
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
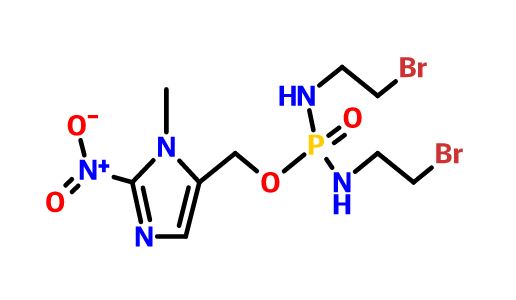

Evofosfamide, HAP-302 , TH-302, TH 302
эвофосфамид , إيفوفوسفاميد , 艾伏磷酰胺 ,
- Molecular Formula C9H16Br2N5O4P
- Average mass 449.036 Da
(1-Methyl-2-nitro-1H-imidazol-5-yl)methyl N,N’-bis(2-bromoethyl)phosphorodiamidate
TH-302 is a nitroimidazole-linked prodrug of a brominated derivative of an isophosphoramide mustard previously used in cancer drugs
- Originator Threshold Pharmaceuticals
- Developer Merck KGaA; Threshold Pharmaceuticals
- Class Antineoplastics; Nitroimidazoles; Phosphoramide mustards; Small molecules
- Mechanism of Action Alkylating agents
- Orphan Drug Status Yes – Soft tissue sarcoma; Pancreatic cancer
- On Fast track Pancreatic cancer; Soft tissue sarcoma
- Suspended Glioblastoma; Leukaemia; Malignant melanoma; Multiple myeloma; Non-small cell lung cancer; Solid tumours
- Discontinued Pancreatic cancer; Soft tissue sarcoma
Most Recent Events
- 01 Aug 2016 Threshold plans a clinical trial for Solid tumours
- 01 Aug 2016 Threshold announces intention to submit NDA to the Pharmaceuticals and Medical Device Agency in Japan
- 16 Jun 2016 Merck KGaA terminates a phase II trial in Soft tissue sarcoma (Combination therapy, Inoperable/Unresectable, Metastatic disease, Late-stage disease) in Japan (IV) due to negative results from the phase III SARC021 trial (NCT02255110)
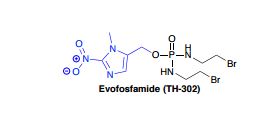
Evofosfamide (first disclosed in WO2007002931), useful for treating cancer.

Threshold Pharmaceuticals and licensee Merck Serono are codeveloping evofosfamide, the lead in a series of topoisomerase II-inhibiting hypoxia-activated prodrugs and a 2-nitroimidazole-triggered bromo analog of ifosfamide, for treating cancer, primarily soft tissue sarcoma and pancreatic cancer (phase 3 clinical, as of April 2015).
In November 2014, the FDA granted Fast Track designation to the drug for the treatment of previously untreated patients with metastatic or locally advanced unresectable soft tissue sarcoma.
Evofosfamide (INN,[1] USAN;[2] formerly known as TH-302) is an investigational hypoxia-activated prodrug that is in clinical development for cancer treatment. The prodrug is activated only at very low levels of oxygen (hypoxia). Such levels are common in human solid tumors, a phenomenon known as tumor hypoxia.[3]
Evofosfamide is being evaluated in clinical trials for the treatment of multiple tumor types as a monotherapy and in combination with chemotherapeutic agents and other targeted cancer drugs.
Dec 2015 : two Phase 3 trials fail, Merck will not apply for a license
Collaboration
Evofosfamide was developed by Threshold Pharmaceuticals Inc. In 2012, Threshold signed a global license and co-development agreement for evofosfamide with Merck KGaA, Darmstadt, Germany (EMD Serono Inc. in the US and Canada), which includes an option for Threshold to co-commercialize evofosfamide in the United States. Threshold is responsible for the development of evofosfamide in the soft tissue sarcoma indication in the United States. In all other cancer indications, Threshold and Merck KGaA are developing evofosfamide together.[4] From 2012 to 2013, Merck KGaA paid 110 million US$ for upfront payment and milestone payments to Threshold. Additionally, Merck KGaA covers 70% of all evofosfamide development expenses.[5]
Mechanism of prodrug activation and Mechanism of action (MOA) of the released drug[edit]
Evofosfamide is a 2-nitroimidazole prodrug of the cytotoxin bromo-isophosphoramide mustard (Br-IPM). Evofosfamide is activated by a process that involves a 1-electron (1 e−) reduction mediated by ubiquitous cellular reductases, such as the NADPH cytochrome P450, to generate a radical anion prodrug:
- A) In the presence of oxygen (normoxia) the radical anion prodrug reacts rapidly with oxygen to generate the original prodrug and superoxide. Therefore, evofosfamide is relatively inert under normal oxygen conditions, remaining intact as a prodrug.
- B) When exposed to severe hypoxic conditions (< 0.5% O2; hypoxic zones in many tumors), however, the radical anion undergoes irreversible fragmentation, releasing the active drug Br-IPM and an azole derivative. The released cytotoxin Br-IPM alkylates DNA, inducing intrastrand and interstrand crosslinks.[6]
Evofosfamide is essentially inactive under normal oxygen levels. In areas of hypoxia, evofosfamide becomes activated and converts to an alkylating cytotoxic agent resulting in DNA cross-linking. This renders cells unable to replicable their DNA and divide, leading to apoptosis. This investigational therapeutic approach of targeting the cytotoxin to hypoxic zones in tumors may cause less broad systemic toxicity that is seen with untargeted cytotoxic chemotherapies.[7]
The activation of evofosfamide to the active drug Br-IPM and the mechanism of action (MOA) via cross-linking of DNA is shown schematically below:

Drug development history
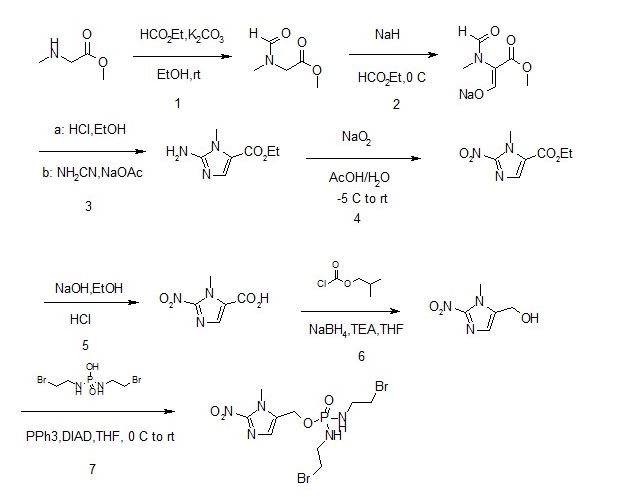
Phosphorodiamidate-based, DNA-crosslinking, bis-alkylator mustards have long been used successfully in cancer chemotherapy and include e.g. the prodrugs ifosfamide andcyclophosphamide. To demonstrate that known drugs of proven efficacy could serve as the basis of efficacious hypoxia-activated prodrugs, the 2-nitroimidizole HAP of the active phosphoramidate bis-alkylator derived from ifosfamide was synthesized. The resulting compound, TH-281, had a high HCR (hypoxia cytotoxicity ratio), a quantitative assessment of its hypoxia selectivity. Subsequent structure-activity relationship (SAR) studies showed that replacement of the chlorines in the alkylator portion of the prodrug with bromines improved potency about 10-fold. The resulting, final compound is evofosfamide (TH-302).[8]
Synthesis
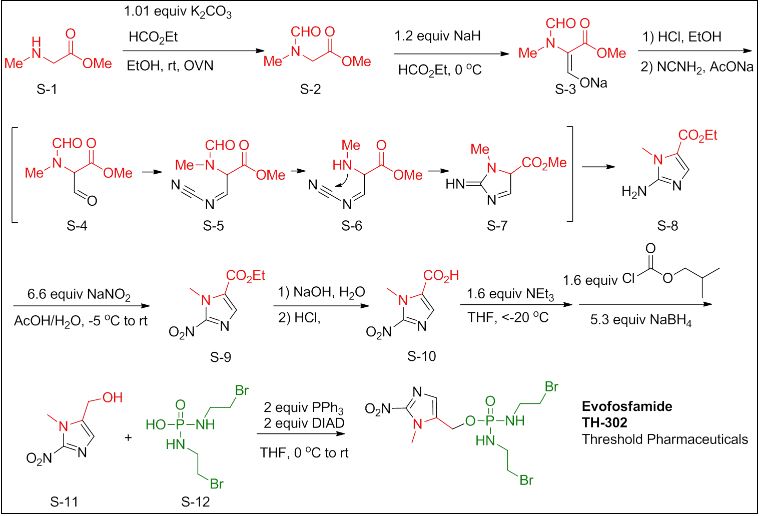
Evofosfamide can be synthesized in 7 steps.[9][10]
- CPhI.cn: Synthetic routes to explore anti-pancreatic cancer drug Evofosfamide, 22 Jan 2015
- Synthetic route Reference: International patent application WO2007002931A2
Formulation
The evofosfamide drug product formulation used until 2011 was a lyophilized powder. The current drug product formulation is a sterile liquid containing ethanol,dimethylacetamide and polysorbate 80. For intravenous infusion, the evofosfamide drug product is diluted in 5% dextrose in WFI.[11]
Diluted evofosfamide formulation (100 mg/ml evofosfamide, 70% ethanol, 25% dimethylacetamide and 5% polysorbate 80; diluted to 4% v/v in 5% dextrose or 0.9% NaCl) can cause leaching of DEHP from infusion bags containing PVC plastic.[12]
Clinical trials
Overview and results
Evofosfamide (TH-302) is currently being evaluated in clinical studies as a monotherapy and in combination with chemotherapy agents and other targeted cancer drugs. The indications are a broad spectrum of solid tumor types and blood cancers.
Evofosfamide clinical trials (as of 21 November 2014)[13] sorted by (Estimated) Primary Completion Date:[14]
Both, evofosfamide and ifosfamide have been investigated in combination with doxorubicin in patients with advanced soft tissue sarcoma. The study TH-CR-403 is a single arm trial investigating evofosfamide in combination with doxorubicin.[35] The study EORTC 62012 compares doxorubicin with doxorubicin plus ifosfamide.[36] Doxorubicin and ifosfamide are generic products sold by many manufacturers.Soft tissue sarcoma
The indirect comparison of both studies shows comparable hematologic toxicity and efficacy profiles of evofosfamide and ifosfamide in combination with doxorubicin. However, a longer overall survival of patients treated with evofosfamide/doxorubicin (TH-CR-403) trial was observed. The reason for this increase is probably the increased number of patients with certain sarcoma subtypes in the evofosfamide/doxorubicin TH-CR-403 trial, see table below.
However, in the Phase 3 TH-CR-406/SARC021 study (conducted in collaboration with the Sarcoma Alliance for Research through Collaboration (SARC)), patients with locally advanced unresectable or metastatic soft tissue sarcoma treated with evofosfamide in combination with doxorubicin did not demonstrate a statistically significant improvement in OS compared with doxorubicin alone (HR: 1.06; 95% CI: 0.88 – 1.29).
Metastatic pancreatic cancer
Both, evofosfamide and protein-bound paclitaxel (nab-paclitaxel) have been investigated in combination with gemcitabine in patients with metastatic pancreatic cancer. The study TH-CR-404 compares gemcitabine with gemcitabine plus evofosfamide.[39] The study CA046 compares gemcitabine with gemcitabine plus nab-paclitaxel.[40] Gemcitabine is a generic product sold by many manufacturers.
The indirect comparison of both studies shows comparable efficacy profiles of evofosfamide and nab-paclitaxel in combination with gemcitabine. However, the hematologic toxicity is increased in patients treated with evofosfamide/gemcitabine (TH-CR-404 trial), see table below.
In the Phase 3 MAESTRO study, patients with previously untreated, locally advanced unresectable or metastatic pancreatic adenocarcinoma treated with evofosfamide in combination with gemcitabine did not demonstrate a statistically significant improvement in overall survival (OS) compared with gemcitabine plus placebo (hazard ratio [HR]: 0.84; 95% confidence interval [CI]: 0.71 – 1.01; p=0.0589).
Drug development risks
Risks published in the quarterly/annual reports of Threshold and Merck KGaA that could affect the further development of evofosfamide (TH-302):
The evofosfamide formulation that Threshold and Merck KGaA are using in the clinical trials was changed in 2011[43] to address issues with storage and handling requirements that were not suitable for a commercial product. Additional testing is ongoing to verify if the new formulation is suitable for a commercial product. If this new formulation is also not suitable for a commercial product another formulation has to be developed and some or all respective clinical phase 3 trials may be required to be repeated which could delay the regulatory approvals.[44]
Even if Threshold/Merck KGaA succeed in obtaining regulatory approvals and bringing evofosfamide to the market, the amount reimbursed for evofosfamide may be insufficient and could adversely affect the profitability of both companies. Obtaining reimbursement for evofosfamide from third-party and governmental payors depend upon a number of factors, e.g. effectiveness of the drug, suitable storage and handling requirements of the drug and advantages over alternative treatments.
There could be the case that the data generated in the clinical trials are sufficient to obtain regulatory approvals for evofosfamide but the use of evofosfamide has a limited benefit for the third-party and governmental payors. In this case Threshold/Merck KGaA could be forced to provide supporting scientific, clinical and cost effectiveness data for the use of evofosfamide to each payor. Threshold/Merck KGaA may not be able to provide data sufficient to obtain reimbursement.[45]
Each cancer indication has a number of established medical therapies with which evofosfamide will compete, for example:
- If approved for commercial sale for pancreatic cancer, evofosfamide would compete with gemcitabine (Gemzar), marketed by Eli Lilly and Company; erlotinib (Tarceva), marketed by Genentech and Astellas Oncology; protein-bound paclitaxel (Abraxane), marketed by Celgene; and FOLFIRINOX, which is a combination of generic products that are sold individually by many manufacturers.
- If approved for commercial sale for soft tissue sarcoma, evofosfamide could potentially compete with doxorubicin or the combination of doxorubicin and ifosfamide, generic products sold by many manufacturers.[46]
Threshold relies on third-party contract manufacturers for the manufacture of evofosfamide to meet its and Merck KGaA’s clinical supply needs. Any inability of the third-party contract manufacturers to produce adequate quantities could adversely affect the clinical development and commercialization of evofosfamide. Furthermore, Threshold has no long-term supply agreements with any of these contract manufacturers and additional agreements for more supplies of evofosfamide will be needed to complete the clinical development and/or commercialize it. In this regard, Merck KGaA has to enter into agreements for additional supplies or develop such capability itself. The clinical programs and the potential commercialization of evofosfamide could be delayed if Merck KGaA is unable to secure the supply.[47]
History
| Date | Event |
|---|---|
| Jun 2005 | Threshold files evofosfamide (TH-302) patent applications in the U.S.[48] |
| Jun 2006 | Threshold files an evofosfamide (TH-302) patent application in the EU and in Japan[49] |
| Sep 2011 | Threshold starts a Phase 3 trial (TH-CR-406) of evofosfamide in combination with doxorubicin in patients with soft tissue sarcoma |
| Feb 2012 | Threshold signs an agreement with Merck KGaA to co-develop evofosfamide |
| Apr 2012 | A Phase 2b trial (TH-CR-404) of evofosfamide in combination with gemcitabine in patients with pancreatic cancer meets primary endpoint |
| Jan 2013 | Merck KGaA starts a global Phase 3 trial (MAESTRO) of evofosfamide in combination with gemcitabine in patients with pancreatic cancer |
| Dec 2015 | two Phase 3 trials fail, Merck will not apply for a license |
CLIP

CLIP
Efficient synthesis of 2-nitroimidazole derivatives and the bioreductive clinical candidate Evofosfamide (TH-302)
E-mail: stuart.conway@chem.ox.ac.uk
DOI: 10.1039/C5QO00211G
http://pubs.rsc.org/en/content/articlelanding/2015/qo/c5qo00211g/unauth#!divAbstract
http://www.rsc.org/suppdata/c5/qo/c5qo00211g/c5qo00211g1.pdf
Hypoxia, regions of low oxygen, occurs in a range of biological environments, and is involved in human diseases, most notably solid tumours. Exploiting the physiological differences arising from low oxygen conditions provides an opportunity for development of targeted therapies, through the use of bioreductive prodrugs, which are selectively activated in hypoxia. Herein, we describe an improved method for synthesising the most widely used bioreductive group, 2-nitroimidazole. The improved method is applied to an efficient synthesis of the anti-cancer drug Evofosfamide (TH-302), which is currently in Phase III clinical trials for treatment of a range of cancers.


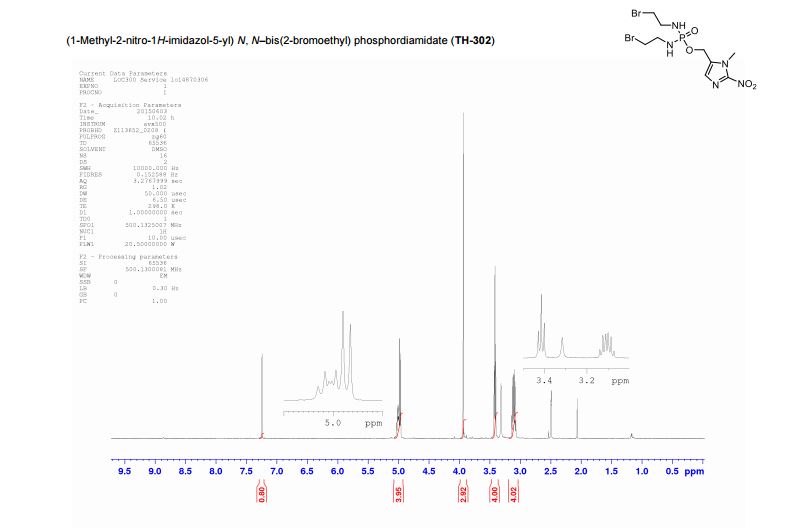
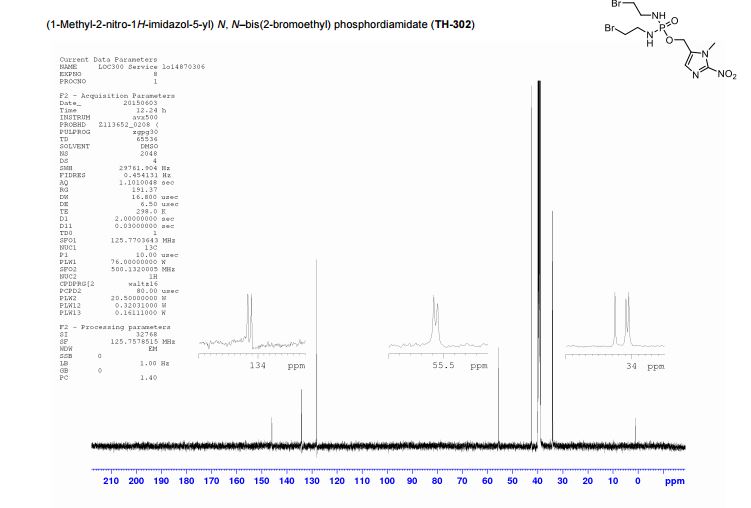
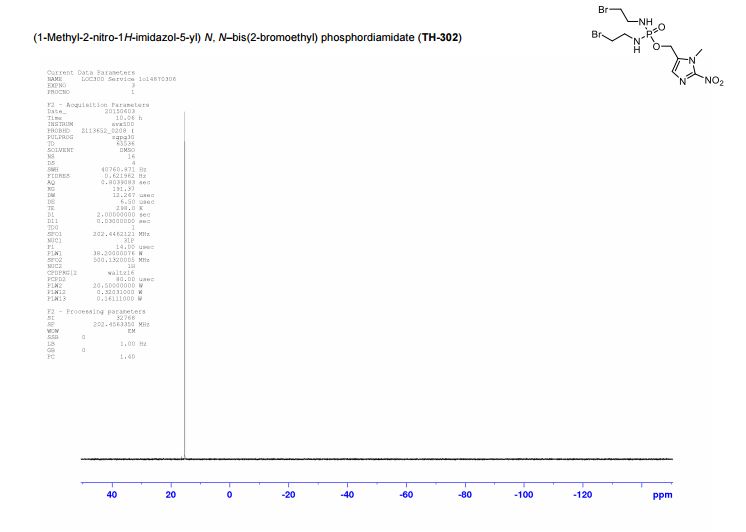
(1-Methyl-2-nitro-1H-imidazol-5-yl)-N,N–bis(2-bromoethyl) phosphordiamidate (TH- 302)
The residue was then purified by semi-preparative HPLC on a Phenomenex Luna (C18(2), 10 µm, 250 × 10 mm) column, eluting with H2O and methanol (50 – 70% methanol over 10 min, then 1 min wash with methanol, 5 mL/min flow rate) to afford TH-302 as a yellow gum: vmax (solid) cm-1 : 3212 (br), 1489 (m), 1350 (m), 1105 (m), 1004 (s); δH (DMSO-D6, 400 MHz) 7.25 (1H, s, CH), 5.10–4.90 (2H, m, NHCH2CH2Br), 4.98 (2H, d, J 7.8, CH2O), 3.94 (3H, s, CH3), 3.42 (4H, t, J 7.0, NHCH2CH2Br), 3.11 (4H, dt, J 9.8, 7.2, NHCH2CH2Br); δC (DMSO-D6, 126 MHz) 146.1, 134.2 (d, J 7.5, OCH2CN), 128.2, 55.6 (d, J 4.6, CH2O), 42.7, 34.2 (d, J 26.4, CH2Br), 34.1; δP (DMSO-D6, 202 MHz) 15.4; HRMS m/z (ESI− ) [found; (M-H)− 447.9216, C9H16 79Br81BrN5O4P requires (M-H)− 447.9213]; m/z (ESI+ ) 448.0 ([M-H]− , 60%, [C9H15 79Br81BrN5O4P] − ), 493.9 ([M+formate] − , 100%, [C10H17 79Br81BrN5O6P] − ). These data are in good agreement with the literature values.4
4 J.-X. Duan, H. Jiao, J. Kaizerman, T. Stanton, J. W. Evans, L. Lan, G. Lorente, M. Banica, D. Jung, J. Wang, H. Ma, X. Li, Z. Yang, R. M. Hoffman, W. S. Ammons, C. P. Hart and M. Matteucci, J. Med. Chem., 2008, 51, 2412–2420.
J. Med. Chem., 2008, 51, 2412–2420/……………….1-Methyl-2-nitro-1H-imidazol-5-yl)methyl N,N-bis(2-bromoethyl)
phosphordiami-date (3b). Compound 3b was synthesized by a procedure similar to that described for 3a and obtained as an off-white solid in 47.6% yield.
1H NMR (DMSO-d6) δ: 7.22 (s, 1H), 5.10–5.00 (m, 2H), 4.97 (d, J ) 7.6 Hz, 2H), 3.94 (s, 3H), 3.42 (t, J ) 7.2 Hz, 4H), and 3.00–3.20 (m, 4H).
13C NMR (DMSOd6)δ: 146.04, 134.16 (d, J ) 32 Hz), 128.17, 55.64, 42.70, 34.33,and 34.11 (d, J ) 17.2 Hz).
31P NMR (DMSO-d6) δ: -11.25.
HRMS: Calcd for C9H16N5O4PBr2, 446.9307; found, 446.9294.
CLIP
Synthesis Route reference WO2007002931A2
Med J.. Chem. 2008, 51, 2412-2420
From compound S-1 starting aminoacyl protection is S-2 , a suspension of NaH grab α -proton, offensive, ethyl, acidification, introduction of an aldehyde group, S-3followed by condensation with the amino nitrile, off N- acyl ring closure, migration rearrangement amino imidazole compound S-. 8 , the amino and sodium nitrite into a diazonium salt, raising the temperature, nitrite anion nucleophilic attack diazonium salt obtained nitro compound S-9, under alkaline conditions ester hydrolysis gives acid S-10 , followed by NEt3 under the action of isobutyl chloroformate and the reaction mixed anhydride formed by of NaBH 4 reduction to give the alcohol S-. 11 , [use of NaBH 4 reduction of the carboxyl group is another way and the I 2 / of NaBH 4 ] , to give S-11 later, the DIAD / PPh3 3 under the action via Mitsunobu linking two fragments obtained reaction Evofosfamide
.
PATENT
http://www.google.co.in/patents/WO2015051921A1?cl=en
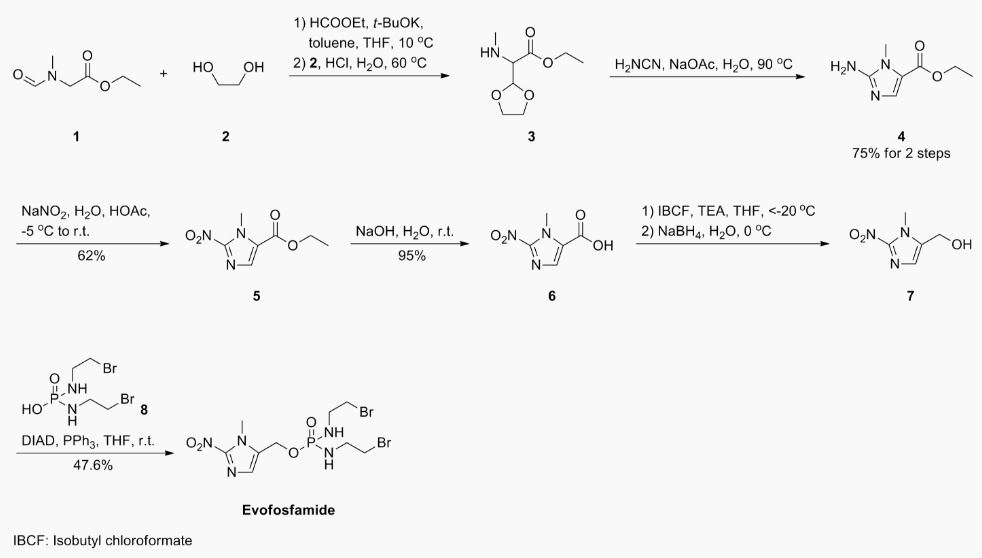
EXAMPLE 1

1
N-Formylsarcosine ethyl ester 1 (1 ,85 kg) was dissolved in toluene (3,9 kg) and ethyl formate (3,28 kg) and cooled to 10 °C. A 20 wt-% solution of potassium tert-butoxide (1 ,84 kg) in tetrahydrofuran (7,4 kg) was added and stirring was continued for 3h. The reaction mixture was extracted 2x with a solution of sodium chloride in water (10 wt-%) and the combined water extracts were washed lx with toluene.
Aqueous hydrogen chloride (25% wt-%; 5,62 kg) was added to the aqueous solution, followed by ethylene glycol (2,36 kg). The reaction mixture was heated to 55-60 °C for lh before only the organic solvent residues were distilled off under vacuum.
Aqueous Cyanamide (50 wt-%, 2,16 kg) was then added at 20 °C, followed by sodium acetate (3,04 kg). The resulting reaction mixture was heated to 85-90 °C for 2h and cooled to 0-5 °C before a pH of ~ 8-9 was adjusted via addition of aqueous sodium hydroxide (32% wt-%; 4,1 kg). Compound 3 (1,66 kg; 75%) was isolated after filtration and washing with water.
Ή-NMR (400 MHz, d6-DMSO): δ= 1,24 (3H, t, J= 7,1 Hz); 3,53 (3H, s); 4,16 (2H, q, J= 7,0 Hz) ; 6,15 (s, 2 H); 7,28 (s, 1H).
HPLC (Rt = 7,7 min): 97,9% (a/a).
HPLC data was obtained using Agilent 1100 series HPLC from agilent technologies using an Column: YMC-Triart CI 8 3μ, 100 x 4,6 mm Solvent A: 950 ml of ammonium acetate/acetic acid buffer at pH = 6 + 50 ml acetonitril; Solvent B: 200 ml of ammonium acetate/acetic acid buffer at pH = 6 + 800 ml acetonitril; Flow: 1,5 ml/min; Gradient: 0 min: 5 % B, 2 min: 5 % B, 7 min: 20 % B, 17 min: 85% B, 17, 1 min: 5% B, 22 min: 5% B.
PATENT
WO2007002931
http://www.google.com/patents/WO2007002931A2?cl=en
Example 8
Synthesis of Compounds 25, 26 [0380] To a solution of 2-bromoethylammmonium bromide (19.4 g) in DCM (90 mL) at – 1O0C was added a solution OfPOCl3 (2.3 mL) in DCM (4 mL) followed by addition of a solution of TEA (14.1 mL) in DCM (25 mL). The reaction mixture was filtered, the filtrate concentrated to ca. 30% of the original volume and filtered. The residue was washed with DCM (3×25 mL) and the combined DCM portions concentrated to yield a solid to which a mixture of THF (6 mL) and water (8 mL) was added. THF was removed in a rotary evaporator, the resulting solution chilled overnight in a fridge. The precipitate obtained was filtered, washed with water (10 mL) and ether (30 mL), and dryed in vacuo to yield 2.1 g of:

Isophosphoramide mustard

can be synthesized employing the method provided in Example 8, substituting 2- bromoethylammmonium bromide with 2-chloroethylammmonium chloride. Synthesis of Isophosphoramide mustard has been described (see for example Wiessler et al., supra).
The phosphoramidate alkylator toxin:

was transformed into compounds 24 and 25, employing the method provided in Example 6 and the appropriate Trigger-OH.
Example 25
Synthesis of l-N-methyl-2-nitroimidazole-5-carboxylis acid

A suspension of the nitro ester (39.2 g, 196.9 rnmol) in IN NaOH (600 mL) and water (200 mL) was stirred at rt for about 20 h to give a clear light brown solution. The pH of the reaction mixture was adjusted to about 1 by addition of cone. HCl and the reaction mixture extracted with EA (5 x 150 mL). The combined ethyl acetate layers were dried over MgS O4 and concentrated to yield l-N-methyl-2-nitroimidazole-5-carboxylis acid (“nitro acid”) as a light brown solid (32.2 g, 95%). Example 26
Synthesis of l-N-methyl-2-nitroimidazole-5-carboxylis acid

A mixture of the nitro acid (30.82 g, 180.23 mmol) and triethylamine (140 niL, 285 mmol) in anhydrous THF (360 mL) was stirred while the reaction mixture was cooled in a dry ice-acetonitrile bath (temperature < -20 0C). Isobutyl chloroformate (37.8 mL, 288 mmol) was added drop wise to this cooled reaction mixture during a period of 10 min and stirred for 1 h followed by the addition of sodium borohydride (36 g, 947 mmol) and dropwise addition of water during a period of 1 h while maintaining a temperature around or less than O0C. The reaction mixture was warmed up to O0C. The solid was filtered off and washed with THF. The combined THF portions were evaporated to yield l-N-methyl-2- nitroimidazole-5-methanol as an orange solid (25 g) which was recrystallized from ethyl acetate.
PATENT
WO-2015051921

EXAMPLE 1

1
N-Formylsarcosine ethyl ester 1 (1 ,85 kg) was dissolved in toluene (3,9 kg) and ethyl formate (3,28 kg) and cooled to 10 °C. A 20 wt-% solution of potassium tert-butoxide (1 ,84 kg) in tetrahydrofuran (7,4 kg) was added and stirring was continued for 3h. The reaction mixture was extracted 2x with a solution of sodium chloride in water (10 wt-%) and the combined water extracts were washed lx with toluene.
Aqueous hydrogen chloride (25% wt-%; 5,62 kg) was added to the aqueous solution, followed by ethylene glycol (2,36 kg). The reaction mixture was heated to 55-60 °C for lh before only the organic solvent residues were distilled off under vacuum.
Aqueous Cyanamide (50 wt-%, 2,16 kg) was then added at 20 °C, followed by sodium acetate (3,04 kg). The resulting reaction mixture was heated to 85-90 °C for 2h and cooled to 0-5 °C before a pH of ~ 8-9 was adjusted via addition of aqueous sodium hydroxide (32% wt-%; 4,1 kg). Compound 3 (1,66 kg; 75%) was isolated after filtration and washing with water.
Ή-NMR (400 MHz, d6-DMSO): δ= 1,24 (3H, t, J= 7,1 Hz); 3,53 (3H, s); 4,16 (2H, q, J= 7,0 Hz) ; 6,15 (s, 2 H); 7,28 (s, 1H).
HPLC (Rt = 7,7 min): 97,9% (a/a).
PATENT
WO 2016011195
http://google.com/patents/WO2016011195A1?cl=en
Figure 1 provides the differential scanning calorimetry (DSC) data of crystalline solid form A of TH-302.
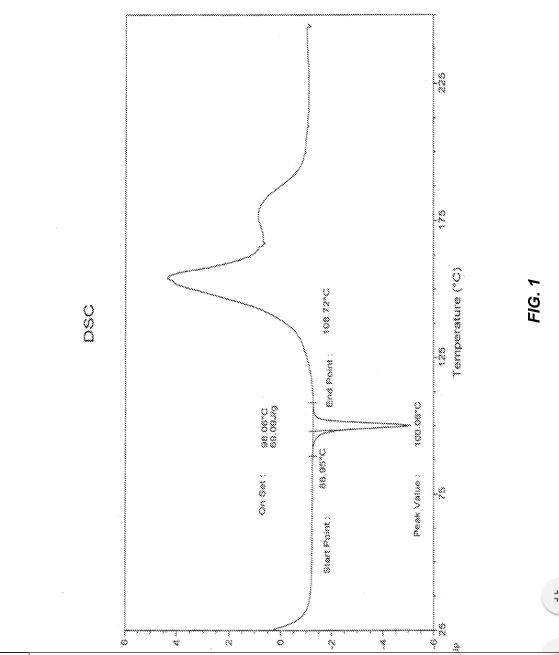
Figure 2 shows the 1H-NMR of crystalline solid form A of TH-302.

Figure 5 shows the Raman Spectra of TH-302 (Form A)

Scheme 1 illustrates a method of preparing TH-302.
Scheme 1: Process for the Preparation of TH-302 
NaOH (RGT)
Step 1. Imidazole Purified water (SLV)
Carboxylic Acid IPC: NMT 1.0% SM by HPLC
HCI (RGT)
IPC: pH 1.0 ± 0.5
IPC: NMT 1.0% water by KF

TH-302
MW = 449.0
SM = Starting Material INT = Intermediate IPC = In-process Control RGT = Reagent SLV = Solvent MW = Molecular Weight LOD = Loss on drying NMT = Not more than NLT = Not less than
TH-302 can be prepared by hydro lyzing (l-methyl-2-nitro-lH-imidazol-5-yl) ethyl ester above for example under aqueous conditions with a suitable base catalyst (e.g. NaOH in water at room temperature). The imidazole carboxylic acid prepared by this method can be used without further purification. However, it has been found that treating the dried crude intermediate product with a solvent such as acetonitrile, ethyl acetate, n-heptane, acetone, dimethylacetamide, dimethylformamide, 1, 4-dioxane, ethylene glycol, 2-propanol, 1-propanol, tetrahydrofuran (1 : 10 w/v) or combinations thereof in a vessel with heating, followed by cooling and filtration through a filtration aid with acetone decreased the number and levels of impurities in the product. The number and levels of impurities could be further reduced by treating the dried crude product with water (1 :5.0 w/v) in a vessel with heating followed by cooling and filtration through a filtration aid with water.
The carboxylic acid of the imidazole can then be reduced using an excess of a suitable reducing agent (e.g. sodium borohydride in an appropriate solvent, typically aqueous. The reaction is exothermic (i.e. potentially explosive) releasing borane and hydrogen gases over several hours. It was determined that the oxygen balance of the product imidazole alcohol is about 106.9, which suggests a high propensity for rapid decomposition. It has been found that using NaOH, for example 0.01M NaOH followed by quenching the reaction with an acid. Non-limiting examples of acids include, but are not limited to water, acetic acid, hydrobromic acid, hydrochloric acid, sodium hydrogen phosphate, sulfuric acid, citric acid, carbonic acid, phosphoric acid, oxalic acid, boric acid and combinations thereof. In some embodiments, the acid may diluted with a solvent, such as water and/or tetrahydrofuran. In some embodiments, acetic acid or hydrochloric acid provide a better safety profile, presumably because it is easier to control the temperature during the addition of the reducing agent and the excess reducing agent is destroyed after the reaction is complete. This also results in improved yields and fewer impurities, presumably due to reduced impurities from the reducing agent and decomposition of the product. Using this process, greater than 98.5% purity could be achieved for this intermediate. The formation of ether linkage can be accomplished by treating the product imidazole alcohol with solution of N,N’-Bis(2-bromoethyl)phosphorodiamidic acid (Bromo IPM), a trisubstituted phosphine and diisopropyl azodicarboxylate in tetrahydrofuran at room temperature to afford TH-302. It has been found that by recrystallizing the product from a solvents listed in the examples, one could avoid further purfication by column chromatography, which allowed for both reduced solvent use especially on larger scales.
Scheme 2 illustrates an alternative method of preparing TH-302.
Scheme 2: Process for the Preparation of TH-302
(SM)
ethylamine mide (SM) 04.9 ) SLV) , RGT) ter by KF
NT)

MW = 449.0
Example 1: Synthesis of TH-302
Step 1 – Preparation intermediate imidazole carboxylic
I T)

Crude imidazole carboxylic acid ethyl ester (1 : 1.0 w/w) was taken in water (1 : 10.0 w/v) at 25± 5°C and cooled to 17± 3°C. A 2.5 N sodium hydroxide solution (10 V) was added slowly at 17±3°C. The reaction mass was warmed to 25±5°C and monitored by HPLC. After the completion of reaction, the reaction mass was cooled to 3±2°C and pH of the reaction mass adjusted to 1=1=0.5 using 6 M HC1 at 3±2°C. The reaction mass was then warmed to 25±5°C and extracted with ethyl acetate (3 x 10 V). The combined organic layers
were washed with water (1 x 10 V) followed by brine (1 x 10 V). The organic layer was dried over sodium sulfate (3 w/w), filtered over Celite and concentrated. n-Heptane (1.0 w/v) was added and the the reaction mixture was concentrated below 45°C to 2.0 w/v. The reaction mass was cooled to 0±5°C. The solid was filtered, and the bed was washed with n-heptane (1 x 0.5 w/v) and dried at 35±5°C. In a vessel, acetone (1 : 10 w/v) was added. Dry crude imidazole carboxylic acid (ICA) from 1.12 was added to the acetone. The mixture was warmed to 45±5°C and was stirred for 30 minutes. The mass was cooled to 28±3°C and filtered through a Celite bed. The filter bed was washed with 1 : 1.0 w/v of acetone. Water (1 :5.0 w/v) was added to the filtrate and the mixture was concentrated. The concentrated mass was cooled to 5±5°C and stirred for 30 minutes. The material was filtered and the solid was washed 2 x 1 : 1.0 w/v of water at 3±2°C. The product was dried for 2 hours at 25±5°C and then at 45±5°C. As can be seen below, the number and levels of impurities are decreased.
Table I: Purity and Impurity Profile Comparison of Typical Crude ICA and Purified
ICA


Imidazole alcohol:
CI^Oi-Bu
T
o

Imidazole carboxylic acid (1.0 w/w) was taken in tetrahydrofuran (10 w/v) under nitrogen atmosphere at 25±5°C. The reaction mass was cooled to -15±5°C. Triethylamine (1 : 1.23 w/v) was added slowly over a period of 1 hour maintaining the temperature at – 15±5°C. The reaction mass was stirred at -15±5°C for 15-20 min. Isobutylchloroformate (1 : 1.14 w/v) was added slowly over a period of 1 hour maintaining the temperature at – 15±5°C. The reaction mass was stirred at -15±5°C for 30-40 min. A solution of sodium borohydride (1 : 1.15 w/w) in 0.01 M aqueous sodium hydroxide (2.2 w/v) was divided into 6 lots and added to the above reaction mass while maintaining the temperature of the reaction mass between 0±10°C for 40-60 min for each lot. The reaction mass was warmed to 25±5°C and stirred until imidazole carboxylic acid content < 5.0 % w/w. The reaction mass was filtered and the bed was washed with tetrahydrofuran (1 :2.5 w/v). The filtrate was quenched with 10 % acetic acid in water at 25±5°C. Reaction mass stirred for 50-60 minutes at 25±5°C. The filtrate was concentrated below 45°C until no distillate was observed. The mass was cooled to 5±5°C and stirred for 50-60 minutes. The reaction mass was filtered and the solid was taken in ethanol (1 :0.53 w/v). The reaction mass was cooled 0±5°C and stirred for 30-40 min. The solid was filtered and the bed was washed ethanol (1 :0.13 w/v). The solid was dried at 40±5 °C.
Step 3 – Synthesis of intermediate Br-IPM:
P
o
M 
W = 286.7 MW = 204.9 Purified water (SLV, RGT)
Acetone (SLV)
IPC: NMT 1.0% water by KF
2-Bromoethylamine hydrobromide (1 : 1.0 w/w) and POBr^ (1 :0.7 w/w) were taken in DCM (1 :2 w/v) under nitrogen atmosphere. The reaction mixture was cooled to -70±5°C. Triethylamine (1 : 1.36 w/v) in DCM (1 :5 w/v) was added to the reaction mass at -70±5°C. The reaction mass was stirred for additional 30 min at -70±5°C. Reaction mass was warmed to 0±3°C and water (1 :1.72 w/v) was added. The reaction mixture was stirred at 0±3°C for 4 hrs. The solid obtained was filtered and filter cake was washed with ice cold water (2 x 1 :0.86 w/v) and then with chilled acetone (2 x 1 :0.86 w/v). The solid was dried in at 20±5°C.
Step 4 Synthesis ofTH-302

TH-302
MW = 449.0
Imidazole alcohol (IA) (1 : 1.0 w/w), Bromo-IPM (1 :2.26 w/w) and
triphenylphosphine (1 :2.0 w/w) were added to THF (1 : 13.5 w/v) at 25±5°C. The reaction
mass was cooled to 0±5°C and DIAD (1.5 w/v) was added. The reaction mixture warmed to 25±5°C and stirred for 2 hours. Progress of the reaction was monitored by HPLC. Solvent was removed below 50°C under vacuum. Solvent exchange with acetonitrile (1 :10.0 w/v) below 50°C was performed. The syrupy liquid was re-dissolved in acetonitrile (1 : 10.0 w/v) and the mixture was stirred at -20±5°C for 1 hour. The resulting solid was filtered and the filtrate bed was washed with chilled acetonitrile (1 : 1.0 w/v). The acetonitrile filtrate was concentrated below 50°C under vacuum. The concentrated mass was re-dissolved in ethyl acetate (1 : 10.0 w/v) and concentrated below 50°C under vacuum. The ethyl acetate strip off was repeated two more times. Ethyl acetate (1 : 10.0 w/v) and silica gel (230-400 mesh, 1 :5.3 w/w) were added to the concentrated reaction mass. The mixture was concentrated below 40°C under vacuum. n-Heptane (1 :5.0 w/v) was charged to the above mass and the mixture was evaporated below 40°C under vacuum. n-Heptane (1 :5.0 w/v) was again added to the above mass and the solid was filtered and the bed was washed with n-heptane (1 : 1.0 w/v). The solid was suspended in a mixture oftoluene (1 :7.1 w/v) and n-heptane (1 :21.3 w/v), stirred at 35±5°C for 15-20 minutes, filtered off and the bed was washed with n-heptane
(1 : 1.0 w/v). The solid was re-suspended in a mixture of toluene (1 : 10.6 w/v) and n-heptane (1 : 10.6 w/v), stirred at 35±5°C for 15-20 minutes, filtered off and the bed was washed with n-heptane (1 : 1.0 w/v). The solid was suspended in acetone (1 : 19.0 w/v), stirred at 35±5°C for 15-20 minutes, filtered off and the bed was washed with acetone (1 : 1.0 w/v). The acetone washes were repeated 3 more times. Filtrates from the above acetone washings were combined and concentrated below 40°C under vacuum. The residue dissolved in ethyl acetate (1 : 10.0 w/v) and concentrated below 40°C under vacuum. The ethyl acetate strip off was repeated one more time. The residue was re-dissolved in ethyl acetate (1 :5.5 w/v), cooled to 0±3°C and stirred at 0±3°C for 2 h and then at -20±5°C for 2 h. The solid was filtered and the solid was washed with ethyl acetate (1 :0.10 w/v). The solid was dissolved in ethyl acetate (1 : 10.0 w/v) at 50±5°C and the resulting solution was filtered through a cartridge filter. The filtrate was concentrated to ~4.0 w/w and stirred at 0±3°C for 4 hours. The solid was filtered and washed with ethyl acetate (1 :0.10 w/v). The crystallization from ethyl acetate was repeated and TH-302 was dried at 25±5°C. Table 2 shows how the process reduces solvent use.
Table 2: Solvent and Silica Gel Usage for 10 kg Column and 10 kg Column-free Purification

“Amounts are estimated from a 5 kg batch
b Amounts are estimated
Example 2: Synthesis ofTH-302 using alternative procedure to purify ICA:
Crude ICA was prepared according to the method described in Example 1. In a vessel, water (1 :7.0 w/v) was added. Dry crude ICA was added to the water. The reaction mixture was heated to 85±5°C until a clear solution was obtained. The reaction mass was cooled to 20±5°C and filtered through a Celite bed. The filter bed was washed with 2 x 5.0 of n-heptane. The material was dried for 2 hours at 25±5°C and then 45±5°C. As can be seen below, the number and levels of impurities decreased.
Table 3: Purity and Impurity Profile Comparison of Typical Crude ICA and Purified
ICA

Example 3: Synthesis ofTH-302 using alternative procedure to purify ICA:
Crude ICA was prepared according to the method described in Example 1. In a vessel
ethanol (1 :30.0 w/v) and ICA (1 : 1.0 w/w) were mixed. The reaction mixture was stirred at
25±5°C for 30 minutes and filtered. Water (1 :50.0 w/v) was added and the mixture was
stirred at 50±5°C for 30 minutes. The reaction mass was cooled to 20±5°C and filtered. The isolated solid was dried at 25±5°C for 24 hours. As can be seen below, the number and levels
of impurities generally decreased.
Table 4: Purity and Impurity Profile Comparison of Typical Crude ICA and Purified
ICA

Example 4: Synthesis ofTH-302 using alternative procedure to purify ICA:
Crude ICA was prepared according to the method described in Example 1. In a vessel
acetonitrile (1 :20.0 w/v) and ICA (1 : 1.0 w/w) were mixed at 25±5°C for one hour. The
reaction mixture was filtered and the solution was concentrated to ~ 6 volumes. The mixture
was then cooled to 0±5°C, stirred at this temperature for one hour and filtered. The isolated
solid was dried at 25±5°C for 24 hours. As can be seen below the number of impurities
decreased and except for TH-2717, the amounts also decreased.
Table 5: Purity and Impurity Profile Comparison of Typical Crude ICA and Purified
ICA

Example 5: Synthesis ofTH-302 using alternative procedure to purify ICA:
Crude ICA is prepared according to the method described in Example 1 and purified by treatment with dimethylacetamide and water.
Example 6: Synthesis ofTH-302 using alternative procedure to purify ICA:
Crude ICA is prepared according to the method described in Example 1 and purified by treatment with dimethylforamide and water.
Example 7: Synthesis ofTH-302 using alternative procedure to purify ICA:
[0109] Crude ICA is prepared according to the method described in Example 1 and purified by crystallization from a 1,4-dioxane and water mixture.
Example 8: Synthesis ofTH-302 using alternative procedure to purify ICA:
Crude ICA is prepared according to the method described in Example 1 and purified by crystallization from a mixture of ethylene glycol and water.
Example 9: Synthesis ofTH-302 using alternative procedure to purify ICA:
Crude ICA is prepared according to the method described in Example 1 and purified by treatment with 2-propanol and water.
Example 10: Synthesis ofTH-302 using alternative procedure to purify ICA:
[0112] Crude ICA is prepared according to the method described in Example 1 and purified by treatment with 1-propanol and water.
Example 11: Synthesis ofTH-302 using alternative procedure to purify ICA:
[0113] Crude ICA is prepared according to the method described in Example 1 and purified by crystallization from a mixture of tetrahydrofuran and water.
Example 12: Synthesis ofTH-302 using alternative procedure to quench IA:
[0114] The reduction of ICA to IA was carried out according to Example 1 except that after reaction completion and filtration of the inorganics, the filtrate was quenched with 1.5 M hydrochloric acid.
Example 13: Synthesis ofTH-302 using alternative procedure to quench IA:
[0115] The reduction of ICA to IA was carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate was quenched with 1.5 M
hydrobromic acid.
Example 14: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA was carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate was quenched with
hydrobromic acid in acetic acid.
Example 15: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA was carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate was treated with sodium
hydrogen phosphate.
Example 16: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA was carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate was quenched with 10% acetic
acid in tetrahydrofuran.
Example 17: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA was carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate was quenched with water.
Example 18: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA is carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate is quenched with sulfuric acid.
Example 19: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA is carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate is quenched with citric acid.
Example 20: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA is carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate is treated with carbonic acid.
Example 21: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA is carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate is treated with phosphoric
acid.
Example 22: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA is carried out according to Example 1 except that after
reaction completion and filtration of the inorganics, the filtrate is quenched with oxalic acid.
Example 23: Synthesis ofTH-302 using alternative procedure to quench IA:
The reduction of ICA to IA is carried out according to Example 1 except that after reaction completion and filtration of the inorganics, the filtrate is quenched with boric acid.
Example 24: Synthesis ofTH-302 using alternative procedure to purify TH-302:
[0126] Coupling of bromo-IPM and IA was performed according to Example 1 except that after concentration of the reaction mixture, ethyl acetate (1 : 10 w/v) was added to the concentrated mass. The mixture was stirred at -55±5°C for 2 hours. The resulting solid was filtered and washed with chilled EtOAc (1 :2.0 w/v). The solid was reslurried in ethyl acetate (1 : 10 w/v) at -55±5°C for 2 hours, filtered and the solid was washed with chilled ethyl acetate (1 : 1.0 w/v). The filtrates from both filtrations were combined and treated with silica gel (1 :5.3 w/w) of silica gel (230-400 mesh). The mixture was concentrated below 40°C under vacuum. n-Heptane (1 :5.0 w/v) was again added to the above mass and the solid was filtered and the bed was washed with n-heptane (1 : 1.0 w/v). The solid was suspended in a mixture of toluene (1 :7.1 w/v) and n-heptane (1 :21.3 w/v), stirred at 35±5°C for 15-20 minutes, filtered off and the bed was washed with n-heptane (1 : 1.0 w/v). The solid was re-suspended in a mixture of toluene (1 : 10.6 w/v) and n-heptane (1 :10.6 w/v), stirred at 35±5°C for 15-20 minutes, filtered off and the bed was washed with n-heptane (1 : 1.0 w/v). The solid was suspended in acetone (1 : 19.0 w/v), stirred at 35±5°C for 15-20 minutes, filtered off and the bed was washed with acetone (1 : 1.0 w/v). The acetone washes were repeated 3 more times. Filtrates from the above acetone washings were combined and concentrated below 40°C under vacuum. The residue dissolved in ethyl acetate (1 :5.5 w/v), cooled to 0±3°C and stirred at 0±3°C for 2 h and then at -20±5°C for 2 h. The solid was filtered and the solid was washed with ethyl acetate (1 :0.10 w/v). The solid was dissolved in ethyl acetate (1 :27 w/v), stirred at 50±5°C and filtered through Celite. The filtrate was concentrated to ~4.0 w/w and stirred at 0±5°C for 4 hours. The recrystallization from ethyl acetate was repeated and TH- 302 was dried at 25±5°C. Table 4 shows how the process reduced solvent use.
Table 4: Estimated Solvent and Silica Gel Usage for Column and 10 kg Column-free
(EtOAc) Purification

References
- WHO Drug Information; Recommended INN: List 73
- Adopted Names of the United States Adopted Names Council
- Duan J; Jiao, H; Kaizerman, J; Stanton, T; Evans, JW; Lan, L; Lorente, G; Banica, M; et al. (2008). “Potent and Highly Selective Hypoxia-Activated Achiral Phosphoramidate Mustards as Anticancer Drugs”. J. Med. Chem. 51 (8): 2412–20. doi:10.1021/jm701028q.PMID 18257544.
- Jump up^ Threshold Pharmaceuticals and Merck KGaA Announce Global Agreement to Co-Develop and Commercialize Phase 3 Hypoxia-Targeted Drug TH-302 – Press release from 3 February 2012
- Jump up^ Threshold Pharmaceuticals Form 8-K from 3 Nov 2014
- Jump up^ Weiss, G.J., Infante, J.R., Chiorean, E.G., Borad, M.J., Bendell, J.C., Molina, J.R., Tibes, R., Ramanathan, R.K., Lewandowski, K., Jones, S.F., Lacouture, M.E., Langmuir, V.K., Lee, H., Kroll, S., Burris, H.A. (2011) Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of TH-302, a Hypoxia-Activated Prodrug, in Patients with Advanced Solid Malignancies. Clinical Cancer Research 17, 2997–3004.doi:10.1158/1078-0432.CCR-10-3425
- J. Thomas Pento (2011). “TH-302”. Drugs of the Future. 36 (9): 663–667.doi:10.1358/dof.2011.036.09.1678337.
- Jump up^ Duan J; Jiao, H; Kaizerman, J; Stanton, T; Evans, JW; Lan, L; Lorente, G; Banica, M; et al. (2008). “Potent and Highly Selective Hypoxia-Activated Achiral Phosphoramidate Mustards as Anticancer Drugs”. J. Med. Chem. 51 (8): 2412–20. doi:10.1021/jm701028q.PMID 18257544.
- Jump up^ CPhI.cn: Synthetic routes to explore anti-pancreatic cancer drug Evofosfamide, 22 Jan 2015
- Synthetic route Reference: International patent application WO2007002931A2
- Jump up^ FDA Advisory Committee Briefing Materials Available for Public Release, TH-302: Pediatric oncology subcommittee of the oncologic drugs advisory committee (ODAC) meeting, December 4, 2012
- Jump up^ AAPS 2014 – Measurement of Diethylhexyl Phthalate (DEHP) Leached from Polyvinyl Chloride (PVC) Containing Plastics by Infusion Solutions Containing an Organic Parenteral Formulation – Poster W4210, Nov 5, 2014
- Jump up^ ClinicalTrials.gov
- The Primary Completion Date is defined as the date when the final subject was examined or received an intervention for the purposes of final collection of data for the primary outcome.
- Jump up^ Detailed Results From Positive Phase 2b Trial of TH-302 in Pancreatic Cancer at AACR Annual Meeting – Press release from 30 March 2012
- Jump up^ TH-302 Plus Gemcitabine vs. Gemcitabine in Patients with Untreated Advanced Pancreatic Adenocarcinoma. Borad et al. Presentation at the European Society for Medical Oncology (ESMO) 2012 Congress, September 2012. (Abstract 6660)
- Stifel 2014 Healthcare Conference; Speaker: Harold Selick – 18 November 2014
- Updated Phase 2 Results Including Analyses of Maintenance Therapy With TH-302 Following Induction Therapy With TH-302 Plus Doxorubicin in Soft Tissue Sarcoma – Press release from 15 November 2012
- TH-302 Maintenance Following TH-302 Plus Doxorubicin Induction: The Results pf a Phase 2 Study of TH-302 in Combination with Doxorubicin in Soft Tissue Sarcoma. Ganjoo et al. Connective Tissue Oncology Society (CTOS) 2012 Meeting, November 2012
- Jump up^ Chawla, S.P., Cranmer, L.D., Van Tine, B.A., Reed, D.R., Okuno, S.H., Butrynski, J.E., Adkins, D.R., Hendifar, A.E., Kroll, S., Ganjoo, K.N., 2014. Phase II Study of the Safety and Antitumor Activity of the Hypoxia-Activated Prodrug TH-302 in Combination With Doxorubicin in Patients With Advanced Soft Tissue Sarcoma. Journal of Clinical Oncology 32, 3299–3306.doi:10.1200/JCO.2013.54.3660
- Jump up^ Follow-Up Data From a Phase 1/2 Clinical Trial of TH-302 in Solid Tumors – Press release from 12 October 2010
- TH-302 Continues to Demonstrate Promising Activity in Pancreatic Cancer Phase 1/2 Clinical Trial – Press release from 24 January 2011
- Jump up^ TH-302, a tumor selective hypoxia-activated prodrug, complements the clinical benefits of gemcitabine in first line pancreatic cancer. Borad et al. ASCO Gastrointestinal Cancers Symposium, January 2011
- Jump up^ Stifel 2014 Healthcare Conference; Speaker: Harold Selick – 18 November 2014
- Jump up^ Borad et al., ESMO Annual Meeting, October 2010
- Jump up^ Video interview of Stefan Oschmann, CEO Pharma at Merck – Merck Serono Investor & Analyst Day 2014 – 18 Sept 2014 – 2:46 min – Youtube
- Jump up^ The Phase 3 Trial of TH-302 in Patients With Advanced Soft Tissue Sarcoma Will Continue as Planned Following Protocol-Specified Interim Analysis – Press release from 22 September 2014
- Jump up^ Threshold Pharmaceuticals’ Partner Merck KGaA, Darmstadt, Germany, Completes Target Enrollment in the TH-302 Phase 3 MAESTRO Study in Patients With Locally Advanced or Metastatic Pancreatic Adenocarcinoma – Press release from 3 November 2014
- Data From Ongoing Phase 1/2 Trial of TH-302 Plus Bevacizumab (Avastin(R)) in Patients With Recurrent Glioblastoma – Press release from 30 May 2014
- Jump up^ Phase 1/2 Study of Investigational Hypoxia-Targeted Drug, TH-302, and Bevacizumab in Recurrent Glioblastoma Following Bevacizumab Failure. Brenner, et al. 2014 ASCO, 7 – 30 May 2014
- Jump up^ Phase 1/2 Interim Data Signaling Activity of TH-302 Plus Bevacizumab (Avastin(R)) in Patients With Glioblastoma – Press release from 17 November 2014
- Jump up^ Threshold Pharmaceuticals’ Partner Merck KGaA, Darmstadt, Germany, Completes Target Enrollment in the TH-302 Phase 3 MAESTRO Study in Patients With Locally Advanced or Metastatic Pancreatic Adenocarcinoma – Press release from 3 November 2014
- Jump up^ Stifel 2014 Healthcare Conference; Speaker: Harold Selick – 18 November 2014
- Jump up^ Stifel 2014 Healthcare Conference; Speaker: Harold Selick – 18 November 2014
- Jump up^ Chawala SP, et al. J Clin Oncol. 2014 (54) 3660 doi:10.1200/JCO.2013.54.3660
- Jump up^ Judson I, et al. Lancet Oncol. 2014 Apr;15(4):415-23doi: 10.1016/S1470-2045(14)70063-4
- Jump up^ Judson I, et al. Lancet Oncol. 2014 Apr;15(4):415-23doi: 10.1016/S1470-2045(14)70063-4
- Jump up^ Chawala SP, et al. J Clin Oncol. 2014 (54) 3660 doi:10.1200/JCO.2013.54.3660
- Jump up^ Borad, M. J. et al. Randomized Phase II Trial of Gemcitabine Plus TH-302 Versus Gemcitabine in Patients With Advanced Pancreatic Cancer. Journal of Clinical Oncology (2014). doi: 10.1200/JCO.2014.55.7504
- Jump up^ Von Hoff, D. D. et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. New England Journal of Medicine 369, 1691–1703 (2013). doi:10.1056/NEJMoa1304369
- Jump up^ Von Hoff, D. D. et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. New England Journal of Medicine 369, 1691–1703 (2013). doi:10.1056/NEJMoa1304369
- Jump up^ Borad, M. J. et al. Randomized Phase II Trial of Gemcitabine Plus TH-302 Versus Gemcitabine in Patients With Advanced Pancreatic Cancer. Journal of Clinical Oncology (2014). doi: 10.1200/JCO.2014.55.7504
- Jump up^ Threshold Pharmaceuticals 10-K Annual report 2011 from 15 Mar 2012
- Jump up^ Threshold Pharmaceuticals 10-Q Quarterly report Q3/2014 from 3 Nov 14
- Jump up^ Threshold Pharmaceuticals Form 8-K from 9 Oct 14
- Jump up^ Threshold Pharmaceuticals Form 8-K from 9 Oct 14
- Threshold Pharmaceuticals Form 8-K from 9 Oct 14
- Phosphoramidate alkylator prodrugs US8003625B2,US8507464B2, US8664204B2
- Phosphoramidate alkylator prodrugs EP1896040B1and JP5180824B2
| WO2007002931A2 * | Jun 29, 2006 | Jan 4, 2007 | Threshold Pharmaceuticals, Inc. | Phosphoramidate alkylator prodrugs |
| WO2008083101A1 * | Dec 21, 2007 | Jul 10, 2008 | Threshold Pharmaceuticals, Inc. | Phosphoramidate alkylator prodrugs for the treatment of cancer |
| WO2010048330A1 * | Oct 21, 2009 | Apr 29, 2010 | Threshold Pharmaceuticals, Inc. | Treatment of cancer using hypoxia activated prodrugs |
| WO2015051921A1 * | Oct 10, 2014 | Apr 16, 2015 | Merck Patent Gmbh | Synthesis of 1-alkyl-2-amino-imidazol-5-carboxylic acid ester via calpha-substituted n-alkyl-glycine ester derivatives |
| Reference | ||
|---|---|---|
| 1 | * | DUAN, J.-X. ET AL.: “Potent and Highly Selective Hypoxia-Activated Achiral Phosphoramidate Mustards as Anticancer Drugs“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 51, 2008, pages 2412 – 2420, XP008139620, DOI: doi:10.1021/jm701028q |
 |
|
| Names | |
|---|---|
| IUPAC name
(1-Methyl-2-nitro-1H-imidazol-5-yl)methyl N,N’-bis(2-bromoethyl)phosphorodiamidate
|
|
| Other names
TH-302; HAP-302
|
|
| Identifiers | |
| 918633-87-1 |
|
| ChemSpider | 10157061 |
| Jmol-3D images | Image |
| PubChem | 11984561 |
| Properties | |
| C9H16Br2N5O4P | |
| Molar mass | 449.04 g·mol−1 |
| 6 to 7 g/l | |
///////////Orphan Drug Status, soft tissue sarcoma, Pancreatic cancer, Fast track, TH-302, TH 302, эвофосфамид , إيفوفوسفاميد , 艾伏磷酰胺 , Evofosfamide, 918633-87-1, PHASE 3
O=[N+]([O-])c1ncc(COP(=O)(NCCBr)NCCBr)n1C


{kind=link}