PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
28 Mar 2022No recent reports of development identified for phase-I development in Peripheral-T-cell-lymphoma in China (IV, Injection)
26 Jan 2022ZIOPHARM Oncology is now called Alaunos Therapeutics
11 Dec 2021Safety and efficacy data from a phase II trial in Peripheral T-cell lymphoma presented at the 63rd American Society of Hematology Annual Meeting and Exposition (ASH-2021)
Darinaparsin is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects. Compared to inorganic arsenic compounds such as arsenic trioxide (As2O3), darinaparsin appears to exhibit a wide therapeutic window.
Darinaparsin, also know as ZIO-101 and SP-02, is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects.
Darinaparsin is an organic arsenical composed of dimethylated arsenic linked to glutathione, and is being investigated for antitumor properties in vitro and in vivo. While other arsenicals, including arsenic trioxide, have been used clinically, none have shown significant activity in malignancies outside of acute promyelocytic leukemia. Darinaparsin has significant activity in a broad spectrum of hematologic and solid tumors in preclinical models. Here, we review the literature describing the signaling pathways and mechanisms of action of darinaparsin and compare them to mechanisms of cell death induced by arsenic trioxide. Darinaparsin has overlapping, but distinct, signaling mechanisms. We also review the current results of clinical trials with darinaparsin (both intravenous and oral formulations) that demonstrate significant antitumor activity.
[0071] Sterile water (15.5 L) and ethyl alcohol (200 proof, 15.5 L) were charged in a reaction flask prior to the addition of L-glutathione (3.10 kg). While being stirred, the reaction mixture was cooled to 0-5 °C prior to the addition of triethylamine (1.71 L). Stirring was continued until most of the solids were dissolved and the solution was filtered. After filtration, the reaction mixture was cooled to 0-5 °C prior to the addition of chlorodimethylarsine (1.89 kg) over 115 minutes while maintaining the temperature at 0-5 °C. Stirring continued at 0-5 °C for 4 hours before acetone (30.6 L) was added over 54 minutes while maintaining the temperature at 0-5 °C. The suspension was stored at 0-5°C overnight prior to filtration. The solid was collected in a filter funnel, washed successively with ethyl alcohol (200 proof, 13.5 L) and acetone (13.5 L) and dried in suction for 23 minutes. A second similar run was performed and the collected solids from both runs were combined. Ethyl alcohol (200 proof, 124 L) and the combined solids (11.08 kg) were charged in a vessel. The slurry was stirred at ambient temperature for 2 hours before filtration, washing successively with ethyl alcohol (200 proof, 27 L) and acetone (27 L) and dried in suction for 60 minutes. The resulting solid was transferred to drying trays and dried in a vacuum oven at ambient temperature for 66 hours to provide darinaparsin as a solid with the differential scanning calorimetry (DSC) thermogram of Figure 1, with an extrapolated onset temperature at about 191.36° C and a peak temperature at about 195.65° C.
PATENT
WO 2010021928
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109°C was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
SGLU-1: Glutathione (14.0 g, 45.6 mmol) was stirred rapidly in glyme while dimethylchoroarsine (6.5 g, 45.6 mmol) was added dropwise. Pyridine (6.9 g, 91.2 mmol) was then added to the slurry and the mixture was subsequently heated to reflux. The heat was removed immediately and the mixture stirred at room temperature for 4 h. Isolation of the resultant insoluble solid and recrystallization from ethanol afforded 4 as the pyridine hydrochloride complex (75% yield). mp 115-118°C; NMR (D20) δ1.35 (s, 6H), 1.9-4.1 (m’s, 10H), 7.8-9.0 (m, 5H); mass spectrum (m/e) 140, 125, 110, 105, 79, 52, 45, 36.
PATENT
WO 2009075870
Step 1
Example 1. Preparation of Dimethylchloroarsine (DMCA). A 3-neck round-bottom flask (500 mL) equipped with mechanical stirrer, inlet for nitrogen, thermometer, and an ice bath was charged with cacodylic acid (33 g, 0.23 mol) and cone. hydrochloric acid (67 mL). In a separate flask, a solution of SnCl2·2H2O (54 g, 0.239 mol) in cone. hydrochloric acid (10 mL) was prepared. The SnCl2·2 H2O solution was added to the cacodylic acid in HCl solution under nitrogen while maintaining the temperature between 5 °C and 10 °C. After the addition was complete, the ice bath was removed and the reaction mixture was stirred at ambient temperature for 1 h. The reaction mixture was transferred to a separatory funnel and the upper layer (organic) collected. The bottom layer was extracted with dichloromethane (DCM) (2 × 25 mL). The combined organic extract was washed with 1 N HCl (2 × 10 mL) and water (2 × 20 mL). The organic extract was dried over MgSO4 and DCM was removed by rotary evaporation (bath temperature 80 °C, under nitrogen, atmospheric pressure). The residue was further distilled under nitrogen. Two tractions of DMCA were collected. The first fraction contained some DCM and the second fraction was of suitable quality (8.5 g, 26% yield). The GC analysis confirmed the identity and purity of the product.
Step 2
Example 3. Preparation of S-Dimethylarsinoglutathione (SGLU-1). In a 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel and thermometer under an inert atmosphere was prepared a suspension of glutathione (114.5 g, 0.37 mol) in a 1:1 (v/v) mixture of water/ethanol (1140 mL) and cooled to below 5 °C. The mixture was treated slowly (over 15 min) with triethylamine (63.6 mL, 0.46 mol) while maintaining the temperature below 20 °C. The mixture was cooled to 4 °C and stirred for 15 min and then the traces of undissolved material removed by filtration. The filtrate was transferred in a clean 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel, nitrogen inlet, and thermometer and DMCA (70 g, 0.49 mol) (lot # 543-07-01-44) was added slowly while maintaining the temperature at 3-4°C. The reaction mixture was stirred at 1-4°C for 4 h, and acetone (1.2 L) was added over a period of 1 h. The mixture was stirred for 90 min between 2 and 3°C and the resulting solid was isolated by filtration. The product was washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and the wet solids were suspended in ethanol 200 Proof (2000 mL). The product was isolated by filtration, washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and dried in vacuum for 2 days at RT to give 115 g (75%) of SGLU-1, HPLC purity > 99.5% (in process testing).
PATENT
WO 2007027344
Example 2 Preparation of S-Dimethylarsinoglutathione A 5 L, three necked round bottom flask was equipped with a mechanical stirrer assembly, thermometer, addition funnel, nitrogen inlet, and a drying tube was placed in a cooling bath. A polyethylene crock was charged with glutathione-reduced (200 g) and deionized water (2 L) and stirred under a nitrogen atmosphere to dissolve all solids. The mixture was filtered to remove any insoluble material and the filtrate was transferred to the 5 L flask. While stirring, ethanol, 200 proof (2 L) was added and the clear solution was cooled to 0-5° C. using an ice/methanol bath. Pyridine (120 g) was added followed by a dropwise addition of Me2AsCl (120 g) over a minimum of 1 hour. The reaction mixture was stirred at 0-5° C. for a minimum of 2 hours prior to removal of the cooling bath and allowing the mixture to warm to room temperature under a nitrogen atmosphere with stirring. The reaction mixture was stirred overnight (>15 hrs) at room temperature under a nitrogen atmosphere at which time a white solid may precipitate. The reaction mixture was concentrated to a slurry (liquid and solid) at 35-45° C. using oil pump vacuum to provide a white solid residue. As much water as possible is removed, followed by two coevaporations with ethanol to azeotrope the last traces of water. The white solid residue was slurried in ethanol, 200 pf. (5 L) under a nitrogen atmosphere at room temperature overnight. The white solid was filtered and washed with ethanol, 200 pf. (2×500 mL) followed by acetone, ACS (2×500 mL). The resulting solid was transferred to drying trays and vacuum oven dried overnight at 25-35° C. using oil pump vacuum to provide pyridinium hydrochloride-free S-dimethylarsinoglutathione as a white solid. melting point of 189-190° C.
PATENT
WO 20060128682
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109° C. was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
Pyridine Hydrochloride Free Synthesis of S-Dimethylarsinoglutathione (GLU) Dimethylarsinoglutathione is made using an adapted of Chen (Chen, G. C., et al. Carbohydrate Res. (1976) 50: 53-62) the contents of which are hereby incorporated by reference in their entirety. Briefly, dithiobis(dimethylarsinoglutamine) is dissolved in dichloromethane under nitrogen. Tetramethyldiarsine is added dropwise to the solution and the reaction is stirred overnight at room temperature under nitrogen and then exposed to air for 1 h. The mixture is then evaporated to dryness and the residue is washed with water and dried to give a crude solid that is recrystallized from methanol to give S-dimethylarsinoglutathione.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Solasia Pharma K.K. (TSE: 4597, Headquarters: Tokyo, Japan, President & CEO: Yoshihiro Arai, hereinafter “Solasia”) today announced submission of a New Drug Application (NDA) for its new anti-cancer drug darinaparsin (generic name, development code: SP-02) as a treatment for relapsed or refractory peripheral T-cell lymphoma to the Ministry of Health, Labour and Welfare (MHLW). Based on positive results of R&D on darinaparsin, centered primarily on the results of the Asian Multinational Phase 2 Study (study results released in June 2020), Solasia filed an NDA for the drug with the regulatory authority in Japan ahead of anywhere else in the world.
Solasia expects to obtain regulatory approval in 2022 and to also launch in the same year. If approved and launched, darinaparsin would be the third drug Solasia successfully developed and brought to market since its founding and is expected to contribute to the treatment of PTCL.
Mr. Yoshihiro Arai, President and CEO of Solasia, commented as follows: “No standard treatment has been established for relapsed or refractory PTCL as of yet. I firmly believe that darinaparsin, with its novel mechanism of action that differs from those of already approved drugs, will contribute to patients and healthcare providers at clinical sites as a new treatment option for relapsed or refractory PTCL. Since founding, Solasia has conducted R&D on five pipeline drugs. Of the five, we have successfully developed and brought to market two drugs, i.e., began providing them to patients, and today, we submitted an NDA for our first anti-cancer drug. Under our mission to provide patients with ‘Better Medicine for a Brighter Tomorrow’, we will continue aiming to contribute to patients’ treatment and enhanced quality of life. ”
About darinaparsin (SP-02) Darinaparsin, an organoarsenic compound with anticancer activity, is a novel mitochondrial-targeted agent being developed for the treatment of various hematologic and solid tumors. The proposed mechanism of action of the drug involves the disruption of mitochondrial function, increased production of reactive oxygen species, and modulation of intracellular signal transduction pathways. Darinaparsin is believed to exert anticancer effect by inducing cell cycle arrest and apoptosis. Darinaparsin has been granted orphan drug designation in the US and EU. For more information, please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Asian Multinational Phase 2 Study The Asian Multinational Phase 2 Study was a multinational, multicenter, single-arm, open-label, non-randomized study to evaluate the efficacy and safety of darinaparsin monotherapy in patients with relapsed or refractory PTCL conducted in Japan, Korea, Taiwan, and Hong Kong. (CT.gov Identifier: NCT02653976). Solasia plans to present the results of the study at an international academic conference to be held in the near future.
Anamorelin is a non-peptidic ghrelin mimetic Treatment of cancer anorexia and cancer cachexia
Anamorelin hydrochloride has been submitted New Drug Application (NDA) for the treatment of cachexia in non-small cell lung cancer (NSCLC) patients.
It was originally developed by Novo Nordisk, then it was licensed to Ono and Helsinn Therapeutics for the treatment of cachexia and anorexia in cancer patients.
Anamorelin hydrochloride has been submitted New Drug Application (NDA) for the treatment of cachexia in non-small cell lung cancer (NSCLC) patients.
It was originally developed by Novo Nordisk, then it was licensed to Ono and Helsinn Therapeutics for the treatment of cachexia and anorexia in cancer patients.
On 18 May 2017, the European Medicines Agency recommended the refusal of the marketing authorisation for the medicinal product, intended for the treatment of anorexia, cachexia or unintended weight loss in patients with non-small cell lung cancer. Helsinn requested a re-examination of the initial opinion. After considering the grounds for this request, the European Medicines Agency re-examined the opinion, and confirmed the refusal of the marketing authorisation on 14 September 2017.[8] The European Medicines Agency concluded that the studies show a marginal effect of anamorelin on lean body mass and no proven effect on hand grip strength or patients’ quality of life. In addition, following an inspection at clinical study sites, the agency considered that the safety data on the medicine had not been recorded adequately. Therefore, the agency was of the opinion that the benefits of anamorelin did not outweigh its risks.[9]
The chemical name of anamorelin hydrochloride is 2-Amino-N-((R)-1-((R)-3-benzyl-3-(1,2,2-trimethylhydrazine-1-carbonyl)piperidin-1-yl)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-methylpropanamide hydrochloride corresponding to the molecular formula C31H42N6O3•HCl and has a relative molecular mass 583.16 g/mol and has the following structure:
The structure of the active substance was elucidated by a combination of 1 H-NMR, 13C-NMR, elemental analysis, FT-IR, UV and and mass spectrometry. Anamorelin HCl appears as a white to off-white hygroscopic solid, freely soluble in water, methanol and ethanol, sparingly soluble in acetonitrile and practically insoluble in ethyl acetate, isopropyl acetate and n-heptane. Its pka was found to be 7.79 and the partition coefficient 2.98. It has two chiral centres with the R,R absolute configuration, which is controlled in the active substance specification by chiral HPLC. Based on the presented data, neither anamorelin hydrochloride, nor any of its salts have been previously authorised in medicinal products in the European Union. Anamorelin is therefore considered as a new active substance.
SYN
OPRD
PATENT
WO 9958501
PATENT
WO 2001034593
https://patents.google.com/patent/WO2001034593A1/enExample 1A procedure for the preparation of the compound which is either 2-Amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide
A one-necked round-bottom flask (1 I) equipped with a magnetic stirrer and addition funnel was charged with NaOH-pellets (15,6 g), tetrahydrofuran (400 ml) and ethylnipecotate (50 ml, 324 mmol). To the stirred mixture at room temperature was added dropwise a solution of Boc2O (84,9 g, 389 mmol) dissolved in tetrahydrofuran (150 ml) (1 hour, precipitation of white solid, NaOH-pellets dissolved, exoterm). The mixture was stirred overnight at room temperature. The mixture was added to EtOAc (500 ml) and H2O (2000 ml), and the aqueous layer was re-extracted with EtOAc (2 X 500 ml) and the combined organic layers were washed with brine (100 ml), dried over MgSO4, filtered and concentrated in vacuo to afford piperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester 3-ethyl ester (82,5 g) as a thin yellow oil.1H-NMR (300 MHz, CDCI3): δ 1,25 (t, 3H, CH3); 1 ,45 (s, 9H, 3 X CH3); 2,05 (m, 1H); 2,45 (m, 1H); 2,85 (m, 1 H); 3,95 (d (broad), 1 H); 4,15 (q, 2H, CH2)Step b3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester 3-ethyl ester (racemic mixture)
A three-necked round-bottom flask (2 I) equipped with a magnetic stirrer, thermometer, nitrogen bubbler and addition funnel was evacuated, flushed with nitrogen, charged with anhydrous tetrahydrofuran (500 ml) and cooled to -70 °C. Then lithium diisopropylamine (164 ml of a 2,0 M solution in tetrahydrofuran, 327 mmol) was added. To the stirred solution at -70 °C was added dropwise over 45 min. a solution of piperidine-1 ,3-dicarboxylic acid 1- tert-butyl ester 3-ethyl ester (80 g, 311 mmol) in anhydrous tetrahydrofuran (50 ml) (temperature between -70 °C and -60 °C, clear red solution). The mixture was stirred for 20 min. and followed by dropwise addition over 40 min. of a solution of benzylbromide (37 ml, 311 mmol) in anhydrous tetrahydrofuran (250 ml) (temperature between -70 °C and -60 °C). The mixture was stirred for 1 hour at -70 °C, and then left overnight at room temperature (pale orange).The reaction mixture was concentrated in vacuo to approx. 300 ml, transferred to a separating funnel, diluted with CH2CI2 (900 ml) and washed with H2O (900 ml). Due to poor separation the aqueous layer was re-extracted with CH2CI2 (200 ml), the combined organic layers were washed with aqueous NaHSO4 (200 ml, 10%), aqueous NaHCO3 (200 ml, saturated), H2O (200 ml), brine (100 ml), dried over MgSO4> filtered and concentrated in vacuo to afford an oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The solids formed was removed by filtration, washed with heptane and dried in vacuo to give a racemic mixture of 3-benzylpiperidine-1 ,3-dicarboxylic acid 1-ter–butyl ester 3-ethyl ester (81 ,4 g). ■ HPLC (h8): Rt = 15,79 min.LC-MS: Rt = 7,67 min. (m+1) = 348,0Step c 3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester (racemic mixture)
3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester 3-ethyl ester (81 g, 233 mmol) was dissolved in EtOH (400 ml) and NaOH (400 ml, 16% aqueous solution) in a one neck round- bottom flask (1 L) equipped with a condenser and a magnetic stirrer. The mixture was refluxed for 10 h under nitrogen, and cooled to room temperature, concentrated in vacuo to approx. 600 ml (precipitation of a solid), diluted with H2O (400 ml), cooled in an icebath, and under vigorous stirring acidified with 4 M H2SO4 until pH = 3 (final temperature: 28 °C). The mixture was extracted with EtOAc (2 X 700 ml), and the combined organic layers were washed with brine (200 ml), dried over MgSO4, filtered and concentrated in vacuo to afford an oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The crystals formed were removed by filtration, washed with heptane and dried in vacuo to give a racemic mixture of 3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester (66,0 g)HPLC (h8): Rt = 12,85 min.LC-MS: Rt = 5,97 min. (m+1) = 320,0Chirale HPLC (Chiracel OJ, heptane(92):iPrOH(8):TFA(0,1)): Rt = 8,29 min. 46,5 % Rt = 13,69 min. 53,5 %Step d(3R)-3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-Benzylpiperidine-1,3-dicarboxylic acid 1-tert-butyl ester
(Resolution of 3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester)
3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester (76 g, 238 mmol) was dissolved in EtOAc (3,0 L) in a one neck flask (5L) equipped with magnetic stirring. Then H2O (30 ml), R(+)-1-phenethylamine (18,2 ml, 143 mmol) and Et3N (13,2 ml, 95 mmol) were added and the mixture was stirred overnight at room temperature resulting in precipitation of white crystals (41 ,9 g), which were removed by filtration, washed with EtOAc and dried in vacuo. The precipitate was dissolved in a mixture of aqueous NaHSO4 (300 ml, 10%) and EtOAc (600 ml), layers were separated and the aqueous layer re-extracted with EtOAc (100 ml). The combined organic layers were washed with brine (100 ml), dried over MgSO4 and filtered. The solvent was removed in vacuo to afford a colourless oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The crystals that had been formed were removed by filtration, washed with heptane and dried in vacuo to give one compound which is either (3R)-3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-benzylpiperidine- 1,3-dicarboxylic acid 1-tert-butyl ester (27,8 g).Chirale HPLC (Chiracel OJ, heptane(92):iPrOH(8):TFA(0,1)):Rt = 7,96 min. 95,8 % eeStep e(3R)-3-Benzyl-3-(N,N’1N’-trimethylhvdrazinocarbonyl)piperidine-1-carboxylic acid tert-butyl ester or (3S)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidine-1-carboxylic acid tert-butyl ester
Trimethylhydrazine dihydrochloride (15,3 g, 104 mmol) was suspended in tetrahydrofuran (250 ml) in a one-neck round-bottom flask (1 I) equipped with a large magnetic stirrer, and an addition funnel/nitrogen bubbler. The flask was then placed in a water-bath (temp: 10- 20°C), bromo-rrts-pyrrolydino-phosphonium-hexafluorophosphate (40,4 g, 86,7 mmol) was added, and under vigorous stirring dropwise addition of diisopropylethylamine (59 ml, 347 mmol). The mixture (with heavy precipitation) was stirred for 5 min., and a solution of the product from step d which is either (3R)-3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-benzylpiperidine-1,3-dicarboxylic acid 1-tert-butyl ester (27,7 g, 86,7 mmol) in tetrahydrofuran (250 ml) was added slowly over 1 ,5 hour. The mixture was stirred overnight at room temperature. The reaction was diluted with EtOAc (1000 ml), washed with H2O (500 ml), aqueous NaHSO4, (200 ml, 10%), aqueous NaHCO3 (200 ml, saturated), brine (200 ml), dried over MgSO4, filtered and concentrated in vacuo to afford a thin orange oil. The mixture was dissolved in EtOAc (300 ml), added to SiO2 (150 g) and concentrated in vacuo to a dry powder which was applied onto a filter packed with SiO2 (150 g), washed with heptan (1 I) and the desired compound was liberated with EtOAc (2,5 I). After concentration in vacuo, the product which is either (3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)-piperidine-1- carboxylic acid tert-butyl ester or (3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)- piperidine-1-carboxylic acid tert-butyl ester (49 g) as an orange oil was obtained.HPLC (h8): Rt = 14,33 min.Ste f(3R)-3-Benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-Benzyl-piperidine-3- carboxylic acid trimethylhydrazide
The product from step e which is either (3R)-3-Benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)-piperidine-1 -carboxylic acid tert-butyl ester or (3S)-3-Benzyl-3- (N,N’,N’-trimethylhydrazinocarbonyl)-piperidine-1 -carboxylic acid tert-butyl ester (56,7 g, 100,9 mmol) was dissolved in EtOAc (500 ml) (clear colourless solution) in a one-neck roundbottom flask (2L) equipped with magnetic stirring. The flask was then placed in a waterbath (temp: 10-20 °C), and HCI-gas was passed through the solution for 5 min. (dust- like precipitation). After stirring for 1 hour (precipitation of large amount of white crystals), the solution was flushed with N2 to remove excess of HCI. The precipitate was removed by gentle filtration, washed with EtOAc (2 X 100 ml), and dried under vacuum at 40 °C overnight to give the product which is either (3R)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide (37,0 g).HPLC (h8): Rt = 7,84 min.Step q r(1 R)-2-r(3R)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-((1 H-indol-3- yl)methyl)-2-oxoethvncarbamic acid tert-butyl ester or .(1 R)-2-..3S)-3-Benzyl-3-(N,N’,N’- trimethylhvdrazinocarbonyl)piperidin-1-vn-1-((1 H-indol-3-yl)methyl)-2-oxoethyllcarbamic acid tert-butyl ester
Boc-D-Trp-OH (32,3 g, 106 mmol) was dissolved in dimethylacetamide (250 ml) in a one- neck roundbottom flask (500 ml) equipped with a magnetic stirrer and a nitrogen bubbler. The solution was cooled to 0-5 °C and 1-hydroxy-7-azabenzotriazole (14,4 g, 106 mmol), 1- ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (20,3 g, 106 mmol), N- methylmorpholine (11 ,6 ml, 106 mmol) were added. After stirring for 20 min. at 0-5 °C the product from step f which is either (3R)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide (37,0 g, 106 mmol) and N-methylmorpholine (24,4 ml, 223 mmol) were added. The reaction was stirred overnight at room temperature. The mixture was then added to EtOAc (750 ml) and washed with aqueous NaHSO4 (300 ml, 10 %). The layers were allowed to separate, and the aqueous layer was re-extracted with EtOAc (500 ml). The combined organic layers were washed with H2O (100 ml), aqueous NaHCO3 (300 ml, saturated), H2O (100 ml), brine (300 ml), dried over MgSO4, filtered and concentrated in vacuo to afford the product which is either [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-((1H- indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester or [(1 R)-2-[(3S)-3-benzyl-3- (N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-((1 H-indol-3-yl)methyl)-2- oxoethyljcarbamic acid tert-butyl ester (56,7g) as an orange oil.HPLC (h8): Rt = 14,61 min.LC-MS: Rt = 7,35 min. (m+1 ) = 562,6Step h1 -f(2R)-2-Amino-3-(1 H-indol-3-yl)propionylH3R)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide or 1-f(2R)-2-Amino-3-(1 H-indol-3-yl)propionvn-(3S)-3-benzylpiperidine-3- carboxylic acid trimethylhydrazide
The product from step g which is either [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1 -yl]-1 -((1 H-indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester or [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1- yl]-1-((1 H-indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester (56,7 g, 100,9 mmol) was dissolved in EtOAc (500 ml) (clear colourless solution) in a one-neck round-bottom flask (2L) equipped with magnetic stirring. The flask was then placed in a water-bath (temp: 10-20 °C), and HCI-gas was passed through the solution for 10 min. (heavy precipitation of oil). The mixture was flushed with N2 to remove excess of HCI and then separated into an oil and an EtOAc-layer. The EtOAc-layer was discarded. The oil was dissolved in H2O (500 ml), CH2CI2 (1000 ml), and solid Na2CO3 was added until pH > 7. The layers were separated, and the organic layer was washed with H2O (100 ml), brine (100 ml), dried over MgSO4, filtered and concentrated in vacuo to afford the product which is either 1-[(2R)-2-amino-3-(1 H-indol- 3-yl)propionyl]-(3R)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide or 1-[(2R)-2- amino-3-(1H-indol-3-yl)propionyl]-(3S)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide (27 g) as an orange foam.HPLC (h8): Rt = 10,03 min.Step i(1-r(1 R)-2-r(3R)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-(1H-indol-3- ylmethyl)-2-oxo-ethylcarbamovπ-1 -methylethyl fcarbamic acid tert-butyl ester or1-r(1 R)-2-r(3S)-3-Benzyl-3-(N,N’.N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-(1 H-indol-3- ylmethyl)-2-oxo-ethylcarbamovπ-1-methylethyl)carbamic acid tert-butyl ester
Boc-Aib-OH (11 ,9 g, 58,4 mmol) was dissolved in dimethylacetamide (125 ml) in a one-neck roundbottom flask (500 ml) equipped with a magnetic stirrer and nitrogen bubbler. To the stirred solution at room temperature were added 1-hydroxy-7-azabenzotriazole (7,95 g, 58,4 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (11 ,2 g, 58,4 mmol), and diisopropylethylamine (13,0 ml, 75,8 mmol). After 20 min. (yellow with precipitation) a solution of the product from step h which is either 1-[(2R)-2-amino-3-(1 H-indol-3- yl)propionyl]-(3R)-3-benzylpiperidine-3:carboxylic acid trimethylhydrazide or 1-[(2R)-2- amino-3-(1 H-indol-3-yl)propionyl]-(3S)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide (27,0 g, 58,4 mmol) in dimethylacetamide (125 ml) was added. The reaction was stirred at room temperature for 3 h. The mixture was added to EtOAc (750 ml) and washed with aqueous NaHSO4 (300 ml, 10 %). The layers were allowed to separate, and the aqueous layer was re-extracted with EtOAc (500 ml). The combined organic layers were washed with H2O (100 ml), aqueous NaHCO3 (300 ml, saturated), H2O (100 ml), brine (300 ml), dried over MgSO4, filtered and concentrated in vacuo to approx. 500 ml. Then SiO2 (150 g) was added and the remaining EtOAc removed in vacuo to give a dry powder which was applied onto a filter packed with SiO2 (150 g), washed with heptan (1 L), and the desired compound was liberated with EtOAc (2,5 L). After concentration in vacuo, the product which is either {1-[(1 R)-2-[(3R)-3-benzyl-3-(N, N’, N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1-methylethyl}carbamic acid tert-butyl ester or {1-[(1R)-2-[(3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1 H-indol-3- ylmethyl)-2-oxo-ethylcarbamoyl]-1-methylethyl}carbamic acid tert-butyl ester 33,9 g as an orange foam was obtained.HPLC (h8): Rt = 14,05 min.Step j2-Amino-N-r(1 R)-2-f(3R)-3-benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vπ-1- (1 H-indol-3-ylmethyl)-2-oxoethyll-2-methylpropionamide, fumarate or2-Amino-N-r(1 R)-2-r(3S)-3-benzyl-3-(N1N’1N’-trimethylhvdrazinocarbonyl)piperidin-1-yll-1- (1H-indol-3-ylmethyl)-2-oxoethvπ-2-methylpropionamide, fumarate
The product from step i which is either {1-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1- methylethyl}carbamic acid tert-butyl ester or {1-[(1 R)-2-[(3S)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1 -yl]-1 -(1 H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1 – methylethyljcarbamic acid tert-butyl ester (23,8 g, 36,8 mmol) was dissolved in of EtOAc (800 ml) (clear yellow solution) in a one neck round-bottom flask (1L) equipped with magnetic stirring. The flask was then placed in a water-bath (temp: 10-20 °C), and HCI-gas was passed through the solution for 5 min. (dust-like precipitation). After stirring for 1 hour (precipitation of large amount of yellow powder), the solution was flushed with N2 to remove excess of HCI. The precipitate was removed by gentle filtration and dried under vacuum at 40 °C overnight.The non-crystallinic precipitate was dissolved in H2O (500 ml) and washed with EtOAc (100 ml). Then CH2CI2 (1000 ml) and solid Na2CO3 was added until pH > 7. The 2 layers were separated, and the aqueous layer was e-extracted with CH2CI2 (200 ml). The combined organic layers were washed with brine (100 ml), dried over MgSO4 and filtered. The solvent was evaporated under reduced pressure and redissolved in EtOAc (500 ml) in a one neck round-bottom flask (1 L) equipped with magnetic stirring. A suspension of fumaric acid (3,67 g) in isopropanol (20 ml) and EtOAc (50 ml) was slowly added (5 min.), which resulted in precipitation of a white crystallinic salt. After 1 hour the precipitation was isolated by filtration and dried overnight in vacuum at 40 °C to give the fumarate salt of the compound which is either 2-amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1- yl]-1-(1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide or 2-amino-N-[(1 R)-2-[(3S)-3- benzyl-3-(N,N,,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1 H-indol-3-ylmethyl)-2- oxoethyl]-2-methylpropionamide (13,9 g) as a white powder.HPLC (A1): Rt = 33,61 min.HPLC (B1): Rt = 34,62 min. LC-MS: Rt = 5,09 min. (m+1) = 547,4 ClaimsHide Dependent 1. The compound obtainable by the procedure as described in example 1 , or a pharmaceutically acceptable salt thereof.2. The compound obtainable by the procedure as described in example 1 , and which compound is2-Amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide
or a pharmaceutically acceptable salt thereof.3. A pharmaceutical composition comprising, as an active ingredient, a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof together with a pharmaceutically acceptable carrier or diluent.4. A pharmaceutical composition according to claim 3 for stimulating the release of growth hormone from the pituitary.5. A pharmaceutical composition according to claim 3 or claim 4 for administration to animals to increase their rate and extent of growth, to increase their milk and wool production, or for the treatment of ailments.6. A method of stimulating the release of growth hormone from the pituitary of a mammal, the method comprising administering to said mammal an effective amount of a compound according to any one of claims 1 or 2 or a pharmaceutically acceptable salt thereof, or of a composition according to any one of claims 3 – 5.7. A method of increasing the rate and extent of growth, the milk and wool production, or for the treatment of ailments, the method comprising administering to a subject in need thereof an effective amount of a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof, or of a composition according to any one of claims 3-5.8. Use of a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof for the preparation of a medicament.9. Use according to claim 8 wherein the medicament is for stimulating the release of growth hormone from the pituitary of a mammal.
Growth hormone is a major participant in the control of several complex physiologic processes, including growth and metabolism. Growth hormone is known to have a number of effects on metabolic processes, e.g., stimulation of protein synthesis and free fatty acid mobilization and to cause a switch in energy metabolism from carbohydrate to fatty acid metabolism. Deficiency in growth hormone can result in a number of severe medical disorders, e.g., dwarfism.
The release of growth hormone from the pituitary is controlled, directly or indirectly, by number of hormones and neurotransmitters. Growth hormone release can be stimulated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin. In both cases the hormones are released from the hypothalamus but their action is mediated primarily via specific receptors located in the pituitary. Other compounds which stimulate the release of growth hormone from the pituitary have also been described. For example, arginine, L-3,4-dihydroxyphenylalanine (1-Dopa), glucagon, vasopressin, PACAP (pituitary adenylyl cyclase activating peptide), muscarinic receptor agonists and a synthetic hexapeptide, GHRP (growth hormone releasing peptide) release endogenous growth hormone either by a direct effect on the pituitary or by affecting the release of GHRH and/or somatostatin from the hypothalamus.
The use of certain compounds for increasing the levels of growth hormone in mammals has previously been proposed. For example, U.S. Pat. Nos. 6,303,620 and 6,576,648 (the entire contents of which are incorporated herein by reference), disclose a compound: (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide, having the following chemical structure:
(MOL)(CDX) which acts directly on the pituitary cells under normal experimental conditions in vitro to release growth hormone therefrom. This compound is also known under the generic name “anamorelin.” This growth hormone releasing compound can be utilized in vitro as a unique research tool for understanding, inter alia, how growth hormone secretion is regulated at the pituitary level. Moreover, this growth hormone releasing compound can also be administered in vivo to a mammal to increase endogenous growth hormone release.
Example 1
Crystallization of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide form A
0.0103 g of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide was dissolved in methanol (0.1 mL) in a glass vial. The glass vial was then covered with PARAFILM® (thermoplastic film) which was perforated with a single hole. The solvent was then allowed to evaporate under ambient conditions. An X-ray diffraction pattern showed the compound was crystalline ( FIG. 1).
PATENT
WO 2017067438
https://patents.google.com/patent/WO2017067438A1/enAnamorelin, whose chemical name is: (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2- Trimethylformylhydrazide is a compound that increases mammalian growth hormone levels and has a compound structure as shown in Formula I:
Cancer cachexia is a state of consumption in which patients lose a lot of weight and muscle mass. It is necessary for the treatment of cachexia because it weakens the patient, affects the quality of life and interferes with the patient’s treatment plan. The drug alamorelin produces the same effect as the so-called “starved hormone” ghrelin, which stimulates hunger. Alamolin is a mimetic of ghrelin, which is secreted by the stomach and is a ligand for growth hormone receptors. . Alamolin binds to this receptor, causing the release of growth hormone, causing a metabolic cascade that affects a variety of different factors, including fat-removing body weight, as well as blood sugar metabolism. Therefore, alamorelin can also enhance the appetite of patients and help patients stay healthy. The 2014 European Society of Medical Oncology (ESMO) in Madrid, Spain, announced that Alamolin is expected to be the first drug in history to effectively improve cancer cachexia.Alamolin is a drug developed by Helsinn Therapeutics (Switzerland) from Novo Nordisk for the development of a cachexia and anorexia for patients with cancer, including non-small cell lung cancer. It can also be used to treat hip fractures and preventive diseases. The strength of the elderly and the elderly has continued to decline. In two key, 12-week Phase III clinical trials (ROMANA 1, ROMANA 2), alamorelin can significantly increase the body fat loss, and is generally tolerated; the incidence of serious adverse drug reactions is less than 3%, mainly related to hyperglycemia and diabetes. Compared with the placebo group, alamorelin continued to increase body weight and improve cancer anorexia-cachexia-related symptoms and concerns; however, there was no significant difference in the improvement of grip strength between the alamolin group and the placebo group. Therefore, this product has excellent clinical value and market value.The polymorphic form of the drug free base and its preparation are reported as follows:Synthesis of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylmethyl is disclosed in the patent ZL99806010.0 A method for synthesizing hydrazide, and using [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylmethylcarbonyl)piperidin-1-yl tert-Butyl ester of 1-((1H-indol-3-yl)methyl)-2-oxoethyl]carbamate is dissolved in dichloromethane, then trifluoroacetic acid is added to remove tert-butyl formate After the base, the mixture was concentrated to remove the solvent, and then the product was extracted with dichloromethane, and the obtained extract was concentrated to dryness to give (3R)-1-(2-methylalanyl-D-color ammonia as an amorphous powder. Acyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide.Patent ZL00815145.8 discloses the synthesis of alamorelin and its compounds as pharmaceutically acceptable salts, relating to novel diastereomeric compounds, pharmaceutically acceptable salts thereof, compositions containing them and their use in therapy Lack of use of medical conditions caused by growth hormone. Synthesis of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformyl is disclosed in this patent. The synthesis method of hydrazine, and using [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylmethylcarbonylcarbonyl)piperidin-1-yl] 1-((1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was dissolved in ethyl acetate, and then hydrogen chloride gas was passed to remove the tert-butyl formate protection group. , the solid is dissolved in water, and then the pH is adjusted to about 7 with sodium carbonate, and the product is extracted with dichloromethane; the extract phase is concentrated to obtain (3R)-1-(2-methylalanyl-D-tryptophan). -3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide.Patent WO2006016995 discloses (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylmethyl as a medicament Crystalline polymorphs of hydrazides, methods of producing and separating these polymorphs, and pharmaceutical compositions and drug therapies containing these polymorphs, the crystalline polymorphs for direct application to the pituitary Gland cells release the growth hormone. This patent discloses (4R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone 4 Crystal form: Form A, Form B, Form C and Form D. The patent also provides the preparation of 3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone. The method of crystal form, especially the preparation method of Form C, in which the method of removing the tert-butyl formate protecting group of methanesulfonic acid in methanol is utilized without exception. As a well-known cause in the art, clinical studies have found that mesylate is genotoxic, and its DNA alkylation leads to mutagenic effects, in which methyl methanesulfonate and ethyl methanesulfonate have been reported. (eg document EMEA/44714/2008). The invention adopts hydrochloric acid or hydrogen chloride gas to remove the tert-butyl formate protecting group, avoids the method of removing methanesulfonic acid, thereby avoiding the risk of the genotoxic impurities in the process, and increasing the risk. The safety of the drug.(3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-three prepared by the patent ZL99806010.0 and the patent ZL00815145.8 Methyl formyl hydrazide, no data on the purity of its compounds, we found that (3R)-1-(2-methylalanyl-D-tryptophan)-3 was prepared by this method. -Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide does not help to remove the impurities produced, and the purity of the obtained product is not high, and it is difficult to meet the medicinal requirements. And (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethyl obtained by the preparation method of the present invention. The crystal form of the formyl hydrazide has a purity of 99.8% and a single impurity of less than 0.1%, which fully meets the requirements for medicinal purity. Moreover, the crystal form is stable to conditions such as pressure, temperature, humidity and illumination, and the preparation method is simple in operation and suitable for industrial production.Example 1:300 g of [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylcarbamidocarbonyl)piperidin-1-yl]-1-(( 1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was added to the reaction flask, and then 4 L of dichloromethane was added to the reaction flask, and the raw material was completely dissolved by stirring.Then, the reaction system is cooled to 10 ° C or lower in an ice bath, hydrogen chloride gas is continuously supplied to the reaction liquid, and solids are gradually precipitated, and the reaction is further maintained at about 10 ° C for 3 to 5 hours, and the sample is detected. After the reaction of the raw materials is completed, the reaction system is completed. 1.5 L of water was added thereto, the solid was completely dissolved, and then the pH was adjusted to about 8 with a 20% aqueous sodium hydroxide solution, and the layers were separated; the aqueous phase was extracted once more with dichloromethane, and the organic phases were combined.The organic phase was dried over anhydrous sodium sulfate for 3 hrs, filtered, and then evaporated to ethylamine 3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude 246 g, yield 97.2%. HPLC content (area normalization method) was 96.1%.Example 2:300 g of [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylcarbamidocarbonyl)piperidin-1-yl]-1-(( 1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was added to the reaction flask, 36% concentrated hydrochloric acid was added to the reaction flask, and the reaction system was heated to 40 with stirring. The reaction was carried out at ° C to 50 for 3 hours.Then, the sample is detected. After the reaction of the raw material is completed, the reaction system is cooled to 10 or less, and 2.0 L of dichloromethane is added to the reaction system, and then the pH is adjusted to about 8 with a 20% aqueous sodium hydroxide solution, and the aqueous phase is further separated. It was extracted once with dichloromethane and the organic phases were combined.The organic phase was dried over anhydrous sodium sulfate for 3 hrs, filtered, and then evaporated to ethylamine 3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude 248 g, yield 98%. HPLC content (area normalization method) was 96.2%.Preparation of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone E crystal formExample 3Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10g was added to the reaction flask and 30 ml of N- was added.Methylpyrrolidone, stirred and dissolved completely. Then, 60 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 60 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 20 ° C, filtered, and the filter cake was washed with a mixture of N-methylpyrrolidone / H 2 O; the cake was vacuum dried at about 55 ° C to obtain (3R)-1-(2-methylalanyl) -D-tryptophan)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.5 g), HPLC content (area normalization) 99.72%. The XRD pattern is shown in Fig. 1, the DSC chart is shown in Fig. 2, and the TGA pattern is shown in Fig. 3, where the crystal form is defined as the E crystal form. The DSC of the crystal form has an endotherm at 120.05, the TGA is heated at 60A, and the crystal loss of 5 is about 3.1%. Combined with the Karl Fischer method, the moisture content of the product is determined. 3.1% and 3.2% indicate that the sample is present as a monohydrate.Example 4:Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 30 ml of N,N-dimethylformamide was added, stirred, and dissolved completely. Then, 30 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 50 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 h.Slowly cool to below 10 ° C, filter, filter cake washed with N, N-dimethylformamide / H 2 O mixture; vacuum cake dried at around 55 ° C to obtain (3R)-1-(2-A Alanyl-D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.5 g), HPLC content (area normalization) ) 99.87%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 5:Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 30 ml of dimethyl sulfoxide was added, stirred, and dissolved completely. Then, 40 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 60 ° C, the solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 10 ° C, filtered, and the filter cake was washed with a mixture of dimethyl sulfoxide / H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2-methylalanyl) -D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.1 g), HPLC content (area normalization) 99.61%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 6Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 40 ml of 1,4-dioxane was added, stirred, and dissolved completely. Then, 50 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 70 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 10 ° C, filtered, and the filter cake was washed with a mixture of 1,4-dioxane/H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2-methyl alanyl-D-tryptophan-3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.7 g), HPLC content (area normalization) 99.11%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 7Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 40 ml of N,N-dimethylacetamide was added, stirred, and dissolved completely. Then, 40 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 70 ° C. The solution became cloudy, and was slowly cooled to about 50 ° C. Seed crystals were added thereto, and cooling was continued to gradually precipitate a solid.The reaction system was cooled to about 10 ° C, filtered, and the filter cake was washed with a mixture of N,N-dimethylacetamide/H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2- Methylalanyl-D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.1 g), HPLC content (area normalized) Law) 99.78%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 7Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 50 ml of acetone was added, stirred, and dissolved completely. Then, 70 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 45 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cool to below 10 ° C, filter, filter cake washed with acetone / H 2 O mixture; filter cake vacuum dried at around 50 ° C to obtain (3R)-1-(2-methylalanyl-D-color Aminoacyl-3-phenylmethyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.3 g), HPLC content (area normalization) 98.9%. Upon comparison, it was confirmed that the solid was in the E crystal form.
SYN
Reference:
1. Org. Process Res. Dev.2006, 10, 339–345.
Abstract
The rapid process development of a scaleable synthesis of the pseudotripeptide RC-1291 for preclinical and clinical evaluation is described. By employing a nontraditional N-to-C coupling strategy, the peptide chain of RC-1291 was assembled in high yield, with minimal racemization and in an economical manner by introducing the most expensive component last. A one-pot deprotection/crystallization procedure was developed for the isolation of RC-1291 free base, which afforded the target compound in excellent yield and with a purity of >99.5% without chromatographic purification.
(R,R)-2-Amino-N-[2-[3-benzyl-3-(N,N′,N′-trimethyl-hydrazinocarbonyl)piperidin-1-yl]-1-(1H-indol-3-ylmethyl)- 2-oxo-ethyl]-2-methyl-propionamide (1). Crude 7 (911 g; 1.28 mol theoretical)10 was dissolved in methanol (4.12 L) in a 22-L round-bottom flask equipped with a mechanical stirrer, a temperature probe, a reflux condenser, a gas (N2) inlet, and an addition funnel. The solution was heated to 55 °C; then methanesulfonic acid (269.5 g, 2.805 mol) was added over a period of 15 min. (Caution: gas evolution!) The solution was then heated to 60 °C for a period of 1 h, after which HPLC analysis showed that no 7 remained. The temperature of the reaction mixture was increased to reflux (68-72 °C) over a period of 35 min, while simultaneously adding a solution of KOH (85%, 210.4 g, 3.187 mol) in water (4.12 L). The clear, slightly yellow solution was then allowed to cool to 20 °C at a rate of 5 °C/h. The free base of RC1291 (1) crystallized as a pale-yellow solid, which was isolated by filtration. The filter cake was washed with two portions of 50% aqueous methanol (500 mL each) and then dried under high vacuum at 20 ( 5 °C to afford 1 as an off-white, crystalline solid (595 g, 85% yield for two steps, >99.5% AUC by HPLC).
HRMS (ESI) calcd for C31H43N6O3 [M + H]+ 547.3397, found 547.3432.
Cachexia and muscle wasting are very common among patients suffering from cancer, chronic obstructive pulmonary disease, and other chronic diseases. Ghrelin stimulates growth hormone secretion via the ghrelin receptor, which subsequently leads to increase of IGF-1 plasma levels. The activation of the GH/IGF-1 axis leads to an increase of muscle mass and functional capacity. Ghrelin further acts on inflammation, appetite, and adipogenesis and for this reason was considered an important target to address catabolic conditions. We report the synthesis and properties of an indane based series of ghrelin receptor full agonists; they have been shown to generate a sustained increase of IGF-1 levels in dog and have been thoroughly investigated with respect to their functional activity.
Patent
https://patents.google.com/patent/EP2838892A1/enGrowth hormone is a major participant in the control of several complex physiologic processes including growth and metabolism. Growth hormone is known to have a number of effects on metabolic processes such as stimulating protein synthesis and mobilizing free fatty acids, and causing a switch in energy metabolism from carbohydrate to fatty acid metabolism. Deficiencies in growth hormone can result in dwarfism and other severe medical disorders.The release of growth hormone from the pituitary gland is controlled directly and indirectly by a number of hormones and neurotransmitters. Growth hormone release can be stimulated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin.The use of certain compounds to increase levels of growth hormone in mammals has previously been proposed. Anamorelin is one such compound. Anamorelin is a synthetic orally active compound originally synthesized in the 1990s as a growth hormone secretogogue for the treatment of cancer related cachexia. The free base of anamorelin is chemically defined as:® (3R) 1 -(2-methylaIanyl~D ryptophyl)~3-(phenylraethyl)~3~piperidineearboxylie acid 1 ,2,2trimethyihydrazide,* 3-{(2R)-3-{(3R)-3-benzyi-3-| (trimethylhydrazino)carbonyi]piperidin-l»yl}-2-[(2»met hylaianyl)amino]-3-ox.opropyi}-IH-indole, or• 2-Amino-N-[(lR)-2-[(3R)-3~benzyWcarbony piperidin- 1 -yl] – 1-( 1 H-indol-3 -yl^^and has the below chemical structure; U.S. Patent No. 6,576,648 to Artkerson reports a process of preparing anamorelin as the fumarate salt, with the hydrochloride salt produced as an intermediate in Step (j) of Example 1 . U.S. Patent No. 7,825, 138 to Lorimer describes a process for preparing crystal forms of the free base of anamorelin.There is a need to develop anamorelin monohydrochloride as an active pharmaceutical ingredient with reduced impurities and improved stability over prior art forms of anamorelin hydrochloride, such as those described in U.S. Patent No, 6,576,648, having good solubility, bioavailability and processabi!ity. There is also a need to develop methods of producing pharmaceutically acceptable forms of anamorelin monohydrochloride thai have improved yield over prior art processes, reduced residual solvents, and controlled distribution of chloride content,it has unexpectedly been discovered that the process of making the hydrochloride salt of anamorelin described in Step (j) of U.S. Patent No. 6.576,648 can result in excessive levels of chloride in the final product, and that this excess chloride leads to the long-term instability of the final product due at least, partially to an increase in the amount of the less stable dihydrochloride salt of anamorelin. Conversely, because anamorelin free base is less soluble in water than the hydrochloride salt, deficient chloride content in the final product can lead to decreased solubility of the molecule. The process described in U.S. Patent No, 6,576,648 also yields a final product that contains more than 5000 ppm (0.5%) of residual solvents, which renders the product less desirable from a pharmaceutical standpoint, as described in CH Harmonized Tripartite Guideline. See Impurities; Guideline for residual solvents Q3C(R3). in order to overcome these problems, methods have been developed which, for the first time, allow for the efficient and precise control of the reaction between anarnorehn tree base and hydrochloric acid in situ, thereby increasing the yield of anarnorehn monohydrochioride from the reaction and reducing the incidence of unwanted anamorelin dihydroeh ride. According to the method, the free base of anamorelin is dissolved in an organic solvent and combined with water and hydrochloric acid, with the molar ratio of anarnorehn and chloride tightly controlled to prevent an excess of chloride in the final product. The water and hydrochloric acid can be added either sequentially or at the same time as long as two separate phases are formed. Without wishing to be bound by any theory, it is believed thai as the anamorelin free base in the organic phase is protonated by the hydrochloric acid it migrates into the aqueous phase. The controlled ratio of anamorelin free base and hydrochloric acid and homogenous distribution in the aqueous phase allows for the controlled formation of the monohydrochioride salt over the dihydrochloride, and the controlled distribution of the resulting chloride levels within individual batches and among multiple batches of anamorelin monohydrochioride.Thus, in a fust embodiment the invention provides methods for preparing anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride comprising: (a) dissolving anamorelin free base in an organic solvent to form a solution; (b) mixing said solution with water and hydrochloric acid for a time sufficient to: (i) react said anamorelin free base with said hydrochloric acid, and (ii) form an organic phase and an aqueous phase; (c) separating the aqueous phase from the organic phase; and (d) isolating anamorelin monohydrochioride from the aqueous phase.In a particularly preferred embodiment, the molar ratio of anamorelin to hydrochloric acid used in the process is less than or equal to 1 : 1 , so as to reduce the production of anamorelin dihydrochloride and other unwanted chemical species. Thus, for example, hydrochloric acid can be added at a molar ratio of from 0,90 to 1 ,0 relative to said anamorelin, from 0.90 to 0.99, or from 0.93 to 0.97.n another particularly preferred embodiment, the anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride is isolated from the aqueous phase via spray drying, preferably preceded by distillation. This technique has proven especially useful in the manufacture of anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride because of the excellent reduction in solvent levels observed, and the production of a stable amorphous form of anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride. In other embodiments, the invention relates to the various forms of anamorelin monohvdrochloride and compositions comprising anamorelin monohvdrochloride produced by the methods of the present invention. In a first embodiment, which derives from the controlled chloride content among batches accomplished by the present methods, the invention provides anamorelin monohvdrochloride or a composition comprising anamorelin monohydrochloride having an inter-batch chloride content of from 5.8 to 6.2%, preferably from 5.8 to less than 6.2%. Alternatively, the invention provides anamorelin monohydrochloride or a composition comprising anamorelin monohydrochloride having a molar ratio of chloride to anamorelin less than or equal to 1 : 1 , such as from 0.9 to 1.0 or 0.99, in yet another embodiment the invention provides an amorphous form of anamorelin monohydrochloride or a composition comprising anamorelin monohydrochloride. Further descriptions of the anamorelin monohydrochloride and compositions comprising the anamorelin monohydrochloride are given in the detailed description which follows.EXAMPLE 1 . PREPARATION OF ANAMOREUN HYDROCHLORIDEVarious methods have been developed to prepare the hydrochloric acid salt of anarnorelin, with differing results.In a first method, which is the preferred method of the present invention, anarnorelin free base was carefully measured and dissolved in isopropyl acetate. Anarnorelin free base was prepared according to known method (e.g., U.S. Patent No, 6,576,648). A fixed volume of HCl in water containing various molar ratios (0.80, 0,95, 1.00 or 1.05) of HCl relative to the anarnorelin free base was then combined with the anamorelin/isopropyl acetate solution, to form a mixture having an organic and an aqueous phase, The aqueous phase of the mixture was separated from the organic phase and the resulting aqueous phase was concentrated by spray drying to obtain the batches of anarnorelin monohydrochloride (or a composition comprising anarnorelin monohydrochloride ) shown in Table 1 A.Approximately 150mg of the resulting spray dried sample of anarnorelin monohydrochloride (or composition comprising anarnorelin monohydrochloride) was accurately weighed out and dissolved in methanol (50mL). Acetic acid (5mL) and distilled water (5mL) were added to the mixture. The resulting mixture was potentiometricaJ ly titrated using 0,0 IN silver nitrate and the e dpoint was determined. A blank determination was also performed and correction was made, if necessary. The chloride content in the sample was calculated by the following formula. This measurement method of chloride content was performed without any cations other than proton (! ! ‘ ).Chloride content (%) = VxNx35.453x l 00x l 00/{Wx[1 00-(water content (%))-(residual solvent (%))]}V: volume at the endpoint (ml.)N; actual normality of 0.01 mol/L silver nitrate35.453 : atomic weight of ChlorineW: weight of sample (mg)TABLE 1 AHCl Chloride ContentThis data showed that anamorelin monohydrochlonde produced by a fixed volume of HCl in water containing 0.80 or 1 .05 molar equivalents of HC1 relative to anamorelin free base had levels of chloride thai were undesirable, and associated with product instability as shown in Example 3.Alternatively, a fixed volume of HCl in water containing 0.95 moles of HCl relative to anamorelin free base was used to prepare anamorelin monohydrochlonde (or composition comprising anamorelin monohydrochloride) as follows. Anamorelin free base (18.8g, 34.4mmoi) and isopropyl acetate (341.8g) were mixed in a 1000 mL flask. The mixture was heated at 40±5°C to confirm dissolution of the crystals and then cooled at 25±5°C. Distilled water (22.3g) and 3.6% diluted hydrochloric acid (33. Ig, 32.7mmoL 0.95 equivalents) were added into the flask and washed with distilled water. After 30 minutes stirring, the reaction was static for more than 15 minutes and the lower layer (aqueous layer) was transferred into a separate 250mL flask. Distilled water was added to the flask and concentrated under pressure at 50i5cC. The resulting aqueous solution was then filtered and product isolated by spray drying to afford anamorelin monohydrochlonde A (the present invention).The physical properties of anamorelin monohydrochloride A were compared to anamorelin monohydrochloride produced by a traditional comparative method (“anamorelin monohydrochloride B”) (comparative example). Anamorelin mono hydrochloride B in the comparative example was produced by bubbling HCl gas into isopropyl acetate to produce a 2M solution of HCl, and reacting 0.95 molar equivalents of the 2M HCl in isopropyl acetate with anamorelin free base. The physical properties of anamorelin monohydrochloride B are reported in Table IB. This data shows that when 0.95 equivalents of HCl is added to anamorelin free base, the chloride content (or amount of anamorelin dihydrochloride) is increased, even when a stoichiometric ratio of hydrochloride to anamorelin of less than 1 ,0 is used, possibly due to uncontrolled precipitation. In addition, this data shows that the concentration of residual solvents in anamorelin monohydrochloride B was greater than the concentration in anamorelin monohydrochloride A, TABLE I B A similar decrease in residual solvent concentration was observed when 2-methyltetrahydrofuran was used as the dissolving solvent for anamorelin free base instead of isopropvi acetate in the process for preparing spray dried anamorelin monohydrochloride A (data not reported).The residual solvent (organic volatile impurities) concentration (specifically isopropyl acetate) of anamorelin monohydrochloride in TABLE IB was measured using gas chromatography (GC-2010, Shimadzu Corporation) according to the conditions shown in TABLE 1 C,
^ Currow DC, Abernethy AP (April 2014). “Anamorelin hydrochloride in the treatment of cancer anorexia-cachexia syndrome”. Future Oncology. 10 (5): 789–802. doi:10.2217/fon.14.14. PMID24472001.
^ Jump up to:abc Garcia JM, Polvino WJ (June 2009). “Pharmacodynamic hormonal effects of anamorelin, a novel oral ghrelin mimetic and growth hormone secretagogue in healthy volunteers”. Growth Hormone & IGF Research. 19 (3): 267–73. doi:10.1016/j.ghir.2008.12.003. PMID19196529.
^ Jump up to:ab Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, Friend J (January 2015). “Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials”. The Lancet. Oncology. 16 (1): 108–16. doi:10.1016/S1470-2045(14)71154-4. PMID25524795.
^ Jump up to:ab Garcia JM, Friend J, Allen S (January 2013). “Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study”. Supportive Care in Cancer. 21 (1): 129–37. doi:10.1007/s00520-012-1500-1. PMID22699302. S2CID22853697.
^ Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, Fearon KC (April 2016). “Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials”. The Lancet. Oncology. 17 (4): 519–531. doi:10.1016/S1470-2045(15)00558-6. PMID26906526.
Olanexidine gluconate was approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jul 03, 2015. It was developed and marketed as Olanedine® by Otsuka in Japan.
Olanexidine gluconate is an antiseptic/disinfectant compound with potent bactericidal activity against Gram-negative and Gram-positive bacteria, for use in preparing patients for surgery and preventing of postoperative bacterial infections.
Olanedine® is available as topical solution (1.5%), containing 3 g/200 mL, 0.15 g/10 mL and 0.375 g/25 mL, and the recommendation is applying appropriate amount of the drug.
A 7.00-kg quantity of Compound (4) (54.16 mol) was dissolved in 105 liters of ethyl acetate, and the resulting mixture was cooled to 5°C or below. A 2.66-kg quantity of concentrated sulfuric acid (27.12 mol) was added thereto dropwise at a temperature of 4O0C or below while stirring. To the thus- obtained suspension of 1/2 sulfate of Compound (4) was added 5.06 kg of sodium dicyanamide (56.83 mol), and the resulting suspension was heated under reflux for 7 hours. The reaction solution was cooled to 400C or below, and 70 liters of water was added thereto. Subsequently, the resulting solution was heated to 80 to 900C (internal temperature) to distill the ethyl acetate off. The remaining liquid was cooled to 400C or below, and 70 liters of toluene was then added thereto, followed by the extraction of 1-cyano — 3-n-octyl guanidine at about 500C. The extracted toluene layer was washed with 35 liters of water at about 500C and cooled to 100C or below, followed by stirring for about 30 minutes. The resulting precipitated crystals were separated and washed with 7 liters of toluene. The resulting crystals were dried at 400C for 7.5 hours, yielding l-cyano-3-n- octylguanidine. 2007/067107
-16-
Yield: 9.11 kg (The yield was 85.7% based on the Compound(4).) White crystals having a melting point of 69 to 740C (no clear melting point was observed)
Reference Example 2: Acidolysis of 1- (3,4-dichlorobenzyl) -5- octylbiguanide dihydrochloride
A 1-g quantity of 1- (3, 4-dichlorobenzyl) -5-octyl biguanide dihydrochloride was dissolved in 15 ml of 10% ethanol, followed by refluxing for 5 hours. HPLC analysis was conducted under the conditions described below.
The yield of 1-[N- (3,4-dichlorobenzyl) carbamoyl-3- octyl]guanidine (holding time: 9.84 minutes) was 0.91%, and the yield of 1- (N-octyl-carbamoyl) -3- (3, 4-dichlorobenzyl) guanidine
(holding time: 10.54 minutes) was 0.22%.
HPLC analysis conditions:
Column: YMC AM302 4.6 mm I. D. x 150 mm
Eluate: MeCN/0.05 M aqueous solution of sodium 1- octanesulfonate/acetic acid = 700/300/1
Detector: UV 254 nm
The physical property values of the resulting 1-[N- (3,4- dichlorobenzyl) carbamoyl-3-octyl] guanidine were as follows: NMR (DMSO-de) δ: 0.86 (3H, t, J = 6.0 Hz), 1.07-1.35 (1OH, m) , 1.35-1.49 (2H, m) , 2.95-3.15 (2H, m) , 4.12 (2H, d, J = 6.3 Hz), 6.78-7.40 (4H, m) , 7.23 (IH, dd, J = 2.1 Hz, J = 8.4 Hz), 7.46 (IH, d, J = 2.1 Hz), 7.54 (IH, d, J = 8.4 Hz)
The physical property values of the resulting 1- (N-octyl- carbamoyl) -3- (3, 4-dichlorobenzyl) guanidine were as follows: NMR (DMSO-d6) δ: 0.85 (3H, t, J = 6.6 Hz), 1.02-1.40 (12H, m) , 2.89-2.95 (2H, m) , 4.33 (2H, bs) , 5.76-7.00 (4H, m) , 7.28 (IH, dd, J = 2.1 Hz, J = 8.1 Hz), 7.52 (IH, d, J = 2.1 Hz), 7.58 (IH, d, J = 8.1 Hz)
Example 1: 1- (3, 4-dichlorobenzyl) -5-octylbiguanide monohydrochloride 1/2 hydrate
A 9.82-g quantity of Compound (2) (0.05 mol) and 10.63 g of 3, 4-dichlorobenzylamine (0.05 mol) were added to 49 ml of butyl acetate, followed by refluxing for 6 hours. The reaction solution was concentrated under reduced pressure, and a mixture of 12 ml of water and 47 ml of isopropyl alcohol was added and dissolved into the remainder. To the thus-obtained solution was added, dropwise, 10.13 g of concentrated hydrochloric acid. The resulting mixture was stirred at 28 to 300C for 30 minutes, and the precipitated crystals were then filtered out. The thus- obtained crystals were washed with a small amount of isopropyl alcohol, yielding 23.42 g of (non-dried) 1- (3, 4-dichlorobenzyl) – 5-octylbiguanide dihydrochloride. The resulting crystals were suspended in 167 ml of water without drying, the suspension was then stirred at 25 to 27°C for 2 hours, followed by separation of the crystals by filtration. The thus-obtained crystals were washed with a small amount of water and dried at 400C for 20 hours, yielding 17.05 g of 1- (3, 4-dichlorobenzyl) -5-octyl biguanide monohydrochloride 1/2 hydrate having a purity of 99.9% at a yield of 81.6%.
Example 2 : 1- (3, 4-dichlorobenzyl) -5-octylbiguanide dihydrochloride
A 100-g quantity of Compound (4) (0.774 mol) was dissolved in 1 liter of n-butyl acetate, and 37.6 g of concentrated sulfuric acid (0.383 mol) was added thereto while stirring. To the thus-obtained suspension of 1/2 sulfate of Compound (4) was added 68.9 g of sodium dicyanamide (0.774 mol), 7107
-18- and the resulting suspension was heated under reflux for 3 hours. The reaction solution was cooled to about 200C, and the organic layer thereof was sequentially washed with about 500 ml each of (i) 5% hydrochloric acid, (ii) 5% aqueous caustic soda solution, (iii) 5% aqueous sodium bicarbonate solution, and (iv) water.
To the thus-obtained n-butyl acetate solution of Compound (2) were added 118.5 g of Compound (3) (0.673 mol) and then 58.4 ml of concentrated hydrochloric acid while stirring. The reaction solution was heated, and about 800 ml of n-butyl acetate was distilled off under atmospheric pressure (ordinary pressure) , followed by heating the reaction solution under reflux for 3.5 hours . Subsequently, the reaction solution was cooled to about 400C, and 900 ml of isopropanol, 100 ml of water, and 134 ml of concentrated hydrochloric acid were added thereto. The mixture was stirred at 60 to 70°C for 1 hour and cooled to 100C or below and the precipitated crystals were then separated. The resulting crystals were washed with 200 ml of isopropanol and dried at 6O0C, yielding 1- (3, 4-dichlorobenzyl) -5-octylbiguanide dihydrochloride. Yield: 243.8 g (The yield was 81.3% based on the Compound (3).) Melting point: 228.90C IR(KBr) spectrum: 2920, 1682, 1634, 1337, 1035, 820, and 640 cm“1
Olanexidine is a compound with high bactericidal activity having the chemical name 1-(3,4-dichlorobenzyl)-5-octylbiguanide. Research has been carried out into bactericides containing, olanexidine hydrochloride as an active ingredient (see Japanese Patent No. 2662343, etc.).
Olanexidine has very poor solubility in water, and hitherto known salts of olanexidine are also poorly soluble in water. For example, the solubility at 0° C. of olanexidine hydrochloride in water has been measured to be less than 0.05% (W/V), and the solubility of free olanexidine is a further order of magnitude less than this. Consequently, sufficient bactericidal activity cannot be expected of an aqueous solution merely having olanexidine dissolved therein, and moreover, depending on the conditions the olanexidine may precipitate out.
In the case of making an aqueous preparation of olanexidine in particular, to make the concentration of the olanexidine sufficient for exhibiting effective bactericidal activity, and to reduce the possibility of the olanexidine precipitating out, it has thus been considered necessary to use a dissolution aid such as a surfactant.
EXAMPLE 1 Preparation of an Aqueous Solution Aqueous Solution 1
20.9 g (50 mmol) of olanexidine hydrochloride hemihydrate was added to 250 mL of a 1 N aqueous sodium hydroxide solution, and the suspension was stirred for 1.5 hours at room temperature (25° C.). The solid was filtered off, and washed with water. The solid obtained was further suspended in 250 mL of purified water, the suspension was stirred for 5 minutes at room temperature, and the solid was filtered off, and washed with water. This operation was carried out once more to remove sodium chloride formed. The solid obtained (free olanexidine) was put into purified water in which 8.9 g (50 mmol) of gluconolactone had been dissolved, and the mixture was stirred at room temperature until the solid dissolved, and then purified water was further added to give a total volume of 300 mL. The concentration of olanexidine in the aqueous solution obtained was measured by using high performance liquid chromatography to be 6% in terms of free olanexidine.
This aqueous solution was still transparent and colorless even after being left for several months at room temperature.
APPROVED JAPAN , 2011-01-21, Chugai (Originator) , Roche,Taisho Toyama
Eldecalcitol was approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on January 21, 2011. It was developed by Chugai Pharmaceutical (a member of Roche) and marketed as Edirol® by Chugai Pharmaceutical and Taisho.
Eldecalcitol is an orally active vitamin D analogue leading to greater absorption of bind calcium. It is usually used to treat osteoporosis.
Edirol® is available as capsule for oral use, containing 0.5 μg or 0.75 μg of free Eldecalcitol, and the recommended dose is 0.75 μg once daily.
ED-71, a vitamin D analog, is a more potent inhibitor of bone resorption than alfacalcidol in an estrogen-deficient rat model of osteoporosis. ED-71, effectively and safely increased lumbar and hip bone mineral density (BMD) in osteoporotic patients who also received vitamin D3 supplementation.
Eldecalcitol is a drug used in Japan for the treatment of osteoporosis.[1] It is an analog of vitamin D.[2] Osteoporosis is a common bone disease among the older generation, with an estimated prevalence of over 200 million people.[1] This condition often results in bone fractures due to abnormally low bone mass density, and is a leading cause of disability, especially among developed countries with longer average life spans. Osteoporosis is more common in women than with men.
Eldecalcitol
Discovery
Chugai Pharmaceutical/Roche are the originators of the medicinal drug eldecalcitol through Taisho Pharmaceutical Holdings and Chugai Pharmaceutical. The trade name of eldecalcitol is Edirol, and its Chemical Abstracts Service (CAS) registry number is 104121-92-8. Eldecalcitol was approved for use in Japan on January 2011. The approval came from the Japanese Ministry of Health, Labor, and Welfare for the objective of a treatment for osteoporosis.[3]
Effects
Clinical trials have suggested that eldecalcitol, a vitamin D analog, has strong effects to reduce calcium reabsorption into the body from bones, therefore increasing bone mineral density, and to increase calcium absorption in intestines.[4] In animals, eldecalcitol inhibits the activity of osteoclasts for the function to reduce bone degradation for calcium, while still able to maintain osteoblast function so as to not hinder bone formation.[5] Unlike other vitamin D analogs, eldecalcitol does not significantly suppress parathyroid hormone levels, promising a better treatment for osteoporosis in comparison to other medications.[6] Bone mineral density increases with eldecalcitol use, in addition to strengthening bone structure. This occurs due to the function of the eldecalcitol drug, which decreases bone reabsorption as observed through a bone reabsorption marker. Bone geometry assessments show that eldecalcitol increases cortical bone area in patients with osteoporosis more so than other vitamin D analogs, such as alfacalcidol. There was also the maintenance of thickness of cortical bone mass, strongly indicating that eldecalcitol improves the strength and mass of bone, specifically cortical bone structure.[7] Adverse effects of eldecalcitol include an increase in blood and urinary calcium levels. Abnormally high levels of calcium can lead to problems associated with hypercalcemia.
Treatment for Osteoporosis
Eldecalcitol can be used for the treatment of hypocalcaemia or osteoporosis. Calcium absorption increases with the presence of eldecalcitol by the body, occurring in the intestines, which is useful for those who have low calcium levels. Eldecalcitol is more often used due to its effects to treat osteoporosis. In the aging population, the bone matrix becomes weakened through untreated osteoporosis. This leads to an increased risk of severe fractures that include spinal and hip fractures in addition to vertebral and wrist fractures. This creates a burden on the health care system due to a decline in the quality of life for the individuals that suffer from this condition. Some risk factors leading to the predisposition of developing osteoporosis are previous incidents of bone fractures and a reduction in bone mineral density.[1] These factors expectantly increase as age increases. Bone health is reliant on maintaining physiologically needed levels of calcium, where the body constantly maintains this calcium homeostasis through osteoblast and osteoclast activity. Osteoblast activity serves this function of maintaining appropriate calcium levels by depositing calcium in bones when blood calcium levels are above normal. In contrast, osteoclasts break down bone tissue to increase blood calcium levels if they are low.[8] This activity is performed after absorption of calcium by the body, which requires the actions of vitamin D. The active metabolite of vitamin D, calcitriol, performs its function through interactions with the calcitriol receptor. This nuclear hormone receptor is responsible for calcium absorption which, in turn, is involving in bone depletion and formation. The new analogs of vitamin D, such as eldecalcitol, are observed to have stronger effects in preventing bone loss, fractures, and falls in comparison to calcitriol.[9] Eldecalcitol is even more effective than its counterpart alfacalcidol, another vitamin D analog. Studies have shown eldecalcitol is more effective than alfacalcidol in preventing vertebral and wrist fractures, and even falls, with osteoporotic patients with vitamin D insufficiencies.[10] Eldecalcitol is also more effective at preventing fractures than vitamin D and calcium supplements.[1] Eldecalcitol increases calcium absorption for vitamin D deficient patients, and therefore could be used for osteoporosis treatment for all age groups.
Pharmacology
Analogs of vitamin D are being explored intensely for their regulatory effects on calcium metabolism with the purpose of treating osteoporosis, a skeletal disease associated with low bone mass and deterioration of bone tissue. Vitamin D is imperative for absorption of calcium to maintain bone strength.
Mechanism of Action
Eldecalcitol is an orally administered drug to patients, which binds to vitamin D receptors and binding protein for the goal of achieving greater specificity to bind calcium for its absorption. This greater affinity is 2.7-fold that of the active vitamin D form of calcitriol. Eldecalcitol is readily absorbed into the body, with a long elimination half-life of over eight hours, reaching maximum absorption in 3.4 hours.[1]
Dosage
Eldecalcitol is present in the form of pills for oral administration. In preclinical models with healthy male volunteers, oral doses of eldecalcitol ranged from 0.1 to 1.0 micrograms once daily to show an increase in bone mineral density.[11] Preclinical trials show improvements for doses at 0.5 and 0.75 micrograms, which are the recommended dosage amounts for the Edirol product as approved by the Japanese Ministry of Health, Labor, and Welfare for treating osteoporosis.[3]
Chemistry
The class of eldecalcitol is a vitamin D3 derivative. This molecule has a molecular weight of 490.71 grams per mole. The eldecalcitol analog of calcitriol, contains a hydroxypropyl group in the lower cyclohexane ring. The synthesis of eldecalcitol incorporates two units assembled together. The IUPAC names include (3S, 4S, 5R)-oct-1-en-7-yne-3,4,5-triol that is fused to a bicyclic system, (R)-6-((1R, 3aR, 7aR, E)-4-(bromomethylene)-7a-methyloctahydro-1H-inden-1-yl)-2-methylheptan-2-ol. The assembly process includes a Diels-Alder reaction to give the fully protected eldecalcitol. In order to get the parent molecule, the hydroxyl groups have to be deprotected. The chemistry of eldecalcitol allows for its binding 2.7-fold more potently than calcitriol. In addition, some vitamin D derivatives have been known to inhibit the serum parathyroid hormone. Eldecalcitol only weakly inhibits the serum parathyroid hormone, making it an even more appealing medicinal drug for its physiological uses in the treatment of osteoporosis.[3] Animal studies of eldecalcitol, in ovariectomized rats, show improvements in bone mass while lowering bone reabsorption to demonstrate its effectiveness in osteoporosis treatment.[5]
■ Diverse and Important Contributions by Medicinal Chemists to the Development of Pharmaceuticals: An Example of Active Vitamin D3 Analog, Eldecalcitol
Noboru Kubodera*
*International Institute of Active Vitamin D Analogs, 35-6, Sankeidai, Mishima, Shizuoka 411-0017, Japan
Abstract
Presented herein are diverse and important contributions by medicinal chemists to different stages of pharmaceutical development. The conceptual elements reviewed, which are intended for young chemists who engage in drug discovery research, draw upon the author’s experience in developing eldecalcitol, an active vitamin D3 analog used to treat osteoporosis. The review covers exploratory research for a lead candidate compound; process development for practical manufacturing; and synthesis of other compounds relevant to the program, such as tritiated compounds, postulated metabolites, and miscellaneous analogs for mode of action studies.
PAPER
Eldecalcitol [1α,25-dihydroxy-2β-(3-hydroxypropoxy)vitamin D3], an analog of calcitriol (1α,25-dihydroxyvitamin D3), possesses a hydroxypropoxy substituent at the 2β-position of calcitriol. Eldecalcitol has potent biological effects on bone disease such as osteoporosis. The marketing of eldecalcitol has very recently started in Japan. In consideration of this, we have been investigating practical synthesis of eldecalcitol for industrial-scale production. Eldecalcitol was initially synthesized in a linear manner. The 27-step linear sequence was, however, suboptimal due to its lengthiness and low overall yield (ca. 0.03%). Next, we developed a convergent approach based on the Trost coupling reaction, in which the A-ring fragment (ene-yne part obtained in 10.4% overall yield) and the C/D-ring fragment (bromomethylene part obtained in 27.1% overall yield) are coupled to produce the triene system of eldecalcitol (15.6%). Although the overall yield of the convergent synthesis was better than that of the linear synthesis, significant improvements were still necessary. Therefore, additional biomimetic studies were investigated. Process development for the practical production of eldecalcitol is described herein.
Hatakeyama, S; Yoshino, M (2010). “Synthesis and preliminary biological evaluation of 20-epieldecalcitol [20-epi-1α,25-dihydroxy-2β-(3-hydroxypropoxy)vitamin D3: 20-epi-ED-71]”. The Journal of Steroid Biochemistry and Molecular Biology121 (1–2): 25–28.doi:10.1016/j.jsbmb.2010.03.041. PMID20304058.
Robichaud; Stamford; Weinstein; McAlpine; Primeau; Lowe; Bernstein; Bronson; Manoj, Desai (2012). Annual Reports in Medicinal Chemistry47 (1st ed.). San Diego: Elsevier Inc. pp. 529–531. ISBN9780123964922.
Nogachi, Y; Kawate, H; Nomura, M; Takayanagi, R (2013). “Eldecalcitol for the treatment of osteoporosis”. Europe PubMed Central8: 1313–1321. doi:10.2147/CIA.S49825.
Smith, S; Doyle, N; Boyer, M; Chouinard, L; Saito, H (2013). “Eldecalcitol, a vitamin D analog, reduces bone turnover and increases trabecular an cortical bone mass, density, and strength in ovariectomized cynomolgus monkeys”. Bone57 (1): 116–122.doi:10.1016/j.bone.2013.06.005. PMID23774444.
Harada, S; Uno, S; Takahashi, F; Saito, H (2010). “Eldecalcitol is less effective in suppressing parathyroid hormone compared to calcitriol in vivo“. The Journal of Steroid Biochemistry and Molecular Biology121 (1–2): 281–283.doi:10.1016/j.jsbmb.2010.04.001. PMID20398764.
Matsuo, K; Irie, N (2008). “Osteoclast-osteoblast communication”. Archives of Biochemistry and Biophysics473 (2): 201–209. doi:10.1016/j.abb.2008.03.027.PMID18406338.
Saito, H; Takeda, S; Amizuka, N (2013). “Eldecalcitol and calcitriol stimulates ‘bone minimodeling,’ focal bone formation without prior bone resorption, in rat trabecular bone”.The Journal of Steroid Biochemistry and Molecular Biology136 (1): 178–182.doi:10.1016/j.jsbmb.2012.10.004.
Matsumoto, T; Ito, M; Hayashi, Y; Hirota, T; Tanigawara, Y; Sone, T; Fukunaga, M; Shiraki, M; Nakamura, T (2011). “A new active vitamin D3 analog, eldecalcitol, prevents the risk of osteoporotic fractures—A randomized, active comparator, double-blind study”. Bone49 (4): 605–612. doi:10.1016/j.bone.2011.07.011. PMID21784190.
Harada, S; Mizoguchi, T; Kobayashi, Y; Nakamichi, Y; Takeda, S; Sakai, S; Takahashi, F; Saito, H; Yasuda, H; Udagawa, N; Suda, T; Takahashi, N (2012). “Daily administration of eldecalcitol (ED-71), an active vitamin D analog, increases bone mineral density by suppressing RANKL expression in mouse trabecular bone”. Journal of Bone and Mineral Research27 (1): 461–473. doi:10.1002/jbmr.555.
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
May 27, 2016
Release
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
Prostate cancer is the second leading cause of death from cancer in U.S. men. In patients with suspected cancer recurrence after primary treatment, accurate staging is an important objective in improving management and outcomes.
“Imaging tests are not able to determine the location of the recurrent prostate cancer when the PSA is at very low levels,” said Libero Marzella, M.D., Ph.D., director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research. “Axumin is shown to provide another accurate imaging approach for these patients.”
Two studies evaluated the safety and efficacy of Axumin for imaging prostate cancer in patients with recurrent disease. The first compared 105 Axumin scans in men with suspected recurrence of prostate cancer to the histopathology (the study of tissue changes caused by disease) obtained by prostate biopsy and by biopsies of suspicious imaged lesions. Radiologists onsite read the scans initially; subsequently, three independent radiologists read the same scans in a blinded study.
The second study evaluated the agreement between 96 Axumin and C11 choline (an approved PET scan imaging test) scans in patients with median PSA values of 1.44 ng/mL. Radiologists on-site read the scans, and the same three independent radiologists who read the scans in the first study read the Axumin scans in this second blinded study. The results of the independent scan readings were generally consistent with one another, and confirmed the results of the onsite scan readings. Both studies supported the safety and efficacy of Axumin for imaging prostate cancer in men with elevated PSA levels following prior treatment.
Axumin is a radioactive drug and should be handled with appropriate safety measures to minimize radiation exposure to patients and healthcare providers during administration. Image interpretation errors can occur with Axumin PET imaging. A negative image does not rule out the presence of recurrent prostate cancer and a positive image does not confirm the presence of recurrent prostate cancer. Clinical correlation, which may include histopathological evaluation of the suspected recurrence site, is recommended.
The most commonly reported adverse reactions in patients are injection site pain, redness, and a metallic taste in the mouth.
Axumin is marketed by Blue Earth Diagnostics, Ltd., Oxford, United Kingdom
The non-natural amino acid [ F]-l-amino-3-fluorocyclobutane-l-carboxylic acid
([18F]-FACBC, also known as [18F]-Fluciclovine) is taken up specifically by amino acid transporters and has shown promise for tumour imaging with positron emission tomography (PET).
A known synthesis of [18F]-FACBC begins with the provision of the protected precursor compound 1 -(N-(t-butoxycarbonyl)amino)-3 –
[((trifluoromethyl)sulfonyl)oxy]-cyclobutane-l-carboxylic acid ethyl ester. This precursor compound is first labelled with [18F]-fluoride:
II before removal of the two protecting groups:
IT III
EP2017258 (Al) teaches removal of the ethyl protecting group by trapping the [18F]- labelled precursor compound (II) onto a solid phase extraction (SPE) cartridge and incubating with 0.8 mL of a 4 mol/L solution of sodium hydroxide (NaOH). After 3 minutes incubation the NaOH solution was collected in a vial and a further 0.8 mL 4 mol/L NaOH added to the SPE cartridge to repeat the procedure. Thereafter the SPE cartridge was washed with 3 mL water and the wash solution combined with the collected NaOH solution. Then 2.2 mL of 6 mol/L HCl was then added with heating to 60°C for 5 minutes to remove the Boc protecting group. The resulting solution was purified by passing through (i) an ion retardation column to remove Na+ from excess NaOH and Cl~ from extra HCl needed to neutralise excess of NaOH to get a highly acidic solution before the acidic hydrolysis step, (ii) an alumina column, and (iii) a reverse-phase column. There is scope for the deprotection step(s) and/or the
purification step in the production of [18F]-FACBC to be simplified.
Example 1: Synthesis of f FIFACBC
No-carrier- added [18F]fluoride was produced via the 180(p,n)18F nuclear reaction on a GE PETtrace 6 cyclotron (Norwegian Cyclotron Centre, Oslo). Irradiations were performed using a dual-beam, 30μΑ current on two equal Ag targets with HAVAR foils using 16.5 MeV protons. Each target contained 1.6 ml of > 96% [180]water (Marshall Isotopes). Subsequent to irradiation and delivery to a hotcell, each target was washed with 1.6 ml of [160]water (Merck, water for GR analysis), giving approximately 2-5 Gbq in 3.2 ml of [160]water. All radiochemistry was performed on a commercially available GE FASTlab™ with single-use cassettes. Each cassette is built around a one-piece-moulded manifold with 25 three-way stopcocks, all made of polypropylene. Briefly, the cassette includes a 5 ml reactor (cyclic olefin copolymer), one 1 ml syringe and two 5 ml syringes, spikes for connection with five prefilled vials, one water bag (100 ml) as well as various SPE cartridges and filters. Fluid paths are controlled with nitrogen purging, vacuum and the three syringes. The fully automated system is designed for single-step fluorinations with cyclotron-produced [18F]fluoride. The FASTlab was programmed by the software package in a step-by-step time-dependent sequence of events such as moving the syringes, nitrogen purging, vacuum, and temperature regulation. Synthesis of
[18F]FACBC followed the three general steps: (a) [18F]fluorination, (b) hydrolysis of protection groups and (c) SPE purification.
Vial A contained K222 (58.8 mg, 156 μπιοΐ), K2C03 (8.1 mg, 60.8 μπιοΐ) in 79.5% (v/v)
MeCN(aq) (1105 μΐ). Vial B contained 4M HC1 (2.0 ml). Vial C contained MeCN
(4.1ml). Vial D contained the precursor (48.4 mg, 123.5 μιηοΐ) in its dry form (stored at -20 °C until cassette assembly). Vial E contained 2 M NaOH (4.1 ml). The 30 ml product collection glass vial was filled with 200 mM trisodium citrate (10 ml). Aqueous
[18F]fluoride (1-1.5 ml, 100-200 Mbq) was passed through the QMA and into the 180-
H20 recovery vial. The QMA was then flushed with MeCN and sent to waste. The trapped [18F]fluoride was eluted into the reactor using eluent from vial A (730 μΐ) and then concentrated to dryness by azeotropic distillation with acetonitrile (80 μΐ, vial C). Approximately 1.7 ml of MeCN was mixed with precursor in vial D from which 1.0 ml of the dissolved precursor (corresponds to 28.5 mg, 72.7 mmol precursor) was added to the reactor and heated for 3 min at 85°C. The reaction mixture was diluted with water and sent through the tC18 cartridge. Reactor was washed with water and sent through the tC18 cartridge. The labelled intermediate, fixed on the tC18 cartridge was washed with water, and then incubated with 2M NaOH (2.0 ml) for 5 min after which the 2M NaOH was sent to waste. The labelled intermediate (without the ester group) was then eluted off the tC18 cartridge into the reactor using water. The BOC group was hydrolysed by adding 4M HC1 (1.4 ml) and heating the reactor for 5 min at 60 °C. The reactor content with the crude [18F]FACBC was sent through the HLB and Alumina cartridges and into the 30 ml product vial. The HLB and Alumina cartridges were washed with water (9.1 ml total) and collected in the product vial. Finally, 2M NaOH (0.9 ml) and water (2.1 ml) was added to the product vial, giving a purified formulation of [18F]FACBC with a total volume of 26 ml. Radiochemical purity was measured by radio-TLC using a mixture of MeCN:MeOH:H20:CH3COOH (20:5:5: 1) as the mobile phase. The radiochemical yield (RCY) was expressed as the amount of radioactivity in the [18F]FACBC fraction divided by the total used [18F]fluoride activity (decay corrected). Total synthesis time was 43 min.
The RCY of [18F]FACBC was 62.5% ± 1.93 (SD), n=4.
/////FDA, diagnostic imaging agent, recurrent prostate cancer, fda 2016, Axumin, marketed, Blue Earth Diagnostics, Ltd., Oxford, United Kingdom, fluciclovine F 18
Date of issue of marketing authorisation valid throughout the European Union
22/05/2017
Contact address:
Blue Earth Diagnostics Ltd
215 Euston Road
London NW1 2BE
United Kingdom
Manufacture, characterisation and process controls
The active substance fluciclovine (18F) is prepared from the precursor AH113487 by nucleophilic substitution
of a triflate group by 18F-fluoride, followed by two deprotection steps. Due to the short half-life of the 18Ffluorine
radioisotope, each batch is prepared on the day of clinical use.
The active substance is prepared in a proprietary automated synthesiser unit. The synthesiser module is
computer-controlled. A fluid path for synthesis is provided in the form of a single use cassette (FASTlab). The
cassette contains 3 reagent vials and 3 solid phase cartridges. Two other reagent vials are supplied
separately as they have a recommended storage temperature of 2-8°C. These 2 vials are inserted into the
cassette on the day of production.
Assessment report
EMA/237809/2017 Page 13/90
Fluciclovine (18F) is produced in a continuous operation from the precursor AH113487. Due to the radioactive
nature of the process, and the short half-life of [18F] fluorine, intermediates are not isolated and there is no
opportunity for operator intervention or in-process testing. Control of the synthesis of fluciclovine (18F) from
the precursor is achieved through the automated synthesis platform, which is pre-programmed with
synthesis parameters optimised for the process. On-board detectors record transfers of radioactivity through
the fluid path at critical points and monitor temperature and pressure as appropriate so that the operator
may track the progress of the synthesis.
The active substance fluciclovine (18F) progressses immediately to purification, formulation and dispensing as
the finished product within a single, continuous operation. Validation of the manufacturing process for
fluciclovine (18F) is therefore described as part of finished product validation.
The characterisation of the active substance is in accordance with the EU guideline on chemistry of new
active substances.
As mentioned, the manufacture of the active substance and finished product takes place in a single,
continuous process. The active substance is not isolated at any point. Therefore, relevant information about
impurities is given only for the finished product.
For the same reason, information for the container closure system is provided only for the finished product.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004197/WC500230836.pdf
Dipeptidyl Peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) is a type π membrane protein that has been referred to in the literature by a wide a variety of names including DPP4, DP4, DAP-IV, FAPβ, adenosine deaminase complexing protein 2, adenosine deaminase binding protein (AD Abp), dipeptidyl aminopeptidase IV; Xaa-Pro-dipeptidyl-aminopeptidase; Gly-Pro naphthylamidase; postproline dipeptidyl aminopeptidase IV; lymphocyte antigen CD26; glycoprotein GPI lO; dipeptidyl peptidase IV; glycylproline aminopeptidase; glycylproline aminopeptidase; X-prolyl dipeptidyl aminopeptidase; pep X; leukocyte antigen CD26; glycylprolyl dipeptidylaminopeptidase; dipeptidyl-peptide hydrolase; glycylprolyl aminopeptidase; dipeptidyl-aminopeptidase IV; DPP ΓV/CD26; amino acyl-prolyl dipeptidyl aminopeptidase; T cell triggering molecule TρlO3; X-PDAP. Dipeptidyl Peptidase IV is referred to herein as “DPP-IV.” [0003] DPP-W is a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV dependent slow release of dipeptides of the type X-GIy or X-Ser has also been reported for some naturally occurring peptides.
DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of disease states, some of which are discussed below.
[0005] DPP-IV is responsible for the metabolic cleavage of certain endogenous peptides (GLP-I (7-36), glucagon) in vivo and has demonstrated proteolytic activity against a variety of other peptides (GHRH, NPY, GLP-2, VIP) in vitro.
GLP-I (7-36) is a 29 amino-acid peptide derived by post-translational processing of proglucagon in the small intestine. GLP-I (7-36) has multiple actions in vivo including the stimulation of insulin secretion, inhibition of glucagon secretion, the promotion of satiety, and the slowing of gastric emptying. Based on its physiological profile, the actions of GLP-I (7-36) are believed to be beneficial in the prevention and treatment of type II diabetes and potentially obesity. For example, exogenous administration of GLP-I (7-36) (continuous infusion) in diabetic patients has been found to be efficacious in this patient population. Unfortunately, GLP-I (7-36) is degraded rapidly in vivo and has been shown to have a short half -life in vivo (t1/2=1.5 minutes).
Based on a study of genetically bred DPP-IV knock out mice and on in vivo I in vitro studies with selective DPP-IV inhibitors, DPP-IV has been shown to be the primary degrading enzyme of GLP-I (7-36) in vivo. GLP-I (7-36) is degraded by DPP-IV efficiently to GLP-I (9-36), which has been speculated to act as a physiological antagonist to GLP-I (7-36). Inhibiting DPP-TV in vivo is therefore believed to be useful for potentiating endogenous levels of GLP-I (7-36) and attenuating the formation of its antagonist GLP-I (9-36). Thus, DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV, in particular diabetes and more particularly, type 2 diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (WG), metabolic acidosis, ketosis, appetite regulation and obesity.
DPP-IV expression is increased in T-cells upon mitogenic or antigenic stimulation (Mattem, T., et al., Scand. J. Immunol, 1991, 33, 737). It has been reported that inhibitors of DPP-IV and antibodies to DPP-IV suppress the proliferation of mitogen-stimulated and antigen-stimulated T-cells in a dose-dependant manner (Schon, E., et al., Biol. Chem., 1991, 372, 305). Various other functions of T-lymphocytes such as cytokine production, IL-2 mediated cell proliferation and B-cell helper activity have been shown to be dependent on DPP-IV activity (Schon, E., et al., Scand. J. Immunol, 1989, 29, 127). DPP-IV inhibitors, based on boroProline, (Flentke, G. R., et al., Proc. Nat. Acad. Set USA, 1991, 88, 1556) although unstable, were effective at inhibiting antigen-induced lymphocyte proliferation and IL-2 production in murine CD4+ T-helper cells. Such boronic acid inhibitors have been shown to have an effect in vivo in mice causing suppression of antibody production induced by immune challenge (Kubota, T. et al, Clin. Exp. Immun., 1992, 89, 192). The role of DPP-IV in regulating T lymphocyte activation may also be attributed, in part, to its cell-surface association with the transmembrane phosphatase, CD45. DPP-IV inhibitors or non-active site ligands may possibly disrupt the CD45-DPP-TV association. CD45 is known to be an integral component of the T-cell signaling apparatus. It has been reported that DPP-IV is essential for the penetration and infectivity of HTV-I and HTV-2 viruses in CD4+ T-cells (Wakselman, M., Nguyen, C, Mazaleyrat, J.-P., Callebaut, C, Krust, B., Hovanessian, A. G., Inhibition of HIV-I infection of CD 26+ but not CD 26-cells by a potent cyclopeptidic inhibitor of the DPP-IV activity of CD 26. Abstract P.44 of the 24.sup.th European Peptide Symposium 1996). Additionally, DPP-IV has been shown to associate with the enzyme adenosine deaminase (ADA) on the surface of T-cells (Kameoka, J., et al., Science, 193, 26 466). ADA deficiency causes severe combined immunodeficiency disease (SCID) in humans. This ADA-CD26 interaction may provide clues to the pathophysiology of SCID. It follows that inhibitors of DPP-TV may be useful immunosuppressants (or cytokine release suppressant drugs) for the treatment of among other things: organ transplant rejection; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; and the treatment of AIDS.
It has been shown that lung endothelial cell DPP-IV is an adhesion molecule for lung-metastatic rat breast and prostate carcinoma cells (Johnson, R. C, et al., J. Cell. Biol, 1993, 121, 1423). DPP-IV is known to bind to fibronectin and some metastatic tumor cells are known to carry large amounts of fibronectin on their surface. Potent DPP-IV inhibitors may be useful as drugs to prevent metastases of, for example, breast and prostrate tumors to the lungs.
High levels of DPP-PV expression have also been found in human skin fibroblast cells from patients with psoriasis, rheumatoid arthritis (RA) and lichen planus (Raynaud, F., et al., J. Cell. Physiol, 1992, 151, 378). Therefore, DPP-TV inhibitors may be useful as agents to treat dermatological diseases such as psoriasis and lichen planus. [0011] High DPP-TV activity has been found in tissue homogenates from patients with benign prostate hypertrophy and in prostatosomes. These are prostate derived organelles important for the enhancement of sperm forward motility (Vanhoof, G., et al., EMr. /.
Clin. Chem. Clin. Biochem., 1992, 30, 333). DPP-IV inhibitors may also act to suppress sperm motility and therefore act as a male contraceptive agent. Conversely, DPP-IV inhibitors have been implicated as novel for treatment of infertility, and particularly human female infertility due to Polycystic ovary syndrome (PCOS, Stein-Leventhal syndrome) which is a condition characterized by thickening of the ovarian capsule and . formation of multiple follicular cysts. It results in infertility and amenorrhea.
DPP-IV is thought to play a role in the cleavage of various cytokines
(stimulating hematopoietic cells), growth factors and neuropeptides.
[0013] Stimulated hematopoietic cells are useful for the treatment of disorders that are characterized by a reduced number of hematopoietic cells or their precursors in vivo. Such conditions occur frequently in patients who are immunosuppressed, for example, as a consequence of chemotherapy and/or radiation therapy for cancer. It was discovered that inhibitors of dipeptidyl peptidase type PV are useful for stimulating the growth and differentiation of hematopoietic cells in the absence of exogenously added cytokines or other growth factors or stromal cells. This discovery contradicts the dogma in the field of hematopoietic cell stimulation, which provides that the addition of cytokines or cells that produce cytokines (stromal cells) is an essential element for maintaining and stimulating the growth and differentiation of hematopoietic cells in culture. (See, e.g., PCT Intl. Application No. PCT/US93/017173 published as WO 94/03055).
[0014] DPP-IV in human plasma has been shown to cleave N-terminal Tyr-Ala from growth hormone-releasing factor and cause inactivation of this hormone. Therefore, inhibitors of DPP-IV may be useful in the treatment of short stature due to growth hormone deficiency (Dwarfism) and for promoting GH-dependent tissue growth or re-growth.
DPP-IV can also cleave neuropeptides and has been shown to modulate the activity of neuroactive peptides substance P, neuropeptide Y and CLIP (Mentlein, R., Dahms, P., Grandt, D., Kruger, R., Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV, Regul. Pept., 49, 133, 1993; Wetzel, W., Wagner, T., Vogel, D., Demuth, H.-U., Balschun, D., Effects of the CLIP fragment ACTH 20-24 on the duration of REM sleep episodes, Neuropeptides, 31, 41, 1997). Thus DPP-IV inhibitors may also be useful agents for the regulation or normalization of neurological disorders.
Several compounds have been shown to inhibit DPP-IV. Nonetheless, a need still exists for new DPP-IV inhibitors that have advantageous potency, stability, selectivity, toxicity and/or pharmacodynamics properties. In this regard, synthetic methods are provided that can be used to make a novel class of DPP-IV inhibitors.
Trelagliptin (Zafatek) is a pharmaceutical drug used for the treatment of type 2 diabetes (diabetes mellitus).[1]
Indications for Medical Use
It is a highly selective dipeptidyl peptidase (DPP-4) inhibitor that is typically used as an add on treatment when the first line treatment of metformin is not achieving the expected glycemic goals; though it has been approved for use as a first line treatment when metformin cannot be used.[1]
Biochemistry
DPP-4 inhibitors activate T-cells and are more commonly known as T-cell activation antigens (specifically CD26).[1][2] Chemically, it is a fluorinated derivative of alogliptin.
Development
Formulated as the salt trelagliptin succinate, it was approved for use in Japan in March 2015.[3] Takeda, the company that developed trelagliptin, chose to not get approval for the drug in the USA and EU.[1] The licensing rights that Takeda purchased from Furiex Pharmaceuticals for DPP-4 inhibitors included a clause specific to development of this drug in the USA and EU.[1] The clause required that all services done for phase II and phase III clinical studies in the USA and EU be purchased through Furiex.[1] Takeda chose to cease development of this drug in the USA and EU because of the high costs quoted by Furiex for these services.[1] Gliptins have been on the market since 2006 and there are 8 gliptins currently registered as drugs (worldwide).[4] Gliptins are an emerging market and are thus being developed at an increasing rate; there are currently two gliptins in advanced stages of development that are expected to be on the market in the coming year.[4]
Gliptins are thought to have cardiovascular protective abilities though the extent of these effects is still being studied.[4] They are also being studied for the ability that this class of drugs has at promoting B-cell survival.[4]
Administration and Dosing
Similar drugs in the same class as trelagliptin are administered once daily while trelagliptin is administered once weekly.[1][5] Alogliptin (Nesina) is the other major DPP-4 inhibitor on the market. It is also owned by Takeda and is administered once daily. A dosing of once per week is advantageous as a reduction in the frequency of required dosing is known to increase patient compliance.[1][2]
Zafatek is administered in the form trelagliptin succinate in a 1:1 mixture of trelagliptin and succinic acid.[6] The drug is marketed with the IUPAC name Succinic acid – 2-({6-[(3R)-3-amino-1-piperidinyl]-3-methyl-2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl}methyl)-4-fluorobenzonitrile (1:1), has a molecular mass of 475.470143 grams/mol, and has the molecular formula | C=22 | H=26 | F=1 | N=5 | O=6 .[6][7]
SYNTHESIS …………….
PAPER
J. Med .Chem.,2011, 54, 510-524
Synthesis started with selective alkylation of chlorouracil 80, followed by methylation provided compound153via152.
The displacement of chloride with 3-(R)-aminopiperidine83afforded trelagliptin154..
The discovery of two classes of heterocyclic dipeptidyl peptidase IV (DPP-4) inhibitors, pyrimidinones and pyrimidinediones, is described. After a single oral dose, these potent, selective, and noncovalent inhibitors provide sustained reduction of plasma DPP-4 activity and lowering of blood glucose in animal models of diabetes. Compounds 13a, 27b, and 27j were selected for development.
2-[6-(3-Aminopiperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluorobenzonitrile, TFA salt (27j)
A mixture of 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol), and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60 °C for 2 h. The mixture was diluted with water and extracted with EtOAc. The organics were dried over MgSO4, and the solvent was removed. The residue was purified by column chromatography to give 0.66 g of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluorobenzonitrile (60%). 1H NMR (400 MHz, CDCl3): δ 7.73 (dd, J = 7.2, 8.4 Hz, 1H), 7.26 (d, J = 4.0 Hz, 1H), 7.11−7.17 (m, 1H), 6.94 (dd, J = 2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [M + H] calcd for C13H9ClFN3O2, 293; found 293.
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluorobenzonitrile (300 mg, 1.0 mmol), 3-(R)-aminopiperidine dihydrochloride (266 mg, 1.5 mmol), and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 °C for 2 h. The final compound (367 mg, 81% yield) was obtained as a TFA salt after HPLC purification. 1H NMR (400 MHz, CD3OD): δ 7.77−7.84 (m, 1H), 7.16−7.27 (m, 2H), 5.46 (s, 1H), 5.17−5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33−3.47 (m, 2H), 3.22 (s, 3H), 2.98−3.08 (m, 1H), 2.67−2.92 (m, 2H), 2.07−2.17 (m, 1H), 1.82−1.92 (m, 1H), 1.51−1.79 (m, 2H). MS (ES) [M + H] calcd for C18H20FN5O2, 357; found, 357.
(R)-2-((6-(3-amino-3-methylpiperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)methyl)-4-fluorobenzonitrile (30). 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-3-methyl-piperidine dihydrochloride (266 mg, 1.4 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 1000C for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.78-7.83 (m, IH), 7.14-7.26 (m, 2H), 5.47 (s, IH), 5.12-5.36 (ABq, 2H, J = 105.2, 15.6 Hz), 3.21 (s, IH), 2.72-3.15 (m, 4H), 1.75-1.95 (m, 4H), 1.39 (s, 3H). MS (ES) [m+H] calc’d for C19H22FN5O2, 372.41; found, 372.41.
Compound 34
4-Fluoro-2-methylbenzonitrile (31). A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32). A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
Alternatively, 32 was made as follows. 4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AJJBN (122 g) and heated to 750C. A suspension of DBH (353 g) in DCE (500 mL) was added at 750C portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AJ-BN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10°C over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20°C over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro-benzonitrile (33). A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 6O0C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=1.2, 8.4Hz, IH), 7.26 (d, J-4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows. To a solution of 6-chloro-3-methyluracil (750 g) and W,iV-diisopropylethylarnine (998 mL) in NMP (3 L) was added (at <30°C over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 6O0C for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5°C for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2 X 2.25 L). The material was then dried in a vacuum oven at 40±5°C for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile (34). 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 1000C for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 6O0C until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 6O0C and the mixture was allowed to cool to <25°C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2 X 3.6 L). The filtrate was concentrated at 450C under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 00C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35°C over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 5O0C (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purifed water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 3O0C over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 6O0C until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57°C. After stirring at 6O0C for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5°C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2 X 4.2 L). The material was then dried in a vacuum oven at 70±5°C (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75°C and filtered through #3 Whatman filter paper. The solution was heated back to 750C and a IM solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20°C/hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
Example 1Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)
Compound I may be prepared by the follow synthetic route (Scheme 1)
A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)
Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)
Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)
Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)
Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile
The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.
TABLE A
Approximate Solubilities of Compound I
Solubility
Solvent
(mg/mL)a
Acetone
2
Acetonitrile (ACN)
<1
Dichloromethane (DCM)
<1
Dimethyl Formamide (DMF)
68
1,4-Dioxane
<1
Ethanol (EtOH)
2
Ethyl Acetate (EtOAc)
<1
di-Ethyl ether
<1
Hexanes
<1
2-Propanol (IPA)
<1
Methanol (MeOH)
20
Tetrahydrofuran (THF)
<1
Toluene
<1
Trifluoroethanol (TFE)
>200
Water (H2O)
51
ACN:H2O (85:15)
101
EtOH:H2O (95:5)
5
IPA:H2O (88:12)
11
aApproximate solubilities are calculated based on the total solvent used to give a solution; actual solubilities may be greater because of the volume of the solvent portions utilized or a slow rate of dissolution. Solubilities are reported to the nearest mg/mL.
Reference Example 2
in the following formula 2, 2 – ((6 – ((3R) -3- amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H ) – yl) shown in the following example of a production process of a methyl) -4-fluoro-benzonitrile succinate (4b).
[Formula 2]
[In the formula 2, 2 – ((6-chloro-3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) – yl) methyl) -4-fluorobenzonitrile (2b) manufacturing process]
ethyl acetate (3.5 vol), 2- (bromomethyl) -4-fluorobenzonitrile (1b) (1 equiv, 1wt.), 6- chloro-3-methyl uracil (1.05 eq, 0.79wt), N- methylpyrrolidone (NMP;.. 3.5 times the amount), diisopropylethylamine (Hunig’s base, 2.1 eq, 1.27wt) was heated to an internal temperature of 60 ~ 70 ℃ a.
The mixture was stirred until 2-4 hours or the completion of the reaction at 60 ~ 70 ℃.
Then cooling the solution to 40 ~ 50 ℃, after stirring at least 30 minutes, 40 ~ 50 ℃ isopropanol (1.5 times) while maintaining, water (3.5 times the amount) was added, then at least one hour stirring did. The solution was cooled to 20 ~ 30 ℃, was then stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, was then stirred for at least 1 hour. The resulting slurry was filtered, washed with 0 ~ 10 ℃ in cold isopropanol (4.0 vol), and vacuum dried at 45 ~ 55 ℃, to give the above compound (2b).
[In the formula 2, 2 – ((6 – ((3R) -3- amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) – yl) methyl) -4-manufacturing process of the fluorobenzonitrile (3b)]
the above compound (2b) (1 eq, 1wt.), (R) -3- aminopiperidine dihydrochloride (1.1 eq, 0.65wt .), potassium carbonate (2.5 equivalents, 1.18wt.), isopropanol (5.0 vol), water (1.5 times) until the completion of the reaction with 65 ~ 75 ℃ (eg, 3 to 7 hours ) was allowed to react. Potassium carbonate in 65 ~ 75 ℃ (7.05 eq, 3.32wt.), Water (5.5 vol) was added, and after stirring for about 30 minutes, the phases were separated at 50 ℃ ~ 70 ℃. The organic solvent was concentrated under reduced pressure to approximately 5 times. And water (5 vol) was added to the solution and concentrated under reduced pressure to approximately 5 times. The solution was stirred for about 40 minutes at 55 ℃ ~ 75 ℃. The solution was cooled to 20 ℃ ~ 30 ℃, was then stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, subsequently stirred for at least 1 hour, the resulting slurry was filtered, washed with 0 ~ 10 ℃ in cold water (2.0 times the amount), 45 ~ 55 ℃ was vacuum dried to give the above compound (3b).
[In the above formula 2, the compound production step of succinate (4b) of (3b)]
Compound (3b), tetrahydrofuran (6.0 vol), isopropanol (3.0 vol), water (0. a 6-fold amount) was heated to 55 ~ 65 ℃. Tetrahydrofuran solution of succinic acid (20 ℃ ~ 30 ℃) was added and the solution was stirred for about 15 minutes and maintained at 55 ~ 65 ℃.
The solution was cooled to 20 ~ 30 ℃, the mixture was stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, was then stirred for at least 1 hour. After the resulting slurry filtered and washed with isopropanol (6.0 vol). The resulting wet crystals were dried at 65 ~ 75 ℃, was obtained succinate of the compound (3b) and (4b) as a white crystalline solid.
2 – ({6 -! [(3R) -3- amino-piperidin-1-yl] -3-methyl-dihydro-pyrimidin _3,4_ _2,4_ dioxo-1 (2 1) – yl} methyl) benzonitrile is an effective DPP-1V inhibitors class of drugs in recent years in Japan, the structural formula
As shown below.
Chinese Patent Application CN1926128 discloses a process for preparing 2_ ({6_ [(3R) -3- amino-piperidin-1-yl] -3-methyl-2,4-dioxo-3,4- dihydropyrimidine-1 (2 1!) – yl} methyl) benzonitrile method, as shown in Scheme I:
Scheme I
In the above reaction scheme, 6-chloro-uracil and 2-bromomethyl-benzene cyanide in a mixed solvent of DMF-DMSO, in the presence of NaH and LiBr alkylation reaction to give compound 2 in a yield of 54%. Compound 2 is further alkylation reaction of compound yield 3 is 72%. The total yield of the compound 4 prepared in 20% yield is low, and the preparation of compound 4 obtained purity is not high, but also the need for further purification, such as recrystallization, column chromatography and other means in order to obtain high-purity suitable Pharmaceutically acceptable 2 – ({6 – [(3R) -3- amino-piperidin-1-yl] -3-methyl-2,4-dioxo-3,4-dihydro-pyrimidin _1 (2! 1) – yl} methyl) benzonitrile compound. Preparation still find more suitable for industrial production, a higher yield of the 2- ({6- [(3R) -3- amino-piperidin-1-yl] -3-methyl-2,4-dioxo -3, (2Η) 4- dihydropyrimidine-1 – yl} methyl) benzonitrile or a salt or the like.
Example 15
(R) -2 – ((6 (3-amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) – yl) methyl) synthesis of 4-fluoro-benzonitrile
100mL four-necked flask of water and isopropanol 1/1 (v / v) mixture 60mL was added, pyridine 21.4μL [d = 0.98, mw.79.10, 0.26mmol], (R) -1- (3- (2 – cyano-5-fluoro-benzyl) -1-methyl-2,6-dioxo-1,2,3,6-tetra-hydro-4-yl) piperidin-3-carboxamide 2.00g [mw.385.39, 5.19mmol] of It was added to the order. Then, iodobenzene diacetate 1.84g [mw.322.10, 5.71mmol] was added, and the mixture was stirred for 3 h at 20 ℃. After volatile components were distilled off under reduced pressure by an evaporator, and the aqueous solution was washed twice with ethyl acetate 20mL. After cooling to near 0 ℃, potassium carbonate 16g added stepwise at 15 ℃ or less, was extracted by the addition of toluene 6mL and isopropanol 6mL. After separation, the organic layer was washed with saturated brine 10mL, adding toluene 6mL after concentration under reduced pressure by an evaporator, and further subjected to vacuum concentration. It was suspended by the addition of toluene 6mL to concentrate, by the addition of n-heptane 6mL, after 1 hour and aged at 0 ℃, reduced pressure filtration, to obtain the desired compound after drying under reduced pressure at 50 ℃. White crystalline powder, 1.6g, 86% yield.
13 C NMR (126 MHz, CDCl 3 ) ppm 28.0, 33.4, 46.1, 51.9, 59.7, 90.8, 114.6,114.7, 115.6, 115.8, 116.4, 135.4, 135.5, 144.6, 152.7, 159.5, 162.9.
Reference Example 4
(R) -2 – ((6 (3-amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) – yl) methyl) synthesis of 4-fluoro-benzonitrile succinate
50mL eggplant-shaped flask (R) -2 – ((6- (3- amino-1-yl) -3-methyl-2,4-dioxo-3,4-dihydro-pyrimidine -1 (2H) – yl) methyl) -4-fluorobenzonitrile 1.0g [mw.357.38, 2.8mmol], it was added tetrahydrofuran 4.5mL and water 2 drops. After heated and dissolved at 65 ℃, was dropped to the solution was dissolved at the same temperature 0.331g succinic acid [mw.118.09, 2.8mmol] with tetrahydrofuran 4mL and isopropanol 2.5mL. Aged for 16 hours at room temperature after stirring for 30 min at 65 ℃, and stirred for a further 2 hours at 0 ℃. The crystallization product was collected by terrorism to vacuum filtration. To obtain the desired compound after drying under reduced pressure at 45 ℃. White crystalline powder, 1.2g, 93% yield.
The present invention provides a process for the preparation of 4-fluoro-2- methylbenzonitrile of Formula (II), and its use for the preparation of trelagliptin or its salts. The present invention provides an efficient, simple, and commercially friendly process for the preparation of 4-fluoro-2-methylbenzonitrile, which is used as an intermediate for the preparation of trelagliptin or its salts. The present invention avoids the use of toxic and hazardous reagents, high boiling solvents, and bromo intermediates such as 2-bromo-5-fluorotoluene, which is lachrymatory in nature and thus difficult to handle at a commercial scale.
Trelagliptin is a dipeptidyl peptidase IV (DPP-IV) inhibitor, chemically designated as 2- [[6-[(3i?)-3 -aminopiperidin- 1 -yl] -3 -methyl -2,4-dioxopyrimidin- 1 -yljmethyl] -4-fluorobenzonitrile, represented by Formula I.
Formula I
Trelagliptin is administered as a succinate salt of Formula la, chemically designated as 2-[[6-[(3i?)-3-aminopiperidin-l-yl]-3-methyl-2,4-dioxopyrimidin-l-yl]methyl]-4-fluorobenzonitrile butanedioic acid (1 : 1).
Formula la
U.S. Patent Nos. 7,795,428, 8,288,539, and 8,222,411 provide a process for the preparation of 4-fluoro-2-methylbenzonitrile by reacting 2-bromo-5-fluorotoluene with copper (I) cyanide in N,N-dimethylformamide.
Chinese Patent No. CN 102964196 provides a process for the preparation of 4-fluoro-2-methylbenzonitrile by reacting 4-fluoro-2-methylbenzyl alcohol with cuprous iodide in the presence of 2,2′-bipyridine and 2,2,6,6-tetramethylpiperidine oxide (TEMPO) in an anhydrous ethanol.
Copper (I) cyanide is toxic to humans, and therefore its use in the manufacture of a drug substance is not advisable. In addition, 2-bromo-5-fluorotoluene is converted to 4-fluoro-2-methylbenzonitrile by refluxing in N,N-dimethylformamide at 152°C to 155°C for 24 hours. This leads to some charring, resulting in a tedious work-up process and low yield. Furthermore, the use of reagents like cuprous iodide, 2,2′-bipyridine, and 2,2,6,6-tetramethylpiperidine oxide (TEMPO) is hazardous and/or environmentally-unfriendly, and therefore their use in the manufacture of a drug substance is not desirable.
The present invention provides an efficient, simple, and commercially friendly process for the preparation of 4-fluoro-2-methylbenzonitrile, which is used as an intermediate for the preparation of trelagliptin or its salts. The present invention avoids the use of toxic and hazardous reagents, high boiling solvents, and bromo intermediates such as 2-bromo-5-fluorotoluene, which is lachrymatory in nature and thus difficult to handle at a commercial scale.
EXAMPLES
Example 1 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (1.38 g) was added to ethanol (10 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (2.76 g) and pyridine (1 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 3 hours. The solvent was recovered up to maximum extent from the reaction mixture under reduced pressure to afford the title compound. Yield: 3.1 g
Example 2: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (5 g) was added to ethanol (37 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (10 g) and N,N-diisopropylethylamine (3.6 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 2 hours. The solvent was recovered up to maximum extent from the reaction mixture under reduced pressure to afford the title compound. Yield: 3.1 g
Example 3 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (10 g) was added to ethanol (40 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (20 g) and N,N-diisopropylethylamine (7.5 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to afford the title compound. Yield: 11.0 g
Example 4: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (50 g) was added to ethanol (500 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (70 g) and N,N-diisopropylethylamine (36 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 6 hours. The solvent was recovered from the reaction mixture under reduced pressure to afford the title compound. Yield: 51.0 g
Example 5 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (20 g) was added to ethanol (200 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (20 g) and N,N-diisopropylethylamine (18 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to obtain a residue. Deionized water (60 mL) was charged into the residue, and then the slurry was stirred at 0°C to 5°C for 1 hour. The solid obtained was filtered, then washed with deionized water (2 x 20 mL). The wet solid was dried in an air oven at 40°C to 45 °C for 4 hours to 5 hours. The crude product obtained was recrystallized in ethanol (50 mL) to afford the pure title compound. Yield: 21.0 g
Example 6: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methyl benzaldehyde (50 g) was added to ethanol (500 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (50 g) and N,N-diisopropylethylamine (46.4 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to obtain a residue. Deionized water (150 mL) was charged to the residue, and then the slurry was stirred at 0°C to 5°C for 1 hour. The solid obtained was filtered, then washed with deionized water (2 x 50 mL). The wet solid was dried in an air oven at 40°C to 45 °C for 4 hours to 5 hours. The crude product obtained was recrystallized in ethanol (200 mL) to afford the pure title compound. Yield: 53.5 g
Example 7: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (3.1 g) and phosphorous pentoxide (1 g) were added to toluene (30 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 24 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (30 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 1.1 g
Example 8: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (3 g) and phosphorous pentoxide (2 g) were added to toluene (30 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 24 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (30 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 1.0 g
Example 9: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (5 g) and concentrated sulphuric acid (2 mL) were added to toluene (100 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 5 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (50 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 3.24 g
Example 10: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (25 g) and concentrated sulphuric acid (35 g) were added to toluene (500 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 6 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (250 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 20.5 g
Example 11 : Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methyl benzaldoxime (5 g) and sodium bisulphate monohydrate (3.1 g) were added to toluene (50 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C, then filtered, and then washed with toluene (10 mL). The filtrate was concentrated under reduced pressure to afford the title compound. Yield: 3.0 g
Example 12: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methyl benzaldoxime (50 g) and sodium bisulphate monohydrate (31.6 g) were added to toluene (500 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C using a Dean-Stark apparatus for 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25 °C to 30°C, then filtered, and then washed with toluene (100 mL). The filtrate was concentrated under reduced pressure to afford a crude product. The crude product obtained was recrystallized in a mixture of toluene (200 mL) and hexane (500 mL) to afford the title compound.
TREATMENT OF GENOTYPED DIABETIC PATIENTS WITH DPP-IV INHIBITORS SUCH AS LINAGLIPTIN [US2013196898]
2010-11-26
2013-08-01
ANTIDIABETIC MEDICATIONS COMPRISING A DPP-4 INHIBITOR (LINAGLIPTIN) OPTIONALLY IN COMBINATION WITH OTHER ANTIDIABETICS [US2012094894]
2010-02-12
2012-04-19
DPP-IV INHIBITORS FOR TREATMENT OF DIABETES IN PEDIATRIC PATIENTS [US2012122776]
2010-01-29
2012-05-17
Patent
Submitted
Granted
LAMINATED TABLET AND MANUFACTURING METHOD THEREFOR [US2014023708]
2012-03-02
2014-01-23
Combination therapy for the treatment of diabetes and related conditions [US2013310398]
2013-07-24
2013-11-21
USE OF KERATINOCYTES AS A BIOLOGICALLY ACTIVE SUBSTANCE IN THE TREATMENT OF WOUNDS, SUCH AS DIABETIC WOUNDS, OPTIONALLY IN COMBINATION WITH A DPP-4 INHIBITOR [US2013315975]
2013-05-23
2013-11-28
USE OF A DPP-4 INHIBITOR IN AUTOIMMUNE DIABETES, PARTICULARLY LADA [US2013317046]
2013-05-21
2013-11-28
USE OF A DPP-4 INHIBITOR FOR MODIFYING FOOD INTAKE AND REGULATING FOOD PREFERENCE [US2013324463]
POLYMORPHS OF SUCCINATE SALT OF 2-[6-(3-AMINO-PIPERIDIN-1-YL)-3-METHYL-2,4-DIOXO-3,4-DIHYDRO-2H-PYRIMIDIN-1-YLMETHY]-4-FLUOR-BENZONITRILE AND METHODS OF USE THEREFOR [US2008227798]
2008-09-18
GPR119 receptor agonists in methods of increasing bone mass and of treating osteoporosis and other conditions characterized by low bone mass, and combination therapy relating thereto [US7816364]
ADMINISTRATION OF DIPEPTIDYL PEPTIDASE INHIBITORS [US2008287476]
2008-11-20
POLYMORPHS OF SUCCINATE SALT OF 2-[6-(3-AMINO-PIPERIDIN-1-YL)-3-METHYL-2,4-DIOXO-3,4-DIHYDRO-2H-PYRIMIDIN-1-YLMETHY]-4-FLUOR-BENZONITRILE AND METHODS OF USE THEREFOR [US8084605]
2008-11-13
2011-12-27
WEEKLY ADMINISTRATION OF DIPEPTIDYL PEPTIDASE INHIBITORS [US8093236]
ADMINISTRATION OF DIPEPTIDYL PEPTIDASE INHIBITORS [US2011077402]
2011-03-31
DPP-IV INHIBITORS FOR USE IN THE TREATMENT OF NAFLD [US2011092510]
2011-04-21
PURIN DERIVATIVES FOR USE IN THE TREATMENT OF FAB-RELATED DISEASES [US2011190322]
2011-08-04
Patent
Submitted
Granted
Administration of Dipeptidyl Peptidase Inhibitors [US2011192748]
2011-08-11
PHARMACEUTICAL COMPOSITION COMPRISING A GLUCOPYRANOSYL-SUBSTITUTED BENZENE DERIVATE [US2011195917]
2011-08-11
DPP-IV INHIBITOR COMBINED WITH A FURTHER ANTIDIABETIC AGENT, TABLETS COMPRISING SUCH FORMULATIONS, THEIR USE AND PROCESS FOR THEIR PREPARATION [US2011206766]
2011-08-25
COMBINATION OF A CERTAIN DPP-4 INHIBITOR AND VOGLIBOSE [US2014343014]
2014-05-16
2014-11-20
CARDIO- AND RENOPROTECTIVE ANTIDIABETIC THERAPY [US2014274889]
2014-03-14
2014-09-18
TREATMENT FOR DIABETES IN PATIENTS INAPPROPRIATE FOR METFORMIN THERAPY [US2014274890]
2014-06-03
2014-09-18
Fused ring compound and use thereof [US2010190747]
2010-07-29
FUSED RING COMPOUND AND USE THEREOF [US2010197683]
GPR119 Receptor Agonists in Methods of Increasing Bone Mass and of Treating Osteoporosis and Other Conditions Characterized by Low Bone Mass, and Combination Therapy Relating Thereto [US8101626]
Molecular Weight: 541.06
Elemental Analysis: C, 46.62; H, 6.89; Cl, 6.55; N, 10.36; O, 23.66; S, 5.93
Acotiamide, also known as YM-443 and Z-338, is a drug approved in Japan for the treatment of postprandial fullness, upper abdominal bloating, and early satiation due to functional dyspepsia. It acts as an acetylcholinesterase inhibitor. Note: The Approved drug API is a cotiamide HCl trihydrate (1:1:3)
Acotiamide hydrochloride (acotiamide; N-[2-[bis(1-methylethyl) amino]ethyl]-2-[(2-hydroxy-4,5-dimethoxybenzoyl) amino] thiazole-4-carboxamide monohydrochloride trihydrate, Z-338) has been reported to improve meal-related symptoms of functional dyspepsia in clinical studies.
Acotiamide (Acofide(®)), an oral first-in-class prokinetic drug, is under global development by Zeria Pharmaceutical Co. Ltd and Astellas Pharma Inc. for the treatment of patients with functional dyspepsia. The drug modulates upper gastrointestinal motility to alleviate abdominal symptoms resulting from hypomotility and delayed gastric emptying. It exerts its activity in the stomach via muscarinic receptor inhibition, resulting in enhanced acetylcholine release and inhibition of acetylcholinesterase activity. Unlike other prokinetic drugs that are utilized in the management of functional dyspepsia, acotiamide shows little/no affinity for serotonin or dopamine D2 receptors. Acotiamide is the world’s first approved treatment for functional dyspepsia diagnosed by Rome III criteria, with its first approval occurring in Japan. Phase III trials in this patient population are in preparation in Europe, with phase II trials completed in the USA and Europe.
SYNTHESIS
EP 0870765; US 5981557; WO 9636619
Acylation of 2-aminothiazole-4-carboxylic acid ethyl ester (I) with 2,4,5-trimethoxybenzoyl chloride (II) produced the corresponding amide (III). The 2-methoxy group of (III) was then selectively cleaved by treatment with pyridine hydrochloride, yielding the 2-hydroxybenzamide (IV). Finally, displacement of the ethyl ester group of (IV) by N,N-diisopropyl ethanediamine (V) upon heating at 120 C furnished the target compound, which was isolated as the corresponding hydrochloride salt.
EP 0994108; WO 9858918
In a closely related procedure, acid chloride (II), prepared by treatment of 2,4,5-trimethoxybenzoic acid (VI) with SOCl2 in hot toluene, was condensed with aminothiazole (I), yielding amide (III). Displacement of the ethyl ester group of (III) by N,N-diisopropyl ethanediamine (V) furnished diamide (VII). Finally, upon formation of the hydrochloride salt of (VII) in isopropanol, the 2-methoxy group was simultaneously cleaved, directly leading to the title compound.
Acotiamide hydrochloride, chemical name: N_ [2_ (diisopropylamino) ethyl] -2- [(2-hydroxy-4,5-dimethoxybenzoyl) amino ] thiazole-4-carboxamide hydrochloride, the following structure:
A test for the amine hydrochloride Japan Zeria Pharmaceutical Company and Astellas jointly developed acetylcholinesterase inhibitor class of prokinetic drugs, namely the treatment of functional dyspepsia drugs, is the world’s first approved specifically for the treatment of FD drugs, in June 2013 for the first time launched in Japan, under the trade name Acofide. Functional dyspepsia (Functional dyspepsia, FD) is a group of common symptoms include bloating, early satiety, burning sensation, belching, nausea, vomiting and abdominal discomfort and so difficult to describe, and no exact organic disease. Organic diseases because of lack of basic, functional dyspepsia harm to patients focus on the performance of gastrointestinal symptoms caused discomfort and possible impact on the quality of life in. Because some patients with functional dyspepsia symptoms caused by eating less, digestion and absorption efficiency is reduced, resulting in varying degrees of malnutrition (including nutrients are not full). With the people’s demands and improve the quality of life for functional dyspepsia know, the number of visits of the disease gradually increased, to become one of the most common disease of Gastroenterology partner waiting group. Such a high prevalence of functional dyspepsia treatment provides a huge market.
The present synthesis method has been reported in less divided into four methods are described below:
1, reference CN1084739C, synthetic route as shown below. Disadvantage of this patent is that: (I) using thionyl chloride and dichloroethane toxic, environmentally damaging substances; (2) demethylation low yield (64.6% to 86 reported in the literature %). Examples reported in this patent first and second step total yield was 84.6% and the total yield of the third-step reaction and recrystallization of 61%, the total yield of 51.6%.
The method, reported in the patent CN1063442C preparation A (page 25) reports (without reference to examples I and 6, referring to its general method). Patent CN102030654B (page 3) above: Step demethylation reaction generates a lot of by-products, it is difficult to take off only a selective protection of hydroxy groups, poor selectivity. Specific synthetic examples are shown below:
Preparation Method B 3 mentioned patent CN1063442C (prepared unprotected, p. 25), where the yield is very low two-step reaction. A test method for the preparation of amines referenced above example (Example 38) A test for specific preparation yield amine not mentioned in the text, but if you use the above method starting materials primary amino side reactions occur. Synthesis of solid concrete
Following is an example:
reported that patent CN101006040B in Method 4. The first step demethylation can also use titanium tetrachloride and aluminum chloride; the second reaction can also be used phenol / thionyl chloride. Synthetic route are higher yield and purity (total yield 73%).
The method of synthesis of the above methods 3 patent CN1063442C reported, though not suitable for the synthesis of amine A test, but may be modified on this basis.
the above patents, CN1084739 reagents using dichloroethane, toxic, environmentally destructive, and the total yield is low, is not conducive to industrial production; patent CN102030654B mentioned Step demethylation The reaction produces a lot of by-products, it is difficult to take off only selective hydroxy protecting group, the reaction selectivity, more side effects.
Example 4
[Amino-N- (2- tert-butoxycarbonyl group -4,5_ dimethoxybenzoyl)] _4_ Preparation of 2-methoxycarbonyl-1,3-thiazole: [0062] Step 1
2-hydroxy-4,5-dimethoxy-benzoic acid (100 g) was dissolved in dry toluene (400 ml) was added Boc20 (132 g) was stirred at rt for 3 hours at room temperature, was added a 10% aqueous citric acid (100 ml) and washed three times with purified water until neutral, dried over anhydrous sodium sulfate was added (20 g) and dried 8 hours, filtered, and the filtrate was added thionyl chloride (64 g) and N, N-dimethyl- carboxamide (0.19 ml), followed by stirring 80 ° C for 4 hours, the compound was added 2-amino-4-methoxycarbonyl-1,3-thiazole (85 g), stirred for 5 hours at 100 ° C, the reaction was completed After cooling to room temperature, the precipitated crystals were collected by filtration, crystals were added to 1.6 liters of water, 400 g of ice was added with stirring, and added a mass ratio of 10% sodium hydroxide aqueous solution adjusted to pH 7.5, followed by stirring for 3 hours at room temperature, filtered The crystals were collected, washed with water, 60 ° C and dried to give the title compound (170 g).
The 2- [N- (2- tert-butoxycarbonyl group -4,5_ dimethoxybenzoyl) amino] _4_ methoxycarbonyl _1,3_ thiazole prepared (170 g) and N , N- diisopropyl-ethylenediamine (162 ml), N, N- dimethylacetamide (162 ml) was stirred at 135 ° C for 8 hours and cooled, 1-butanol (1.7 liters), with 0.5N aqueous sodium hydroxide solution and washed with saturated brine, the mixture was concentrated under reduced pressure, methanol (1.7 l), hydrogen chloride gas under cooling and stirred for 5 hours, the precipitate was collected by filtration, the crystals were washed with 2-propanol and water do recrystallized from a mixed solvent, to give the title compound. Melting point: 160 ° C.
Example 1: A test preparation for the amine hydrochloride
In 500ml reaction flask was added 2,4, 5- trimethoxy benzoic acid (20 g, 94. 3mmol), 200 ml N, N- dimethylformamide. Was added TBTU (30.88 g, 113.2mmol), jealous% was added diisopropylethylamine (14.59g, 113. 2mmol), stirred at room temperature for 2 hours. Was added 2-aminothiazol 4-carboxylic acid methyl ester setback (14. 92 g, 94. 3mmol), DMAP (2. 30g, 18. 9mmol), was heated to 75 ° C, stirred for 24 hours. Added% Jealous diisopropylethylenediamine (27. 16g, 188. 6mmol), and heated to 140 ° C, stirred for 10 hours. After cooling, 400ml of n-butanol was added, stirred, allowed to stand for stratification. Take the upper, washed with saturated brine, 400ml, standing stratification. Take the upper, lower temperatures hydrogen chloride isopropanol solution of 120ml, precipitated solids. Vacuum filter cake into the oven blast 60 ° C and dried for 1 hour. A test was for amine hydrochloride (Compound V) 28. 5g, HPLC purity 99%, yield 62%.
2 Example: A test preparation for the amine hydrochloride
added 2, 4, 5- trimethoxy benzoic acid (20g, 94. 3mmol) in 500ml reaction flask, 200ml% Jealous dimethylacetamide. Was added TBTU (30.88g, 113. 2mmol), was added diisopropylethylamine jealous% (14. 59g, 113. 2mmol)), followed by stirring at room temperature for 2 hours. Was added 2-aminothiazol-4-carboxylate (14. 92g, 94. 3mmol), DMAP (2. 3g, 18. 9mmol), was heated to 75 ° C, stirred for 24 hours. Added% Jealous diisopropylethylenediamine (27. 16g, 188. 6mmol), and heated to 140 ° C, stirred for 10 hours. After cooling, 400ml of n-butanol was added, stirred, allowed to stand for stratification. Take the upper, washed with saturated brine, 400ml, standing stratification. Take the upper, lower temperatures hydrogen chloride isopropanol solution of 120ml, precipitate a solid, vacuum filtration, cake into the oven blast 60 ° C and dried for 1 hour. A test was for amine hydrochloride (Compound V) 31. 7 g, HPLC purity 99%, yield 69%.
A test for the amine hydrochloride (Z-338) is a new Ml Japan Zeria company’s original research, M2 receptor antagonist, for the treatment of functional dyspepsia clinic.
Chinese patent application describes doxorubicin hydrochloride CN200580028537 test for amines (Z-338) preparation, reaction
Process is as follows.
A test for the amine hydrochloride (z-338) Compound Patent Application (CN96194002.6) choosing 2,4,5-trimethoxy benzoic acid as a starting material first with 2-aminothiazol-4-carboxylate reacts 2- [(2-hydroxy-4,5-dimethoxybenzoyl) amino] -1,3-thiazole-4-carboxylate, 2-methyl-benzene and then removed, the yield of this method lower demethylation selectivity bad. So choose the first 2-methyl-removal before subsequent reaction better.
The first patent application CN200580028537 2_ hydroxyl _4,5_ dimethoxy benzoic acid and triphenyl phosphite placed in toluene, was added a few drops of concentrated sulfuric acid as a catalyst under reflux to give the intermediate 2-hydroxy – 4,5-dimethoxy-phenyl benzoate. After the above intermediate with 2-aminothiazol-4-carboxylate in place of toluene, was added triphenyl borate reacted, treated to give 2- [(2-hydroxy-4,5-dimethoxy- benzoyl) amino] -1,3-thiazole-4-carboxylate, and finally with N, N- diisopropylethylamine in toluene diamine salt in the system after the reaction.
[0030] triphosgene dissolved in 90ml CH2Cl2 19.0g placed in a four-necked flask, under N2 stream, the 2_ hydroxyl _4,5_ dimethoxy benzoic acid (22.2g) was dissolved in 150ml CH2Cl2 and 45ml pyridine, at four-necked flask temperature dropped 0_5 ° C under ice-salt bath. Dropping finished within 45min, kept cold stirred lOmin. After warm to room temperature (20 ° C) was stirred for 50min, the reaction was stopped. Pressure filtration, and the filtrate by rotary evaporation at room temperature to a constant weight, adding 35g 2- aminothiazol-4-carboxylate and 240ml 1,2_ dichloroethane and heated to reflux, the reaction 6h. After stopping the cooling, suction filtration, washed with methanol and the resulting solid was refluxed in 40ml, hot filtration to give a white solid 32.18g, yield 85%. M + Na + 361; 2M + Na + 699. [0031] Example 2
triphosgene dissolved in 15ml CH2Cl2 placed 3.0g four-necked flask, under N2 stream, the 2_ hydroxyl _4,5_ dimethoxy benzoic acid (3.0g) was dissolved in pyridine 30ml CH2Cl2 and 61,111, in four-necked flask temperature dropped 0_5 ° C under ice-salt bath. 20min Upon completion, kept cold stirring lh. After warm to room temperature (20 ° C) and stirred overnight, 24h after stopping the reaction. Rotary evaporation at room temperature to a constant weight is added 3.5g 2- aminothiazol-4-carboxylate and 30ml 1,2- dichloroethane burning, heated to reflux, the reaction 6h. The solvent was evaporated after stopping, add 30ml methanol reflux filtration to give a white solid 4.1g, 20ml methanol was added to the mother liquor evaporated leaching and washing a white solid 0.85g. After the merger was solid 4.95g, yield 97%.
The diphosgene 3.0g was dissolved into 15ml CH2Cl2 four-necked flask, under N2 stream, the 2_ hydroxyl _4,5_ dimethoxy benzoic acid (3.0g) was dissolved in 30ml CH2Cl2 and 61,111 pyridine, Under ice-salt bath temperature dropped a four-necked flask 0_5 ° C. 20min Upon completion, kept cold stirring lh. After warm to room temperature (20 ° C) and stirred overnight, 24h after stopping the reaction. Rotary evaporation at room temperature to a constant weight is added 3.5g 2- aminothiazol-4-carboxylate and 30ml 1,2- dichloroethane burning, heated to reflux, the reaction 6h. After the solvent was evaporated and stopped by adding 30ml of methanol was refluxed for leaching to give a white solid 4.57g, yield 89.6%.
triphosgene dissolved in 15ml CH2Cl2 placed 3.0g four-necked flask, under N2 stream, the 2_ hydroxyl _4,5_ dimethoxy benzoic acid (3.0g) `pyridine was dissolved in 30ml CH2Cl2 and 61 111, Under ice-salt bath temperature dropped a four-necked flask 0_5 ° C. 20min Upon completion, kept cold stirring lh. After warm to room temperature (20 ° C) and stirred overnight, 24h after stopping the reaction. Rotary evaporation at room temperature to a constant weight is added 3.7g 2- aminothiazol-4-carboxylic acid ethyl ester and 30ml 1,2- dichloroethane burning, heated to reflux, the reaction 6h. The solvent was evaporated after stopping, add 30ml methanol reflux filtration to give a white solid 3.8g, 20ml methanol was added to the mother liquor evaporated leaching and washing a white solid 0.54g. After the merger was solid 4.34g, yield 81.4%. M + Na + 375.
2 – [(2-hydroxy-4,5-dimethoxybenzoyl) amino] thiazole _4_ _1,3_ carboxylate and 1.5g IOml 1,4- dioxane placed in a four-necked flask, N2 gas shielded at 75 ° C was added dropwise 1.5ml N, N- diisopropyl-ethylenediamine, rose after reflux, the reaction was stirred for 6 hours. The reaction was stopped, the solvent was evaporated to dryness under reduced pressure, 30ml CH2Cl2 was added dissolved in 20ml10% NaCl solution was washed twice, and then the organic solvent was evaporated to dryness. IOml methanol was added, concentrated hydrochloric acid was added to adjust Xeon acidic. Evaporated methanol, washed with acetone to give the product 2.08g, yield 96.3%. M + H 451, MH 449.
2 – [(2-hydroxy-4,5-dimethoxybenzoyl) amino] thiazole _4_ _1,3_ carboxylate and 1.5g IOml 1,4- dioxane placed in a four-necked flask, N2 gas shielded at 75 ° C was added dropwise 1.5ml N, N- diisopropyl-ethylenediamine, rose after reflux, the reaction was stirred for 6 hours. The reaction was stopped, the solvent was evaporated to dryness under reduced pressure, 30ml CH2Cl2 was added dissolved in 20ml10% NaCl solution was washed twice, and then the organic solvent was evaporated to dryness. IOml methanol was added, concentrated hydrochloric acid was added to adjust Xeon acidic. Evaporated methanol, washed with acetone to give the product 1.76g, yield 84.7%.
PAPER
A Three-Step Synthesis of Acotiamide for the Treatment of Patients with Functional Dyspepsia
A three-step synthesis of acotiamide is described. The agent is marketed in Japan for treatment of patients with functional dyspepsia. We designed a one-pot method to prepare the key intermediate 5a from 2 via an acyl chloride and amide and then reacted with 6 to obtain 1 under solvent-free condition. With the use of DCC, the unavoidable impurity 5b was also successfully converted into the desired 1. After isolation of 1, we carried forward to the next step of HCl salt formation, which was proved to be a very effective procedure for the removal of practically all major impurities. The process is cost-effective, simple to operate, and easy to scale-up.
Matsueda K, Hongo M, Tack J, Aoki H, Saito Y, Kato H (January 2010). “Clinical trial: dose-dependent therapeutic efficacy of acotiamide hydrochloride (Z-338) in patients with functional dyspepsia – 100 mg t.i.d. is an optimal dosage”. Neurogastroenterology and Motility : the Official Journal of the European Gastrointestinal Motility Society22 (6): 618–e173. doi:10.1111/j.1365-2982.2009.01449.x. PMID20059698.
: Mayanagi S, Kishino M, Kitagawa Y, Sunamura M. Efficacy of acotiamide in combination with esomeprazole for functional dyspepsia refractory to proton-pump inhibitor monotherapy. Tohoku J Exp Med. 2014;234(3):237-40. PubMed PMID: 25382232.
2: Zai H, Matsueda K, Kusano M, Urita Y, Saito Y, Kato H. Effect of acotiamide on gastric emptying in healthy adult humans. Eur J Clin Invest. 2014 Dec;44(12):1215-21. doi: 10.1111/eci.12367. PubMed PMID: 25370953.
3: Xiao G, Xie X, Fan J, Deng J, Tan S, Zhu Y, Guo Q, Wan C. Efficacy and safety of acotiamide for the treatment of functional dyspepsia: systematic review and meta-analysis. ScientificWorldJournal. 2014;2014:541950. doi: 10.1155/2014/541950. Epub 2014 Aug 12. PubMed PMID: 25197703; PubMed Central PMCID: PMC4146483.
4: Sun Y, Song G, McCallum RW. Evaluation of acotiamide for the treatment of functional dyspepsia. Expert Opin Drug Metab Toxicol. 2014 Aug;10(8):1161-8. doi: 10.1517/17425255.2014.920320. Epub 2014 May 31. PubMed PMID: 24881488.
5: Matsunaga Y, Tanaka T, Saito Y, Kato H, Takei M. [Pharmacological and clinical profile of acotiamide hydrochloride hydrate (Acofide(®) Tablets 100 mg), a novel therapeutic agent for functional dyspepsia (FD)]. Nihon Yakurigaku Zasshi. 2014 Feb;143(2):84-94. Review. Japanese. PubMed PMID: 24531902.
6: Nowlan ML, Scott LJ. Acotiamide: first global approval. Drugs. 2013 Aug;73(12):1377-83. doi: 10.1007/s40265-013-0100-9. Erratum in: Drugs. 2014 Jun;74(9):1059. Nolan, Mary L [corrected to Nowlan, Mary L]. PubMed PMID: 23881665.
8: Nagahama K, Matsunaga Y, Kawachi M, Ito K, Tanaka T, Hori Y, Oka H, Takei M. Acotiamide, a new orally active acetylcholinesterase inhibitor, stimulates gastrointestinal motor activity in conscious dogs. Neurogastroenterol Motil. 2012 Jun;24(6):566-74, e256. doi: 10.1111/j.1365-2982.2012.01912.x. Epub 2012 Mar 19. PubMed PMID: 22429221.
9: Kusunoki H, Haruma K, Manabe N, Imamura H, Kamada T, Shiotani A, Hata J, Sugioka H, Saito Y, Kato H, Tack J. Therapeutic efficacy of acotiamide in patients with functional dyspepsia based on enhanced postprandial gastric accommodation and emptying: randomized controlled study evaluation by real-time ultrasonography. Neurogastroenterol Motil. 2012 Jun;24(6):540-5, e250-1. doi: 10.1111/j.1365-2982.2012.01897.x. Epub 2012 Mar 4. PubMed PMID: 22385472.
10: McLarnon A. Dyspepsia: Acotiamide can relieve symptoms of functional dyspepsia. Nat Rev Gastroenterol Hepatol. 2012 Jan 17;9(2):62. doi: 10.1038/nrgastro.2011.262. PubMed PMID: 22249733.
Approval in Japan for Treating Functional Dyspepsia with Acofide®
Press Release
Tokyo, March 25, 2013 – Zeria Pharmaceutical Co., Ltd. (Tokyo: 4559; “Zeria”) and Astellas Pharma Inc. (Tokyo: 4503; “Astellas”) announced today that as of March 25, Zeria has obtained the marketing approval of Acofide® Tablets 100mg (nonproprietary name: acotiamide hydrochloride hydrate; “Acofide”; Zeria’sdevelopment code: “Z-338”; Astellas’s development code: “YM443”) for the treatment of functional dyspepsia(FD) from the Ministry of Health, Labour and Welfare in Japan. Acofide has been co-developed by both companies.
Acotiamide hydrochloride hydrate is a new chemical entity originated by Zeria, and inhibits peripheralacetylcholinesterase activities. Acetylcholine is an important neurotransmitter to regulate gastrointestinalmotility, and through the inhibition of degradation of acetylcholine, Acofide improves the impaired gastricmotility and delayed gastric emptying, and consequently the subjective symptoms of FD such as postprandialfullness, upper abdominal bloating, and early satiation.
Acofide, the world first FD treatment which demonstrated efficacy in the patients with FD diagnosed by the Rome III, will be launched in Japan ahead of the rest of the world.Also, since Acofide will be the first treatment with FD indication, Zeria and Astellas will co-promote Acofide for the sake of the increase of disease awareness of FD, the prompt market penetration, and the maximization of product potential.
In March 2008, Zeria and Astellas concluded the agreement for the co-development and co-marketing of Acofide and, subsequently conducted the co-development. In September 2010, Zeria submitted the application for marketing approval to the Ministry of Health, Labour and Welfare in Japan.
We believe that Acofide will contribute to alleviate the subjective symptoms and improve QOL of patients with FD.
Indication: Postprandial fullness, upper abdominal bloating, and early satiation due to functional dyspepsia
Dosage regimen: Normally in adults, 100mg of acotiamide hydrochloride hydrate is taken orally three times per day before a meal.
About Functional Dyspepsia (FD)
According to the Rome III, FD is a gastrointestinal disease comprised of subjective symptoms including postprandial fullness, early satiation and epigastric pain without any organic abnormality on gastrointestinal tract. The etiology of FD is still unclear, but it has been shown that delayed gastric emptying is closely associated with FD.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











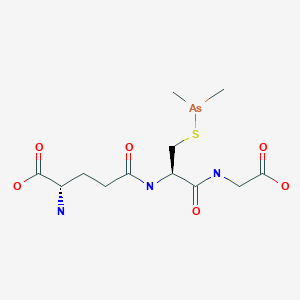
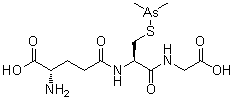








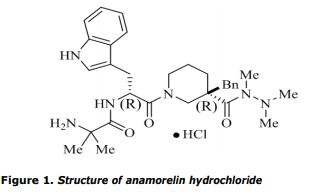























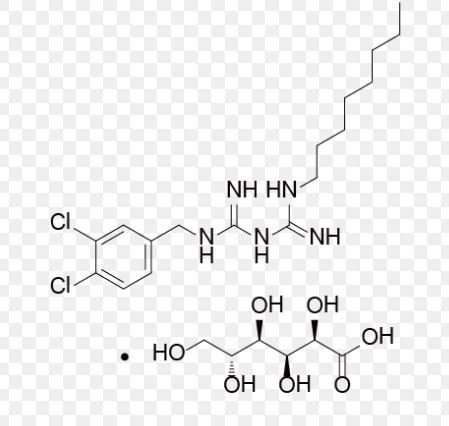





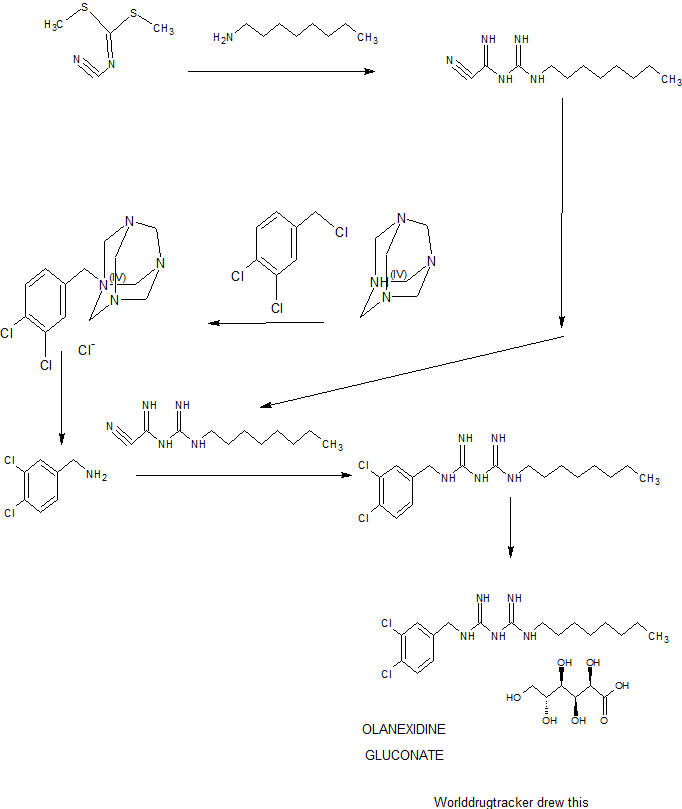


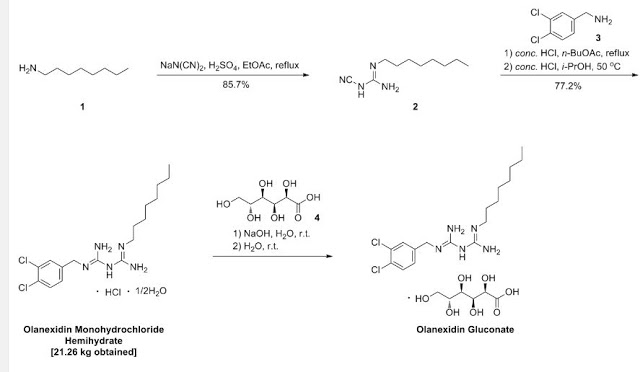
 Eldecalcitol
Eldecalcitol













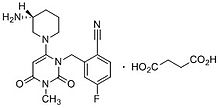










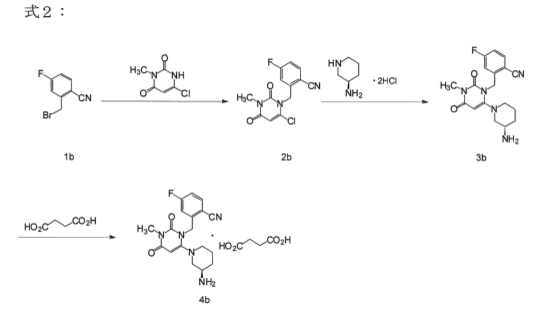




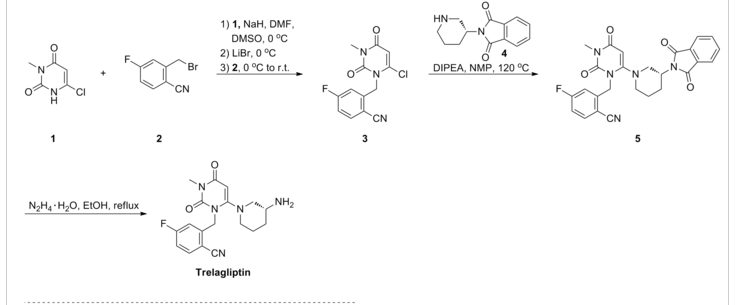
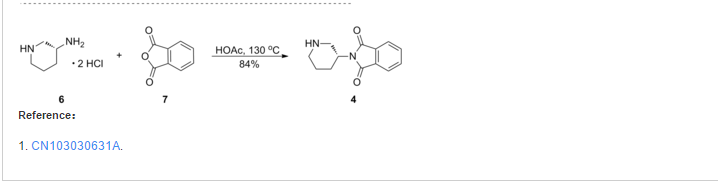
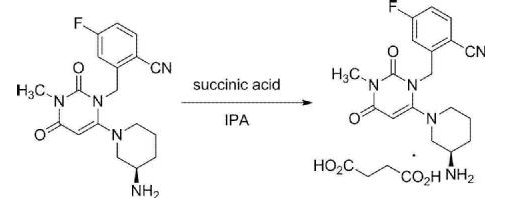







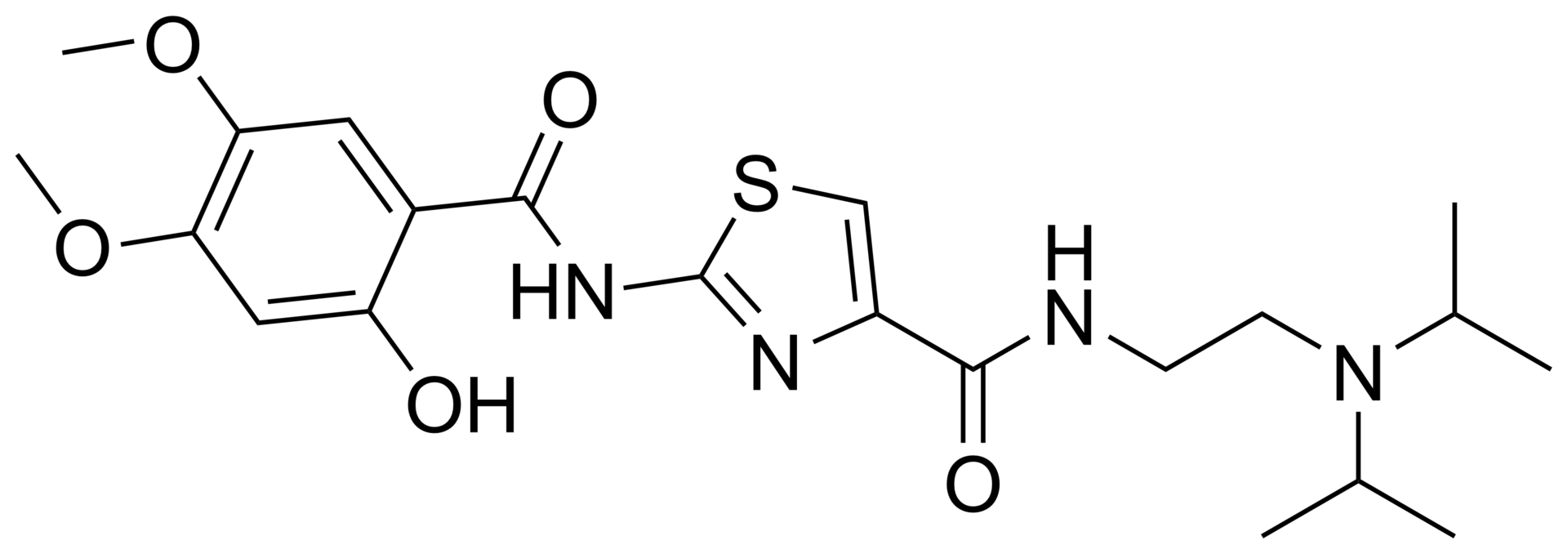





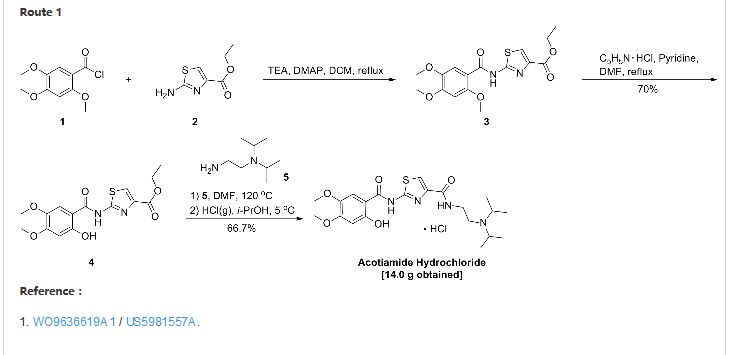

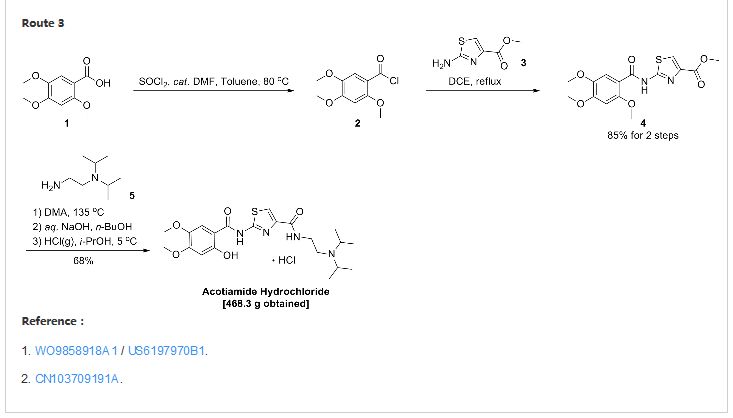






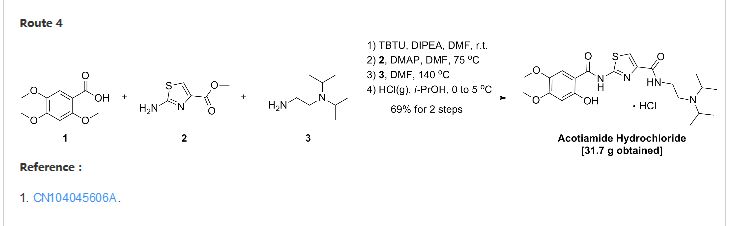
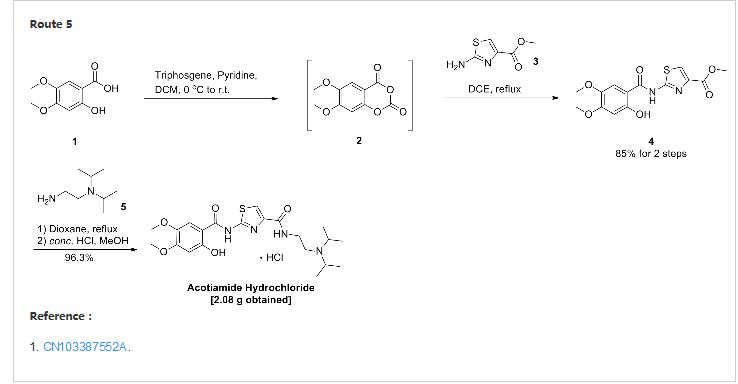





{kind=link}