivity is not absolute and although the cis isomer is obtained in excess, this process still requires its separation from the trans isomer. The separation of the diastereomers Js done by acetylation and chromatographic separation followed by deacetylation. Further separation of the enantiomers of the cis-isomer is not mentioned.
US 6,939,965 discloses the glycosylation of 5-fluoro-cytosine with compound F (configuration: 2R and 2S)

. F
The glycosylation is carried out in the presence of TiCl3(OiPr) which is stereoselective and the cis-2R,5S-isomer is obtained in excess over the trans- 2S,5S-isomer. These diastereomers are then separated by fractional crystallization.
US 6,600,044 relates a method for converting the undesired trans-l,3-oxathiolane nucleoside to the desired cis isomer by a method of anomerizatioή or transglycosylation and the separation of the hydroxy-protected form of cis-, trans- (-)-nucleosides by fractional crystallization of their hydrochloride, hydrobromide, methanesulfonate salts. However, these cis-trans isomers already bear the [R] configuration at C2 and only differ in their configuration at C5; i.e. the isomers are [2R,5R] and [2R,5S]. Hence diastereomeric separation directly yields the desired [2R, 5S] enantiomer of Lamivudine.
In the second approach to prepare enantiomerically pure Lamivudine the resolution of racemic mixtures of nucleosides is carried out. US 5,728,575 provides one such method by using enzyme-mediated enantioselective hydrolysis of esters of the formula

wherein, ‘R’ is an acyl group and ‘Rl ‘ represents the purine or pyrimidine base.
‘R’ may be alkyl carboxylic, substituted alkyl carboxylic and preferably an acyl group that is significantly electron-withdrawing, eg. α-haloesters. After selective hydrolysis, the process involves further separation of the unhydrolyzed ester from the‘ enantiomerically pure 1,3-oxathiolane-nucleoside. Three methods are suggested in this patent, which are:
1. Separation of the more lipophilic unhydrolyzed ester by solvent extraction with one of a wide variety of nonpolar organic solvents.
2. Lyophilization followed by extraction into MeOH or EtOH. 3. Using an HPLC column designed for chiral separations.
In another of its aspects, this patent also refers to the use of the enzyme cytidine- deoxycytidine deaminase, which is enantiomer-specific, Λo catalyze the deamination of the cytosine moiety and thereby converting it to uridine. Thus, the enantiomer that remains unreacted is still basic and can be extracted by using an acidic solution.
However, the above methods suffer from the following drawbacks, (a) Enzymatic hydrolysis sets down limitations on choice of solvents: alcohol solvents cannot be used as they denature enzymes. (b) Lyophilization on an industrial scale is tedious, (c) Chiral column chromatographic separations are expensive.
WO 2006/096954 describes the separation of protected or unprotected enantiomers of the cis nucleosides of below formula by using a chiral acid to form diastereomeric salts that are isolated by filtration. Some of the acids used are R-
(-)-Camphorsulfonic acid, L-(-)-Tartaric acid, L-(-)-Malic acid, et cetera.
However, the configuration of these CIS-nucleosides are [2R,4R] and [2S,4S] as the heterocyclic base is attached at the 4 position of the oxathiolane ring and the overall stereo-structure of the molecule changes from that of the 2,5-substituted oxathiolane ring.

Thus various methods are described for the preparation of Lamivudine. However there is no mention in the prior art about the separation of an enantiomeric pair, either cis-(±) or trans-(±), from a mixture containing cis-[2R,5S], [2S,5R] and trans-[2R,5R], [2S,5S] isomers. Further, there also is a need to provide resolution of the cis-(±) isomers to yield the desired enantiomer in high optical purity.
CN 1223262 (Deng et aϊ) teaches the resolution of a certain class of compounds called Prazoles by using chiral host compounds such as dinaphthalenephenols (BINOL), diphenanthrenols or tartaric acid derivatives. The method consists of the formation of a 1:1 complex between the chiral host (BINOL) and one of the enantiomers, the guest molecule. The other enantiomer remains in solution. (S)- Omeprazole, which is pharmaceutically active as a highly potent inhibitor of gastric acid secretion, has been isolated from its racemic mixture in this manner by using S-BINOL.
BINOL is a versatile chiral ligand that has found its uses in various reactions involving asymmetric synthesis (Noyori, R. Asymmetric Catalysis in Organic
Synthesis) and optical resolution (Cram, D. J. et al J. Org. Chem. 1977, 42, 4173-
4184). Some of these reactions include BINOL-mediated oxidation and reduction reactions, C-C bond formation reactions such as Aldol reaction, Michael addition,
Mannich reaction et cetera (Brunei Chem. Rev. 2005 105, 857-897) and kinetic resolution, resolution by inclusion complexation et cetera.
BINOL, or l,l’-bi-2-Naphthol, being an atropoisomer possesses the property of chiral recognition towards appropriate compounds. One of the uses of BINOL in resolution that is known in literature is in Host-Guest complexation. In one such example, 1,1-binaphthyl derivatives have been successfully incorporated into optically active crown ethers for the enantioselective complexation of amino acid esters and chiral primary ammonium ions (Cram, D. J. Ace. Chem. Res. 1978, 11, 8-14). The chiral ‘host’ is thus able to discriminate between enantiomeric compounds by the formation of hydrogen bonds between the ether oxygen and the enantiomers. The complex formed with one of the isomers, the ‘guest’, will be less stable on steric grounds and this forms the basis for its separation.
It is evident from the literature cited that there exists a need to (a) synthesize Lamivudine by a process requiring less expensive, less hazardous and easily available reagents, and (b) achieve good yields with superior quality of product without resorting to column chromatography as a means of separation, thereby making the process of Lamivudine manufacture more acceptable industrially.
CLIP
ideally, the chemical synthesis of APIs begins from simple, inexpensive building blocks or RMs that are used for multiple purposes and are available in the fine chemicals industry, though some require uncommon RMs that contribute significantly to API manufacturing cost. RMs are converted into APIs by multi-step processes of breaking old chemical bonds and making new ones. A synthesis of 3TC is shown in . In the seven-step sequence, six steps involve breaking existing chemical bonds and creating new ones to build the molecular architecture of the API. The final recrystallization of an API is a critical step; at this stage the crystalline form of the API is determined and related substances (impurities) are removed or reduced to acceptable levels. APIs are often milled in a final step so that their particle size distribution (PSD) falls within specified limits. The crystalline form and PSD of an API must be controlled, because these properties are often critical to the formulation, dissolution, absorption and bioavailability of a drug. Bioavailability is the fraction of a drug dose that reaches systemic circulation (that is, is present in blood plasma) after administration. By definition, a drug is 100% bioavailable when administered by injection; drugs for ART are taken every day and administration by injection is not possible.
The cost of ART is absolutely critical to ensuring access in LMICs. The cost of manufacturing an API is dependent upon the cost of RMs, the cost of overheads and labour (OHL) and volume demand for the product. OHL includes the capital investment to build a manufacturing facility and operating costs, including personnel and energy, waste disposal and the eventual cost of decommissioning of the facility. Increased volume demand generally decreases the cost contribution of RM and OHL. Substantial production volumes are required to obtain full economy of scale . Producing 1–5 metric tons per year is substantially more expensive per kilogram than producing 100 metric tons of an API. There is a practical limit of approximately 50–100 metric tons/year beyond which cost reductions are modest with increased volume, but this practical limit refers to the volumes of drug manufactured in any single manufacturing plant. Exceptions to these generalizations do occur, most often when demand exceeds either the existing manufacturing capacity for a specific API or the availability of critical RMs . Exceptions that have occurred include shortages of β-thymidine for producing AZT and a squeeze on the availability and price of adenine as a starting material for TDF. Another contributor to RM and OHL costs is the efficiency of a chemical synthesis. Since operating costs for a manufacturing facility may be USD2,000/h, the number of steps or processing time for a chemical synthesis affects manufacturing cost. The efficiency of a synthesis is often quoted as an E-factor representing the kilograms of waste produced per kilogram of product manufactured. Waste management is expensive in chemical manufacturing wherever environmental guidelines are both reasonable and followed. From a slightly different perspective, increasing the overall yield of an API synthesis reduces RM use and associated cost for manufacturing.
Jinliang L, Feng LV. inventors; Shanghai Desano Pharmaceutical, assignee. A process for stereoselective synthesis of lamivudine. European Patent Application EP 2161 267 A1. 2007 June 29.

Object of the invention
Thus, one object of the present invention is to provide a process for the synthesis of_Lamivudine which is cost effective, uses less hazardous and easily available reagents, yet achieves good yields with superior quality of product without resorting to column chromatography.
A further object of the present invention is to provide an improved process for the synthesis of Lamivudine, by separating the mixture of diastereomers: Cis-[2R,5S], [2S,5R] from Trans-[2R,5R], [2S,5S] and then resolving the Cis isomers using BINOL to obtain (-)-[2R,5S]^Cis-Lamivudine with at least 99% ee.
This 1,3-oxathiolane compound VIII is further condensed with silylated cytosine in the presence of a Lewis acid such as trimethylsilyliodide to get protected 6-amino-3 – {2-hydroxymethyl- 1 ,3 -oxathiolan-5-yl} -3 -hydropyrimidine- 2-one (compound IX). OH

Cis(±)and Trans (±) racemic mixtures

Lamivudine (-)-[2

Compound (IX) is mixture of following optical isomers

SCHEME 2 The separation of the four-component diastereomeric mixture of isomers bearing the following configuration: trans-[2R,5R], [2S.5S] and cis-[2R,5S], [2S,5R] forms the next step. The separation efficiency of the benzoyl-protected compound
Example 9
Preparation of Lamivudine: (-)-[2R,5S]-4-amino-l-[2-(hydroxymethyl)-l,3- oxathiolan-5 -yl] -2(1 H)-pyrimidin-2-one
Compound I 5mL of cone. HCl was slowly added to a solution of 2Og of Lamivudine-BINOL complex in 100ml of ethylacetate and 10OmL of DM water (pH 2-2.5). The layers. were separated and a 10OmL aliquot of ethylacetate was added to the aqueous layer. The layers were separated again and the aqueous layer was neutralized using 1OmL of 10% aqueous NaOH solution. The solvent was recovered under vacuum at 40-45 0C, the product obtained was dissolved in 160 mL of methanol, filtered, the filtrate was concentrated and 32 mL of water-ethanol mixture (3:1) was added to this product, heated to get a clear solution, cooled to 5 – 10 0C and then filtered. The residue was vacuum dried at 45-50 0C. Yield: 4-5g.
Enantiomeric excess = 99.74 % m.p. = 133-135 °C [<X]D at 25°C = 98.32° (c = 5 water)
1H NMR (DMSO d6): 2.99-3.07 (dd, IH), 3.35-3.38 (dd, IH), 3.72-3.74 (m, 2H), 5.14-5.18 (t, IH), 5.32-5.38 (t, IH), 5.71-5.75 (d, IH), 6.16-6.21 (t, IH), 7.22-
7.27 (d, 2H), 7.80-7.83 (d, IH)
Moisture content: 1.67%
IR (in KBr, cm“1): 3551, 3236, 2927, 1614, 1492, 1404, 1336, 1253, 1146, 1052,
967, 786. MS: M+l =230
XRD [2Θ] (Cu – Ka1=I.54060A, Ka2=1.54443A Kβ= 1.39225A; 4OmA, 45kV):
5.08, 9.89, 10.16, 11.40, 11.65, 12.96, 13.23, 15.26, 15.82, 17.74, 18.74, 18.88,
19.67, 20.69, 22.13, 22.88, 23.71, 25.47, 26.07.
PATENT
http://www.google.com/patents/WO2013021290A1?cl=en



| EP 0382526; EP 0711771; JP 1996119967; JP 2000143662; US 5047407 |

| J Org Chem 1992,57(8),2217-9 |

Lamivudine is a nucleoside reverse transcriptase inhibitor, and is a kind of deoxycytidine analogue, which can inhibit the reproduction of Human immunodeficiency virus (HIV) and hepatitis B virus (HBV), whose chemical name is (2R-cis)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-1H-pyrimidin-2-one, and structural formula is as follows:
-

-
In 1990, Belleau et al firstly reported Lamivudine structure, and BioChem Pharma of Canada firstly developed Lamivudine to be used to treat AIDS ( WO91/17159 ) and hepatitis B ( EP0474119 ), and found that it had distinguished therapeutic effect on hepatitis B. Since Lamivudine has two chiral centers, it has 4 stereisomers, among which the 2R,5S (2R–cis)-isomer is the most potent in anti-HIV and anti-HBV activities, and its cytotoxicity on some cells is lower than its enatiomer or racemic body.
-
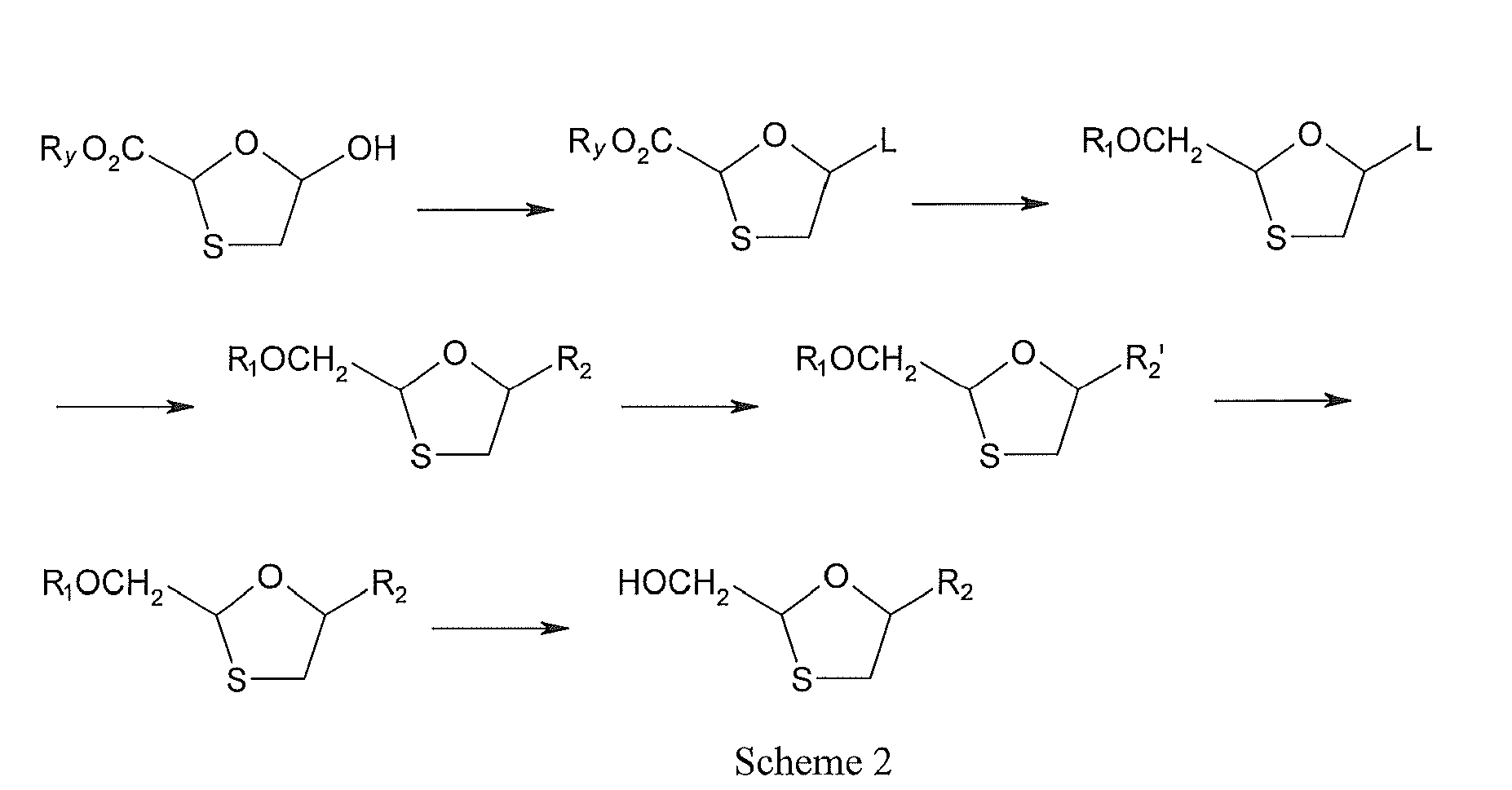
WO94/14802 mentioned two synthetic schemes (see Scheme 1 and Scheme 2):


-
In the above two schemes of this process, chirality was not controlled, and the final product was obtained by column chromatography, thus the yield was low and the requirement on the equipment was high, resulting in that the production cost was high and the operation in the production could not be controlled easily.


The specific reaction scheme is as follows:

synthetic route is preferably as follows:

. The specific reaction scheme is as follows:


The specific reaction scheme is as follows:

Example 8 The preparation of (2R,5S)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolane-5-yl) -2(1H)-pyrimidone (Lamivudine)
-
The compound of Example 7 (41.0g, 0.1mol) and methanol (250ml) were added to a reaction flask, and then stirred to make the compound dissolved in methanol. The mixture was cooled to 0 °C, and then K2CO3 (41.2g, 0.3mol) was added. The mixture was further stirred at room temperature overnight and then was adjusted by 0.1N HCl to a pH of about 7. The mixture was filtered and the solvent was evaporated under reduced pressure from the filtrate, and then to the residue was added 150ml of water. The aqueous layer was extracted by 150ml of toluene (50ml X 3), and then p-nitrobenzoic acid (16.8g, 0.1mol) was added to the aqueous layer and refluxed for 30 minutes, after which, the reaction mixture was cooled and further stirred at 0-5 °C for 2 hours. Then the reaction mixture was filtered and dried to give 31.7g of a white solid.
-
The resulting salt and anhydrous ethanol (120ml) were added to a reaction flask, and warmed to 70-75 °C. Triethylamine (12ml) was added dropwise, and the reaction was conducted at that temperature for 2 hours. Then the mixture was cooled to 50 °C, at which point ethyl acetate (150ml) was added dropwsie. After the addition was complete, the mixture was cooled to 10 °C and further stirred for 4 hours. The mixture was filtered to give 15.6g of Lamivudine, and the yield was 68%. 1H-NMR (DMSO-d6) δ: 7.83(dd, 1H), 7.17∼7.23(dd, 2H), 6.21(t, 1H), 5.72 (dd, 1H), 5.29 (t, 2H), 5.16 (t, 1H), 3.70∼3.74 (m, 2H), 3.32∼3.43 (dd, 1H), 3.01∼3.05(dd, 1H); Elemental analysis: C8H11N3O3S found(%): C 41.85, H 4.88 N 18.25, S 13.94; calculated (%) C 41.91, H 4.84, N 18.33, S13.99.
PAPER

http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
References
- 1″Lamivudine”. The American Society of Health-System Pharmacists. Retrieved 31 July 2015.
- 2
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- 3
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 65. ISBN 9781284057560.
- 4
- Fox Z, Dragsted UB, Gerstoft J, et al. (2006). “A randomized trial to evaluate continuation versus discontinuation of lamivudine in individuals failing a lamivudine-containing regimen: The COLATE trial”. Antiviral Therapy 11 (6): 761–70. PMID 17310820.
- 5
- http://hivdb.stanford.edu/index.html Stanford University Drug Resistance Database.
- 6
- Koziel MJ, Peters MG (2007). “Viral hepatitis in HIV infection”. N Engl J Med 356 (14): 1445–54. doi:10.1056/NEJMra065142. PMID 17409326.
- 7
- “Epivir package insert” (PDF). GlaxoSmithKline. Retrieved January 20, 2011.
- 8
- Curtis, John (June 20, 1998). “Hunting Down HIV”. Yale Medicine.
- 9
- Soderstrom, E John (July 10, 2003). “National Institutes of Health: Moving Research from the Bench to the Bedside”.
- 10
- Cohen, Elizabeth (September 29, 2014). “Doctor treats Ebola with HIV drug in Liberia — seemingly successfully”. CNN.
- 11 HIV drug may stop Ebola. Operonlabs.com, 27 September 2014
External links
- Epivir (manufacturer’s website)
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-amino-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]-1,2-dihydropyrimidin-2-one
|
|
| Clinical data | |
| Trade names | Epivir |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a696011 |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 86% |
| Protein binding | Less than 36% |
| Biological half-life | 5 to 7 hours |
| Excretion | Renal (circa 70%) |
| Identifiers | |
| CAS Number | 134678-17-4 |
| ATC code | J05AF05 (WHO) |
| PubChem | CID 73339 |
| DrugBank | DB00709 |
| ChemSpider | 66068 |
| UNII | 2T8Q726O95 |
| KEGG | D00353 |
| ChEMBL | CHEMBL141 |
| NIAID ChemDB | 000388 |
| Synonyms | L-2′,3′-dideoxy-3′-thiacytidine |
| PDB ligand ID | 3TC (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C8H11N3O3S |
| Molar mass | 229.26 g/mol |

