
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Eribulin
Eribulin mesylate
エリブリンメシル酸塩
CAS 441045-17-6 MESYLATE
C41H63NO14S, 826.00222 g/mol
halichrondrin B analog, B1939, E7389, ER-086526,Halaven
CAS 253128-41-5 FREE FORM
(1S,3S,4R)-3-tert-butoxycarbonylamino-4-hydroxycyclopentanecarboxylic acid methyl ester;
(1S,3S,6S,9S,12S,14R,16R,18S,20R,21R,22S,26R,29S,31R,32S,33R,35R,36S)-20-[(2S)-3-Amino-2-hydroxypropyl]-21-methoxy-14-methyl-8,15-bis(methylene)-2,19,30,34,37,39,40,41-octaoxanonacyclo[24.9.2.13,32.13,33.16,9.112,16.018,22.029,36.031,35]hentetracontan-24-one;
2-(3-Amino-2-hydroxypropyl)hexacosahydro-3-methoxy- 26-methyl-20,27-bis(methylene)11,15-18,21-24,28-triepoxy- 7,9-ethano-12,15-methano-9H,15H-furo(3,2-i)furo(2′,3′-5,6) pyrano(4,3-b)(1,4)dioxacyclopentacosin-5-(4H)-one
(2R,3R,3aS,7R,8aS,9S,10aR,11S,12R,13aR,13bS,15S,18S,21S,24S,26R,28R,29aS)-2-((2S)-3-amino-2-hydroxypropyl)-3-methoxy-26-methyl-20,27-dimethylidenehexacosahydro-11,15:18,21:24,28-triepoxy-7,9-ethano-12,15-methano-9H,15H-furo(3,2-i)furo(2′,3′:5,6)pyrano(4,3-b)(1,4)dioxacyclopentacosin-5(4H)-one methanesulfonate (salt)
11,15:18,21:24,28-Triepoxy-7,9-ethano-12,15-methano-9H,15H-furo(3,2-i)furo(2′,3′:5,6)pyrano(4,3-b)(1,4)dioxacyclopentacosin-5(4H)-one, 2-((2S)-3- amino-2-hydroxypropyl)hexacosahydro-3-methoxy-26-methyl-20,27-bis(methylene)-, 2R,3R,3aS,7R,8aS,9S,10aR,11S,12R,13aR,13bS,15S,18S,21S,24S,26R,28R,29aS)-, methanesulfonate (salt)
![]()
エリブリンメシル酸塩
Eribulin Mesilate

C40H59NO11 CH4O3S : 826
CH4O3S : 826
[441045-17-6]
Eribulin mesylate is the mesylate salt of a synthetic analogue of halichondrin B, a substance derived from a marine sponge (Lissodendoryx sp.) with antineoplastic activity.
E7389 is the mesylate salt of a synthetic analogue of halichondrin B, a substance derived from a marine sponge (Lissodendoryx sp.) with antineoplastic activity. Eribulin binds to the vinca domain of tubulin and inhibits the polymerization of tubulin and the assembly of microtubules, resulting in inhibition of mitotic spindle assembly, induction of cell cycle arrest at G2/M phase, and, potentially, tumor regression.
Halichondrin B, a large polyether macrolide, was isolated 25 years ago from the marine sponge Halichondria okadai

ERBULIN
The anti-cancer drug made from a sea-spongeEribulin is an anticancer drug marketed by Eisai Co. under the trade name Halaven. Eribulin mesylate was approved by the U.S. Food and Drug Administration on November 15, 2010, to treat patients with metastatic breast cancer who have received at least two prior chemotherapy regimens for late-stage disease, including both anthracycline– and taxane-based chemotherapies.[1] It was approved by Health Canada on December 14, 2011 for treatment of patients with metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease. [2]
Eribulin is also being investigated by Eisai Co. for use in a variety of other solid tumors, including non-small cell lung cancer, prostate cancer and sarcoma.[3]
Eribulin has been previously known as E7389 and ER-086526, and also carries the US NCI designation NSC-707389.
Eribulin mesylate is an analogue of halichondrin B, which in 1986 was isolated from the marine sponge Halichondria okadai toxic Pacific.Halichondrin B has a significant anti-tumor activity. The Eribulin synthetically obtained has a simpler but still complex molecular structure.Taxanes such as to inhibit the spindle apparatus of the cell, but it is engaged in other ways.
Drug substance, eribulin mesylate, is a It is a structurally simplified synthetic analogue of halichondrin B, a natural product isolated from the marine sponge Halichondira okadai. Eribulin mesylate is a white powder which is freely soluble in water, methanol, ethanol, 1-octanol, benzyl alcohol, dichloromethane, dimethylsulfoxide, N-methylpyrrolidone and ethyl acetate. It is soluble in acetone, sparingly soluble in acetonitrile, and practically insoluble in tertbutyl methyl ether, n-heptane and n-pentane. Eribulin mesylate is characterized by ion chromatography for counter ion content, and spectroscopic analyses (mass, ultraviolet, nuclear magnetic resonance, single crystal X-ray crystallography, and circular dichroism) for molecular structure and absolute configuration. Bulk drug substance is hygroscopic and sensitive to light, heat, and acid hydrolysis,,,,,,……..http://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/201532orig1s000chemr.pdf


Melvin Yu received his B.S. from MIT, and both his M.A. and Ph.D. degrees from Harvard University while studying under Professor Yoshito Kishi. In 1985, he joined Eli Lilly, and in 1993 he relocated to Eisai Inc. where he led the chemistry team that discovered Halaven. He was then responsible for the initial route nding and synthesis scale-up effort that ultimately provided the rst multi-gram batch of eribulin mesylate. Mel retains a strong interest in natural products as the inspiration of new chemotherapeutic agents, and in this context recently expanded his area of research to include cheminformatics and compound library design.

Wanjun Zheng received a Ph.D. in organic chemistry from Wesleyan University in 1994 under the direction of Professor Peter A. Jacobi working on synthetic methodology development and its application in natural product synthesis. He spent over two years as a postdoctoral research fellow in Harvard University under Professor Yoshito Kishi working on the complete structure determination of maitotoxin. He joined Eisai in 1996 and has since been contributing and leading many drug discovery projects including project in the discovery of Halaven.

Boris M. Seletsky earned his PhD in 1987 from Shemyakin Institute of Bioorganic Chemistry in Moscow, Russia working on new methods in steroid synthesis under direction of Dr George Segal and Professor Igor Torgov. Aer several years of natural product research at the same Institute, he moved on to postdoctoral studies in stereoselective synthesis with Professor Wolfgang Oppolzer at the University of Geneva, Switzerland, and Professor James A. Marshall at the University of South Carolina. Boris joined Eisai in 1994, and has contributed to many oncology drug discovery projects with considerable focus on natural products as chemical leads, culminating in the discovery of Halaven.
PAPER
http://www.sciencedirect.com/science/article/pii/S0960894X0401100X
Volume 14, Issue 22, 15 November 2004, Pages 5551–5554
Macrocyclic ketone analogues of halichondrin B
- a Department of Medicinal Chemistry, Eisai Research Institute, 4 Corporate Drive, Andover, MA 01810, USA
- b Department of Anticancer Research, Eisai Research Institute, 4 Corporate Drive, Andover, MA 01810, USA
- c Advisory Board, Eisai Research Institute, 4 Corporate Drive, Andover, MA 01810, USA


PAPER
From micrograms to grams: scale-up synthesis of eribulin mesylate
E-mail: Melvin_Yu@eisai.com
DOI: 10.1039/C3NP70051H, http://pubs.rsc.org/is/content/articlelanding/2013/np/c3np70051h#!divAbstract
Covering: 1993 to 2002
The synthesis of eribulin mesylate from microgram to multi-gram scale is described in thisHighlight. Key coupling reactions include formation of the C30a to C1 carbon–carbon bond and macrocyclic ring closure through an intramolecular Nozaki–Hiyama–Kishi reaction.


 The synthesis of the C27–C35 tetrahydrofuran fragment.
The synthesis of the C27–C35 tetrahydrofuran fragment.
 The synthesis of the C14–C21 aldehyde subfragment.
The synthesis of the C14–C21 aldehyde subfragment.
CLIP
In 1986, two Japanese chemists Hirata and Uemura [Y. Hirata, D. Uemura, Pure Appl. Chem. 58 (1986) 701.] isolated a naturally-occurring compound from the marine sponge Halichondria okadai (picture above, right). The compound was named Halichondrin B, and it immediately began to generate great excitement when it was realised that it was extremely potent at killing certain types of cancer cells in small-scale tests. As a result of this discovery, it was immediately given top priority to be tested against a wide range of other cancers, and became one of the first compounds to be evaluated using the novel 60-cell line method developed by the US National Cancer Institute (NCI). In this technique, 60 different types of human tumor cells (including leukemia, melanoma and cancers of the lung, colon, brain, ovary, breast, prostate, and kidney) are tested with the potential anti-cancer molecule delivered at a single dose of 10 μM concentration. This process can be run in parallel, with dozens of different molecules being tested against all 60 cancer cell lines at the same time in a huge array. Any molecules which exhibit significant growth inhibition are prioritised, and the test repeated on them, but this time at five different concentration levels.

Halichondrin B – the part of the molecule used to make Eribulin is shown in blue.
Unfortunately, the concentration of Halichondrin B in the sea sponge wasn’t enough to enable commercial production for use in chemotherapy. For example, a ton of sea sponges could only produce 300 mg of Halichondrin B! The race was on to try to synthesise Halichondrin B in the lab, which wasn’t easy due to its large size (molecular weight 1110) and complex structure. However, only 6 years later, chemists at Harvard University published the complete chemical synthesis of this molecule………..T.D. Aicher, K.R. Buszek, F.G. Fang, C.J. Forsyth, S.H. Jung, Y. Kishi, M.C. Matelich, P.M. Scola, D.M. Spero, S.K. Yoon, J. Am. Chem. Soc. 114 (1992) 3162
Although this was a great achievement, Halichondrin B was still far too complex and the sythesis route too expensive to do on a large scale. The molecule needed to be stripped down to its essential components, while keeping, or even improving, its anti-cancer efficacy. Many tests were performed, but eventually the work led to te development of the structurally-simplified and pharmaceutically-optimized analog, which was named Eribulin [3,4]. Eribulin mesylate was approved by the U.S. Food and Drug Administration in 2010, to treat patients with metastatic breast cancer [5], and it is currently being marketed by Eisai Co. under the trade nameHalaven . It is also being investigated for use in a variety of other solid tumors, including lung cancer, prostate cancer and sarcoma .
 ERIBULIN
ERIBULIN
M.J. Towle, K.A. Salvato, J. Budrow, B.F. Wels, G. Kuznetsov, K.K. Aalfs, S. Welsh, W. Zheng, B.M. Seletsk, M.H. Palme, G.J. Habgood, L.A. Singer, L.V. Dipietro, Y. Wang, J.J. Chen, D.A. Quincy, A. Davis, K. Yoshimatsu, Y. Kishi, M.J. Yu, B.A. Littlefield, Cancer Res. 61 (2001) 1013.
M.J. Yu, Y. Kishi, B.A. Littlefield, in D.J. Newman, D.G.I. Kingston, G.M. Cragg, Anticancer agents from natural products, Washington, DC, Taylor and Francis (2005).
http://healthmad.com/conditions-and-diseases/breast-cancer-cure-from-the-sea/
http://www.clinicaltrials.gov/ct2/results?term=eribulin+OR+E7389
M.A. Jordan, L. Wilson, Nature Revs: Cancer 4 (2004) 253.
ERIBULIN
Patent Data
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
Delist Requested |
|---|---|---|---|---|---|---|---|
| N201532 | 001 | 6214865 | Jul 20, 2023 | Y | |||
| N201532 | 001 | 6469182 | Jun 16, 2019 | U – 1096 | |||
| N201532 | 001 | 7470720 | Jun 16, 2019 | Y | |||
| N201532 | 001 | 8097648 | Jan 22, 2021 | U – 1096 |
Exclusivity Data
| Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N201532 | 001 | NCE | Nov 15, 2015 |
The substance inhibits the polymerization of tubulin into microtubules and encapsulates tubulin molecules in non-productive aggregates from. The lack of training of the spindle apparatus blocks the mitosis and ultimately induces apoptosis of the cell. Eribulin differs from known microtubule inhibitors such as taxanes and vinca alkaloids by the binding site on microtubules, also it does not affect the shortening. This explains the effectiveness of the new cytostatic agent in taxane-resistant tumor cell lines with specific tubulin mutations.

Structure and mechanism
Structurally, eribulin is a fully synthetic macrocyclic ketone analogue of the marine sponge natural product halichondrin B,[4][5] the latter being a potent naturally-occurring mitotic inhibitor with a unique mechanism of action found in the Halichondria genus of sponges.[6][7] Eribulin is a mechanistically-unique inhibitor of microtubule dynamics,[8][9] binding predominantly to a small number of high affinity sites at the plus ends of existing microtubules.[10] Eribulin exerts its anticancer effects by triggering apoptosis of cancer cells following prolonged and irreversible mitotic blockade.[11][12]
A new synthetic route to E7389 was published in 2009.[13]
clip
Eisai R&D Management Co., Ltd.
13/9/2013
Halaven is a novel anticancer agent discovered and developed in-house by Eisai and is currently approved in more than 50 countries, including Japan, the United States and in Europe. In Russia, Halaven was approved in July 2012 for the treatment of locally advanced or metastatic breast cancer previously treated with at least two chemotherapy regimens including an anthracycline and a taxane. Approximately 50,000 women in Russia are newly diagnosed with breast cancer each year, with this type of cancer being the leading cause of death in women aged 45 to 55 years. read all at…………………….
http://www.dddmag.com/news/2013/09/eisai-launches-halaven-cancer-drug-russia

Eribulin mesylate (Halaven; Eisai) — a synthetic analogue of the marine natural product halichondrin B that interferes with microtubule dynamics — was approved in November 2010 by the US Food and Drug Administration for the treatment of metastatic breast cancer.
Family members of the product patent, WO9965894, have SPC protection in the EU until 2024 and one of its Orange Book listed filings, US8097648, has US154 extension till January 2021.
The drug also has NCE exclusivity till November 2015.

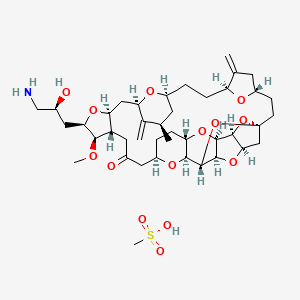
HALAVEN (eribulin mesylate) Injection is a non-taxane microtubule dynamics inhibitor. Eribulin mesylate is a synthetic analogue of halichondrin B, a product isolated from the marine sponge Halichondria okadai. The chemical name for eribulin mesylate is 11,15:18,21:24,28-Triepoxy-7,9-ethano12,15-methano-9H,15H-furo[3,2-i]furo[2′,3′:5,6]pyrano[4,3-b][1,4]dioxacyclopentacosin-5(4H)-one, 2[(2S)-3-amino-2-hydroxypropyl]hexacosahydro-3-methoxy-26-methyl-20,27-bis(methylene)-, (2R,3R,3aS,7R,8aS,9S,10aR,11S,12R,13aR,13bS,15S,18S,21S,24S,26R,28R,29aS)-, methanesulfonate (salt).
It has a molecular weight of 826.0 (729.9 for free base). The empirical formula is C40H59NO11 •CH4O3S. Eribulin mesylate has the following structural formula:
 |
HALAVEN is a clear, colorless, sterile solution for intravenous administration. Each vial contains 1 mg of eribulin mesylate as a 0.5 mg/mL solution in ethanol: water (5:95).


complete syn is available here
http://www.sciencedirect.com/science/article/pii/S0968089611010674

http://www.drugdevelopment-technology.com/projects/halaven-cancer/halaven-cancer1.html

 |
Nitrogen: dark blue, oxygen: red, hydrogen: light blue
graphics: Wurglics, Frankfurt am Main |
clip
Macrocyclization process for preparing a macrocyclic intermediate of halichondrin B analogs, in particular eribulin, from a non-macrocyclic compound, using a carbon-carbon bond-forming reaction.

http://www.pnas.org/content/108/17/6699/F1.expansion.html

http://www.nature.com/nrd/journal/v8/n1/fig_tab/nrd2487_F6.html
UPDATED
Eisai has developed and launched eribulin mesylate for treating breast cancer. Follows on from WO2014208774, claiming use of a combination comprising eribulin mesylate and lenvatinib mesylate, for treating cancer.
Macrocyclization reactions and intermediates useful in the synthesis of analogs of halichondrin B
By: Fang, Francis G.; Kim, Dae-Shik; Choi, Hyeong-Wook; Chase, Charles E.; Lee, Jaemoon
Assignee: Eisai R&D Management Co., Ltd., Japan
The invention provides methods for the synthesis of eribulin or a pharmaceutically acceptable salt thereof (e.g., eribulin mesylate) through a macrocyclization strategy. The macrocyclization strategy of the present invention involves subjecting a non-macrocyclic intermediate to a carbon-carbon bond-forming reaction (e.g., an olefination reaction (e.g., Horner-Wadsworth-Emmons olefination), Dieckmann reaction, catalytic Ring-Closing Olefin Metathesis, or Nozaki-Hiyama-Kishi reaction) to afford a macrocyclic intermediate. The invention also provides compds. useful as intermediates in the synthesis of eribulin or a pharmaceutically acceptable salt thereof and methods for prepg. the same.
CLIPS
http://www.chemistry-blog.com/2012/09/15/from-natural-product-to-pharmaceutical/
In a recent discussion (Nicolau), about the suggested move of Prof. NicoIau from Scripps, the issue of the practicality of natural product total synthesis was raised. Here is a wonderful example of just that very usefulness, a wonderful piece of science extending over many years. It concerns the journey from Halichondrin B to Eribulin (E7389) a novel anti-cancer drug. The two compounds have the following structures:

I think you can see the relationship and as a development chemist I am glad they managed to simplify things (a bit).
Both compounds have an enormous number of possible isomers: Halichondrin B, with 32 stereocenters has 232possible isomers; Eribulin has 19 with 219 isomers (if I have counted correctly, it does not really matter, there are lots of isomers). Remarkable is the fact that only one of these isomers is active in the given area of anti-cancer agents.
An excellent review of the biology and chemistry of these compounds has been published by Phillips etal1. This review is an excellent read and is to be commended. Another one written by Kishi2, is also full of information about the discovery of E7389 and I hope you will all get a chance to read this chapter.
The history of Halichondrin B goes back to 1987 when Blunt2-5 isolated it with other similar compounds from extraction of 200Kg of a sponge. Independently Pettit isolated the same compound from a different species4. The appearance of this compound in different species of sponge may indicate that it is produced by a symbiote.
The biological activity of Halichondrin B is amazing. When evaluated against B-16 melanoma cells it was found to have an IC50 of 0.093ng/mL. Against various cancers, generated in mice, it was shown to be affective at a daily dose of 5ug/kg, which resulted in a doubling of the survival rate. It has also been demonstrated that Halichondrin acts as a microtubule destabiliser and mitoitic spindle poison. It was proven that it is has tremendous in vivo activity against a variety of drug resistant cancers, lung, colon, breast, ovarian to mention a few. Consequently the National Cancer Institute selected it for pre-clinical trials and it’s here that the problems began. According to reference 1 the entire clinical development would require some 10g, and if successful the annual production amount would be between 1-5 kg. Blunt and co-workers managed to isolate 310mg from 1000kg-harvested sponge therefore, the only way to obtain the amounts required is total chemical synthesis. But synthesising 1-5 kg of such a compound would indeed be a mammoth task.
Kishi synthesised this compound7 in 1992 starting from carbohydrate precursors employing the Nozaki-Hiyama-Kishi Ni/Cr reaction, several times, in the long synthetic sequence8, 9. Now as an aside I have used this reaction on scale several times and although it works well its success is very dependant upon the quality of the chromium source and also the presence of other trace transition metals.
In collaboration with Eisai work on the SAR of Halichondrin began. They had a good start: Thanks to the total syntheses of Kishi several advanced intermediates were available for biological screening and one popped out of the screen as being very active:

The first active lead compound
As one can see the complete left hand side of Halichondrin has gone! However, this compound was not active in vivo. Many derivatives and analogues of this compound were prepared: furans, diols, ketones and so on and a lead emerged from this complex SAR study, ER-076349. The vicinal diol was used as a handle for further refinement and lead ultimately to E7389, the clinical candidate.

It can be synthesised in around 35 steps from simple starting materials.
Going through all this work in a few sentences really belittles the tremendous amount of effort that went into discovery and development of this compound and the people involved are to be applauded for their dedication.
Kishi continues to optimise the synthesis of Eribulin as judged by a recent publication10. Where he describes an approach to the amino-alcohol-tetrahydrofuran part of Eribulin (top left fragment, compound 1 below). The retro-synthetic analysis is shown below. The numbering corresponds to that of Eribulin.

The first generation synthesis consisted of 20 steps and delivered compound 1 about 5% yield, the second-generation route was completed in 12 steps with a yield of 48%. One of the highlights includes a remarkable asymmetric hydrogenation11 with Crabtree’s catalyst12:

This selectivity was not just luck; it seems to quite general, at least in this system. I always wonder how long it took them to stumble across this catalyst, but then I suppose that Eisai like most of the large pharma. companies has a hydrogenation group that probably indulges in catalyst screening.
The C34-C35 diol was obtained by a Sharpless asymmetric hydroxylation, here the diastereoisomeric ratio was not very high, only about 3:1 in favour of the desired isomer. Fortunately the undesired isomer could be removedcompletely by crystallisation.
This is a remarkable story and references 1 and 2 are worth reading to obtain the complete picture and learn lots of new chemistry as well. Eisai filed a NDA and the FDA approved the compound in 2010 for the treatment of metastatic breast cancer.

Patent
https://www.google.com/patents/WO2013142999A1?cl=en
EXAM PLE 23 : Preparation of Eribulin :

[00120] Compound E-12A (133 mg, 160 μηιοΙ, 1.0 eq) was dissolved in anhydrous dichloromethane (20 mL) and cooled to 0 °C. To this solution was sequentially added 2,6-lutidine (0.09 m L, 0.8 mmol, 5.0 eq), and trimethyl silyl triflate (TMSOTf) (0.12 m L, 0.64 mmol, 4.0 eq) and the cooling bath was removed . The reaction was stirred at room temperature for 1.5 hours and another portion of 2,6-lutidine (5.0 eq) and TMSOTf (4.0 eq) were added at room temperature. The reaction was further stirred for 1 hour and quenched with water (10 m L). The layers were separated and the organic phase was washed with additional water (2x 10 m L), brine (10 m L), dried over MgS04 and concentrated under reduced pressure. The residue was dissolved in MeOH (10 m L), a catalytic amount of K2C03 was added at room temperature and the resulting mixture was stirred for 2 hours. The reaction was diluted with dichloromethane and quenched with water (10 mL). The layers were separated and the aqueous phase was further extracted with dichloromethane (5 x 10 m L). The combined organic layers were washed with brine (20 m L), dried over MgS04, filtered and concentrated. The residue was dissolved in dichloromethane and purified by column chromatography on silica gel, using 1 : 9 MeOH : CH2CI2 to 1 : 9 : 90 N H4OH : MeOH : CH2CI2 as eluent. The product was afforded as a white amorphous solid (103 mg, 88%) . [00121] EXAMPLE 23 : Preparation of compound of formula 4a

D-Gulonolactone 4a
[00122] The compound of formula 4a was prepared from D-Gulonolactone according to the conditions described in PCT publication number WO 2005/118565. [00123] EXAMPLE 24: Preparation of Eribulin mesylate (3)
[00124] Eribulin mesylate (3) was prepared from Eribulin according to the conditions described in US patent application publication number US
2011/0184190.
PATENT
https://www.google.com/patents/EP2528914A1?cl=en
Halichondrin B analogs, e.g., eribulin or pharmaceutically acceptable salts thereof, can be synthesized from the C14-C35 fragment as described in U.S. Patent No. 6,214,865 and International Publication No. WO 2005/118565. In one example described in these references, the C14-C35 portion, e.g., ER- 804028, of the molecule is coupled to the C1-C13 portion, e.g., ER-803896, to produce ER-804029, and additional reactions are carried out to produce eribulin (Scheme 1):

Scheme 1

eribulin, eribulin mesylate
Scheme 2
ER-804028
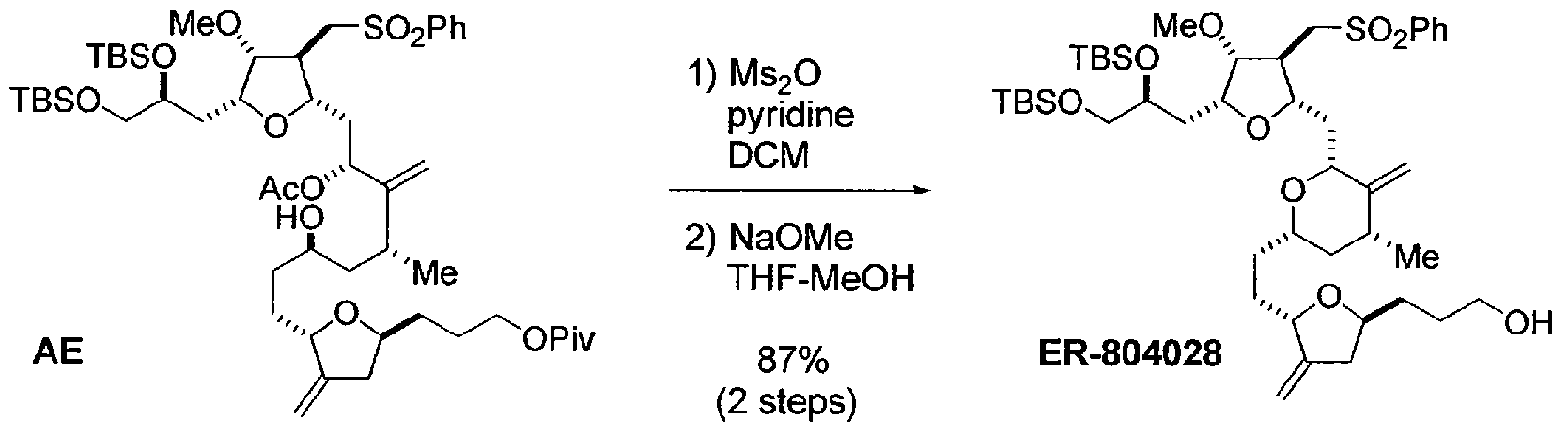
Compound AE (280 mg, 0.281 mmol, 1 eq) was dissolved in CH2C12 and cooled to 0 °C. Pyridine (0.045 ml, 0.56 mmol, 2.0 eq) was added followed by Ms20 (58.8 mg, 0.338 mmol, 1.20 eq). The reaction was allowed to warm to room temperature, and stirring was continued for an additional 1 h. The reaction mixture was cooled to 0 °C, diluted with MTBE (5.6 ml), washed with saturated NaHC03 (0.84 g), and concentrated to give crude product as colorless film. The crude was azeotropically dried with heptane (3 ml χ 2) and re-dissolved in THF (7.0 ml). The mixture was cooled to 0 °C and treated with 25 wt% NaOMe (0.13 ml). After 10 min, the reaction was allowed to warm to room temperature, and stirring was continued for an additional 30 min. The mixture was treated with additional 25 wt% NaOMe (0.045 ml), and stirring was continued for an additional 20 min. The reaction mixture was diluted with heptane (7.0 ml) and washed with water (1.4 ml). The organic layer was separated, sequentially washed with: 1) 20 wt% NH4C1 (0.84 g) and 2) 20 wt% NaCl (3 g), and concentrated to give crude product as brownish oil. The crude was purified by Biotage (Uppsala, Sweden) 12M (heptane-MTBE 2:3 v/v) to give ER-804028 (209 mg, 0.245 mmol, 87%) as pale yellow oil. 1H NMR (400 MHz, CDC13): δ 7.89 (2H, m), 7.64 (IH, m), 7.56 (2H, m), 4.85 (IH, d, J= 1.6 Hz), 4.80 (IH, s), 4.72 (IH, s), 4.61 (IH, d, J= 1.6 Hz), 4.23 (IH, br), 3.91 (IH, m), 3.79 (IH, m), 3.76 (2H, m), 3.63 (IH, m), 3.50-3.60 (4H, m), 3.43 (IH, dd, J= 5.6 Hz, 10.0 Hz), 3.38 (3H, s), 3.32 (IH, m), 2.98 (2H, m), 2.61 (IH, br), 2.56 (IH, m), 2.50 (IH, m), 2.08-2.22 (3H, m), 1.96 (IH, m), 1.84 (IH, m), 1.78 (IH, m), 1.70 (IH, m), 1.42-1.63 (6H, m), 1.28-1.42 (2H, m), 1.01 (3H, d, J= 6.8 Hz), 0.84 (18H, s), 0.05 (3H, s), 0.04 (3H, s), 0.00 (3H, s), -0.01 (3H, s); and 13C NMR (100 MHz, CDC13): δ 150.34, 150.75, 139.91, 134.18, 129.73 (2C), 128.14 (2C), 105.10, 85.97, 80.92, 79.72, 78.50, 77.45, 77.09, 75.53, 71.59, 68.04, 62.88, 58.27, 57.73, 43.51, 42.82, 39.16, 37.68, 35.69, 33.31, 32.41, 31.89, 31.48, 29.79, 26.21 (3C), 26.17 (3C), 18.58, 18.38, 18.13, -3.85, – 4.71, -5.12 (2C).
CLIP
Eribulin mesylate (Halaven)
Eribulin is a highly potent cytotoxic agent approved in the US for the treatment of metastatic breast cancer for patients who have
received at least two previous chemotherapeutic regimens.30 Eribulin was discovered and developed by Eisai and it is currently
undergoing clinical evaluation for the treatment of sarcoma (PhIII) and non-small cell lung cancer which shows progression after platinum-based chemotherapy and for the treatment of prostate cancer (PhII). Early stage clinical trials are also underway to evaluate
eribulin’s efficacy against a number of additional cancers. Eribulin is a structural analog of the marine natural product halichondrin B.
Its mechanism of action involves the disruption of mitotic spindle formation and inhibition of tubulin polymerization which results
in the induction of cell cycle blockade in the G2/M phase and apoptosis.31 Several synthetic routes for the preparation of eribulin have
been disclosed,32–35 each of which utilizes the same strategy described by Kishi and co-workers for the total synthesis of halichondrin B.36 Although the scales of these routes were not disclosed in all cases, this review attempts to highlight what appears to be the production-scale route based on patent literature.37,38 Nonetheless, the synthesis of eribulin represents a significant accomplishment in the field of total synthesis and brings a novel chemotherapeutic option to cancer patients.
The strategy to prepare eribulin mesylate (V) employs a convergent synthesis featuring the following: the late stage coupling of
sulfone 22 and aldehyde 23 followed by macrocyclization under Nozaki–Hiyami–Kishi coupling conditions, formation of a challenging
cyclic ketal, and installation of the primary amine (Scheme 5).Sulfone 22 was further simplified to aldehyde 24 and vinyl triflate 25 which were coupled through a Nozaki–Hiyami–Kishi reaction.
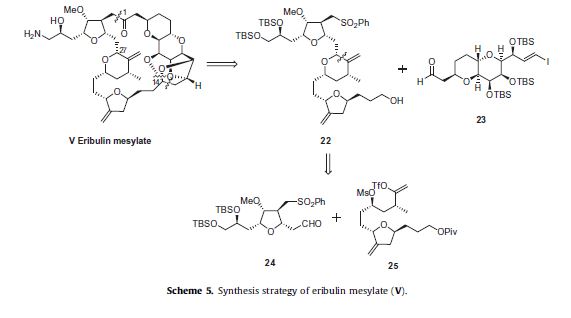
The schemes that follow will describe the preparation of fragments 23, 24 and 25 along with how the entire molecule was assembled.
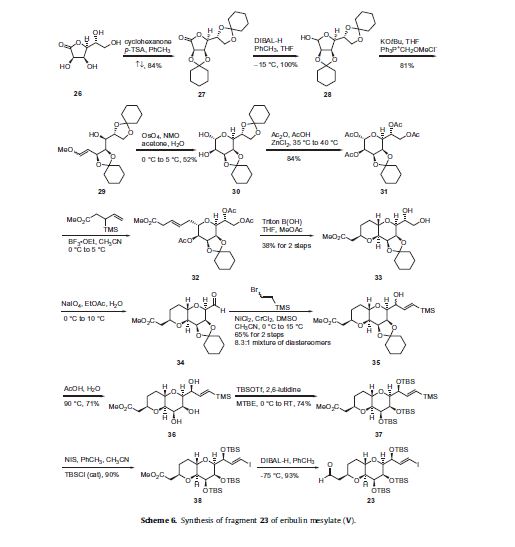
The synthesis of the C1–C13 aldehyde fragment 23 is described in Scheme 6. L-Mannonic acid-lactone 26 was reacted with cyclohexanone in p-toluene sulfonic acid (p-TSA) to give the biscyclohexylidene ketal 27 in 84% yield. Lactone 27 was reduced with
diisobutylaluminum hydride (DIBAL-H) to give lactol 28 followed by condensation with the ylide generated from the reaction of
methoxymethylene triphenylphosphorane with potassium tertbutoxide to give a mixture of E and Z vinyl ethers 29 in 81% yield.
Dihydroxylation of the vinyl ether of 29 using catalytic osmium teteroxide and N-methylmorpholine-N-oxide (NMO) with concomitant cyclization produced diol 30 in 52% yield. Bis-acetonide 30 was then reacted with acetic anhydride in acetic acid in the presence of ZnCl2 which resulted in selective removal of the pendant ketal protecting group. These conditions also affected peracylation, giving rise to tetraacetate 31 in 84% yield. Condensation of 31 with methyl 3-(trimethylsilyl)pent-4-enoate in the presence of boron trifluoride etherate in acetonitrile provided alkene 32. Saponification conditions using Triton B(OH) removed the acetate protecting groups within 32 and presumably induced isomerization of the alkene into conjugation with the terminal ester, triggering an intramolecular Michael attack of the 2-hydroxyl group, ultimately resulting in the bicylic-bispyranyl diol methyl ester 33 as a crystalline solid in 38% yield over two steps. Oxidative cleavage of the vicinal diol of 33 with sodium periodate gave aldehyde 34 which was coupled to (2-bromovinyl)trimethylsilane under Nozaki–Hiyami–Kishi conditions to give an 8.3:1 mixture of allyl alcohols 35 in 65% yield over two steps. Hydrolysis of the cyclohexylidine ketal 35 with aqueous acetic acid followed by recrystallization gave diastereomerically pure triol 36 which was reacted with tert-butyldimethylsilyl triflate (TBSOTf) to afford the tris-TBS ether 37 in good yield. Vinyl silane 37 was treated with NIS and catalytic tert-butyldimethylsilyl chloride (TBSCl) to give vinyl iodide 38 in 90% yield.
Reduction of the ester with DIBAL-H produced the key C1–C14 fragment 23 in 93% yield.
The preparation of the tetra-substituted tetrahydrofuran intermediate 24 is described in Scheme 7. D-Glucurono-6,3-lactone
39 was reacted with acetone and sulfuric acid to give the corresponding acetonide and the 5-hydroxyl group was then removed by converting it to its corresponding chloride through reaction with sulfuryl chloride (SO2Cl2) followed by hydrogenolysis
to give lactone 40 in good overall yield. Reduction of the lactone 40 with DIBAL-H gave the corresponding lactol which was condensed
with (trimethylsilyl)methylmagnesium chloride to afford silane 41. Elimination of the silyl alcohol of 41 was accomplished
under Peterson conditions with potassium hexamethyldisilazide (KHMDS) to afford the corresponding terminal alkene in 94% yield.
The secondary alcohol of this intermediate was alkylated with benzyl bromide to afford ether 42 in 95% yield. Asymmetric dihydroxylation of the alkene of 42 under modified Sharpless conditions using potassium osmate (VI) dehydrate (K2OsO4), potassium
ferricyanide (K3Fe(CN)6) and the (DHQ)2AQN ligand produced the vicinal diol which was then reacted with benzoyl chloride,
N-methylmorpholine, and DMAP to give di-benzoate 43 in excellent yield as a 3:1 mixture of diastereomeric alcohols. Allyl trimethylsilane was added to the acetal of 43 using TiCl3(OiPr) as the Lewis acid to give 44 in 83% yield. Re-crystallization of 44 from
isopropanol and n-heptane afforded 44 in >99.5% de in 71% yield.
Oxidation of the secondary alcohol of 44 under the modified Swern conditions generated the corresponding ketone which was condensed with the lithium anion of methyl phenyl sulfone to give a mixture of E and Z vinyl sulfones 45. Debenzylation of 45 using iodotrimethylsilane (TMSI) followed by chelation-controlled reduction of the vinyl sulfone through reaction with NaBH(OAc)3, and
then basic hydrolysis of the benzoate esters using K2CO3 in MeOH resulted in triol 46 as a white crystalline solid in 57% yield over the
five steps after re-crystallization. The vicinal diol of 46 was protected as the corresponding acetonide through reaction with 2,2-
dimethoxypropane and sulfuric acid and this was followed by methyl iodide-mediated methylation of the remaining hydroxyl
group to give methyl ether 47. The protecting groups within acetonide 47 were then converted to the corresponding bis-tert-butyldimethylsilyl ether by first acidic removal of the acetonide with aqueous HCl and reaction with TBSCl in the presence of imidazole to give bis-TBS ether 48. Then, ozonolysis of the olefin of 48 followed by hydrogenolysis in the presence of Lindlar catalyst afforded the key aldehyde intermediate 24 in 68% yield over the previous five steps after re-crystallization from heptane.
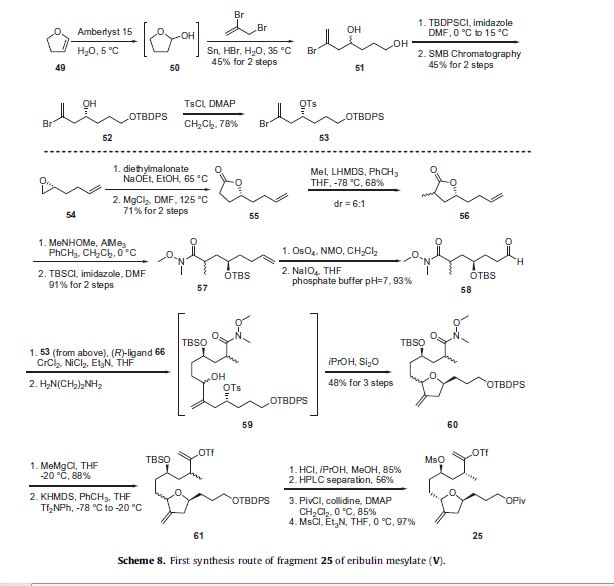
Two routes to the C14–C26 fragment 25 will be described as both are potentially used to prepare clinical supplies of eribulin.
The first route features a convergent and relatively efficient synthesis of 25, however it is limited by the need to separate enantiomers
and mixture of diastereomers via chromatographic methods throughout the synthesis.37 The second route to 25 is a
much lengthier synthesis from a step-counting perspective; however it takes full advantage of the chiral pool of starting materials
and requires no chromatographic separations and all of the products were carried on as crude oils until they could be isolated as
crystalline solids.38 The first route to fragment 25 is described in Scheme 8 and was initiated by the hydration of 2,3-dihydrofuran (49) using an aqueous suspension of Amberlyst 15 to generate the intermediate tetrahydro-2-furanol (50) which was then immediately reacted with 2,3-dibromopropene in the presence of tin and catalytic HBr to afford diol 51 in 45% for the two steps.
The primary alcohol of 51 was selectively protected as its tert-butyldiphenylsilyl ether using TBDPSCl and imidazole and the racemate was then separated using simulated moving bed (SMB) chromatography to give enantiopure 52 in 45% yield over the two steps. The secondary alcohol of 52 was reacted with p-toluenesulfonyl chloride and DMAP to give tosylate 53 in 78% yield which was used as a coupling partner later in the synthesis of this fragment. The synthesis of the appropriate coupling partner was initiated by condensing diethylmalonate with (R)-2-(3-butenyl)oxirane (54), followed by decarboxylation to give lactone 55 in 71% yield for the two step process. Methylation of the lactone with LHMDS and MeI provided 56 in 68% yield as a 6:1 mixture of diastereomers. The lactone 56 was reacted with the aluminum amide generated by the reaction of AlMe3 and N,O-dimethylhydroxylamine to give the corresponding Weinreb amide which was protected as its tert-butyldimethylsilyl ether upon reaction with TBSCl and imidazole to give 57 in 91% yield over the two steps. Dihydroxylation of the olefin of 57 by reaction with OsO4 and NMO followed by oxidative cleavage with NaIO4 gave the desired coupling partner aldehyde 58 in 93% yield. Aldehyde 58 was coupled with vinyl bromide 53 using an asymmetric Nozaki–Hiyami– Kishi reaction using CrCl2, NiCl2, Et3N and chiral ligand 66 (described in Scheme 9 below). The reaction mixture was treated with ethylene diamine to remove the heavy metals and give the secondary alcohol 59. This alcohol was stirred with silica gel in isopropanol to affect intramolecular cyclization to give the tetrahydrofuran 60 in 48% yield over the three step process. The Weinreb amide of 60 was reacted with methyl magnesium chloride to generate the corresponding methyl ketone which was converted to vinyl triflate 61 upon reaction with KHMDS and Tf2NPh. De-silylation of the primary and secondary silyl ethers with methanolic HCl gave the corresponding diol in 85% yield over two steps and the resulting mixture of diastereomers was separated using preparative HPLC to provide the desired diastereomer in 56% yield. The primary alcohol was protected as its pivalate ester with the use of pivaloyl chloride, DMAP and collidine; the secondary alcohol was converted to a mesylate upon treatment with methanesulfonyl chloride (MsCl) and Et3N to give the C15–C27 fragment 25 in high yield.
The preparations of the chiral ligand 66 used in the coupling reaction in Scheme 8 along with the chiral ligand 67 utilized later
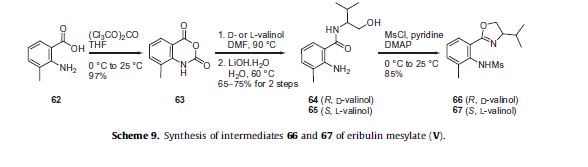
in the synthesis are described in Scheme 9. 2-Amino-3-methylbenzoic acid (62) was reacted with triphosgene to give benzoxazine
dione 63 in 97% yield, which then was reacted with either D- or L-valinol in DMF followed by aqueous LiOH to give alcohols 64
and 65, respectively in 65–75% yield for the two steps. Reaction of alcohol 64 or 65 with MsCl in the presence of DMAP effected formation of the dihydrooxazole ring and mesylation of the aniline to give the corresponding (R)-ligand 66 derived from D-valinol or the (S)-ligand 67 derived from L-valinol, respectively in high yield.
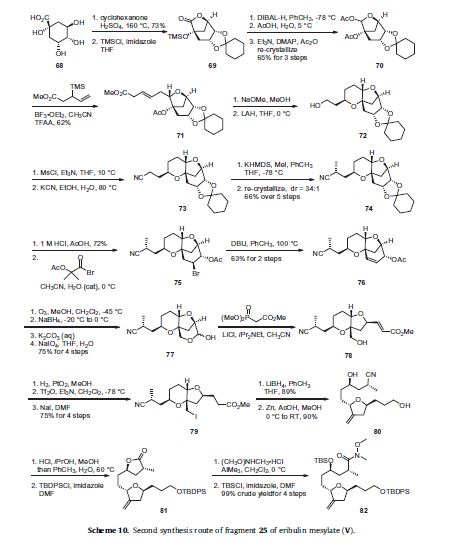
An alternative route to intermediate 25 is described in Scheme 10 and although much lengthier than the route described in
Scheme 8, it avoids chromatographic purifications as all of the products are carried on crude until a crystalline intermediate
was isolated and purified by re-crystallization. Quinic acid (68) was reacted with cyclohexanone in sulfuric acid to generate a protected
bicyclic lactone in 73% yield and the resulting tertiary alcohol was protected as its trimethylsilyl ether 69. Reduction of the
lactone 69 was accomplished with DIBAL-H and the resulting lactol was treated with acetic acid to remove the TMS group and the resulting compound was reacted with acetic anhydride, DMAP and Et3N to give bis-acetate 70 in 65% yield for the three steps after re-crystallization. Methyl 3-(trimethylsilyl)pent-4-enoate was coupled to the acetylated lactol 70 in the presence of boron trifluoride etherate and trifluoroacetic anhydride to give adduct 71 in 62% yield. The acetate of 71 was removed upon reaction with sodium methoxide in methanol and the resulting tertiary alcohol cyclized on to the isomerized enone alkene to give the fused pyran ring. Reduction of the methyl ester with lithium aluminum hydride provided pyranyl alcohol 72. Mesylation of the primary alcohol was followed by displacement with cyanide anion to give nitrile 73.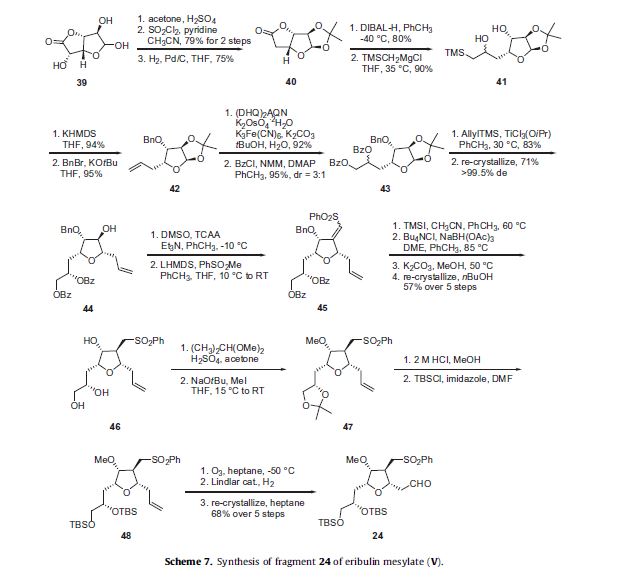
The nitrile was methylated upon reaction with KHMDS and MeI and the resulting product was purified by re-crystallization
to provide nitrile 74 in 66% over the previous five steps in a 34:1 diastereomeric ratio. Acid hydrolysis of the ketal of 74 liberated
the corresponding diol in 72% yield and this was reacted with 2-acetoxy-2-methylpropionyl bromide to give bromo acetate 75.
Elimination of the bromide was accomplished upon treatment with 1,8-diazabicycloundec-7-ene (DBU) to give alkene 76 in 63%
yield for two steps. Ozonolysis of the cyclohexene ring followed by reductive work-up with NaBH4 and basic hydrolysis of the acetate
produced a triol which upon reaction with NaIO4 underwent oxidative cleavage to give cyclic hemiacetal 77 in 75% yield over
the previous four steps. Wittig condensation with carbomethoxymethylene triphenylphosphorane gave the homologated unsaturated
ester 78. Catalytic hydrogenation of the alkene using PtO2 as the catalyst was followed by converting the primary alcohol to the
corresponding triflate prior to displacement with sodium iodide resulted in iodide 79 in 75% yield over four steps. The ester of 79
was reduced to the corresponding primary alcohol upon reaction with LiBH4 in 89% yield and the resulting iodoalcohol was treated
with Zn dust to affect reductive elimination of the iodide and decomposition of the pyran ring system to give the tetrahydrofuran
diol 80 in 90% yield. This diol was treated with methanolic HCl to affect an intramolecular Pinner reaction and this was followed
by protection of the primary alcohol as its tert-butyldiphenylsilyl ether to give lactone 81 The lactone was reacted with the
aluminum amide generated from AlMe3 and N,O-dimethylhydroxylamine and the resulting secondary alcohol was protected as
its tert-butyldimethylsilyl ether to give Weinreb amide 82 in 99% crude yield over four steps. Compound 82 is the diastereomerically
pure version of compound 60 and can be converted to compound 25 by the methods described in Scheme 8 absent the required
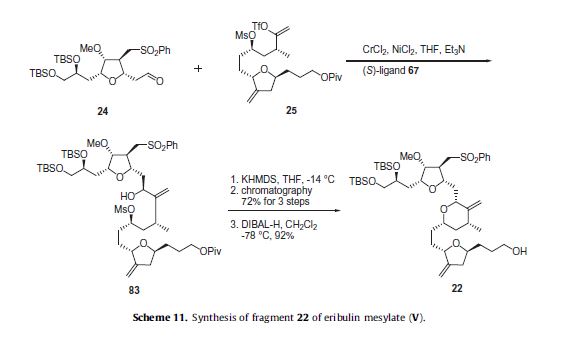
HPLC separation of diastereomers. With the three key fragments completed, the next step was to assemble them and complete the synthesis of eribulin. Aldehyde 24 was coupled to vinyl triflate 25 using an asymmetric Nozaki– Hiyami–Kishi reaction using CrCl2, NiCl2, Et3 N and chiral ligand 67 (Scheme 9) to give alcohol 83 (Scheme 11).
Formation of the THP ring was accomplished by reaction with KHMDS which allowed for displacement of the mesylate with the secondary alcohol and provided the THP containing product in 72% yield for the three steps. The pivalate ester group was removed with DIBAL-H to give the western fragment 22 in 92% yield.
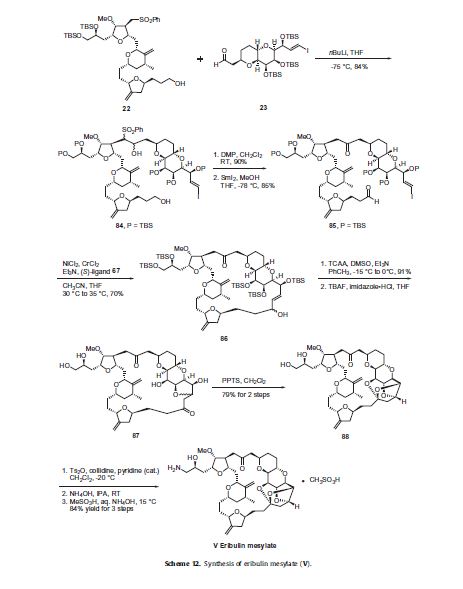
The completion of the synthesis of eribulin is illustrated in Scheme 12. The lithium anion of sulfone 22 generated upon reaction
with nBuLi was coupled to aldehyde 23 to give diol 84 in 84% yield. Both of the alcohol functional groups of 84 were oxidized
using a Dess–Martin oxidation in 90% yield and the resulting sulfone was removed via a reductive cleavage upon reaction with
SmI2 to give keto-aldehyde 85 in 85% yield. Macrocyclization of 85 was accomplished via an asymmetric Nozaki–Hiyami–Kishi
reaction using CrCl2, NiCl2, Et3N and chiral ligand 67 to give alcohol 86 in 70% yield. Modified Swern oxidation of the alcohol provided the corresponding ketone in 91% yield and this was followed by removal of the five silyl ether protecting groups upon reaction with TBAF and subsequent cyclization to provide ketone 87. Compound 87 was treated with PPTS to provide the ‘caged’ cyclic ketal 88 in 79% over two steps. The vicinal diol of 88 was reacted with Ts2O in collidine to affect selective tosylation of the primary alcohol and this crude product was reacted with ammonium hydroxide to install the primary amine to give eribulin which was treated
with methanesulfonic acid in aqueous ammonium hydroxide to give eribulin mesylate (V) in 84% yield over the final three steps.
30. Zheng, W.; Seletsky, B. M.; Palme, M. H.; Lydon, P. J.; Singer, L. A.; Chase, C. E.;
Lemelin, C. A.; Shen, Y.; Davis, H.; Tremblay, L.; Towle, M. J.; Salvato, K. A.;
Wels, B. F.; Aalfs, K. K.; Kishi, Y.; Littlefield, B. A.; Yu, M. J. Bioorg. Med. Chem.
Lett. 2004, 14, 5551.
31. Wang, Y.; Serradell, N.; Bolós, J.; Rosa, E. Drugs Future 2007, 32, 681.
32. Chiba, H.; Tagami, K. J. Synth. Org. Chem. Jpn. 2011, 69, 600.
33. Choi, H.; Demeke, D.; Kang, F.-A.; Kishi, Y.; Nakajima, K.; Nowak, P.; Wan, Z.-
K.; Xie, C. Pure Appl. Chem. 2003, 75, 1.
34. Kishi, Y.; Fang, F.; Forsyth, C. J.; Scola, P. M.; Yoon, S. K. WO 9317690 A1, 1993.
35. Littlefield, B. A.; Palme, M.; Seletsky, B. M.; Towle, M. J.; Yu, M. J.; Zheng, W.
WO 9965894 A1, 1999.
36. Aicher, T. D.; Buszek, K. R.; Fang, F. G.; Forsyth, C. J.; Jung, S. H.; Kishi, Y.;
Matelich, M. C.; Scola, P. M.; Spero, D. M.; Yoon, S. K. J. Am. Chem. Soc. 1992,
114, 3162.
37. Austad, B.; Chase, C. E.; Fang, F. G. WO 2005118565 A1, 2005.
38. Chase, C.; Endo, A.; Fang, F. G.; Li, J. WO 2009046308 A1, 2009.
CLIP
http://www.rsc.org/chemistryworld/2015/06/longest-organic-syntheses-natural-product
Eribulin (Halaven)
Halichondrin B is a wicked molecule. In tests in mice, it is an extremely potent cancer cell killer, active at around 80 picomolar concentration. It also possesses a fiendish macrocyclic polyketide structure, with 32 stereocentres meaning that it could adopt over four billion different isomers – with just one that fights cancer.
 Eribulin is a cut-down derivative of halichondrin B, which maintains most of its activity with significantly reduced complexity
Eribulin is a cut-down derivative of halichondrin B, which maintains most of its activity with significantly reduced complexity
Its power is therefore inherently hard to harness. Halichondrin B was found in various sea sponge species in the 1980s, but getting 400mg of the compound from a tonne of sponge was doing well. Clinical development required at least 10g, and annual production takes kilograms.
Although developing a synthetic route to halichondrin B looked just as tough as trying to extract it from sponges, Yoshito Kishi’s group at Harvard University in the US accepted the challenge. Frank Fang, one of the team, recalls how the Nozaki–Hiyama–Kishi (NHK) coupling reaction would prove critical. ‘Another feature that was impressed upon me was the importance of crystalline intermediates,’ Fang adds. These allowed simple purification by recrystallisation, rather than expensive and time-consuming chromatography.
Published in 1992, their method used several NHK couplings, forming carbon–carbon bonds between multifunctional vinyl halides and aldehydes via a nickel-catalysed, chromium-mediated process.4 The sprawling convergent synthesis, whose longest linear sequence involved 47 steps, prompted Japanese pharmaceutical company Eisai to collaborate with Kishi in exploring halichondrin B’s structure–activity relationship. On screening the team’s intermediates, one featuring the macrocyclic half of halichondrin B proved especially active. A series of medicinal chemistry refinements led to what would eventually becomeeribulin (marketed by Eisai as Halaven), promising a slightly simpler synthesis. It has ‘just’ 19 stereocentres, which along with other structural restrictions cuts the possible number of isomers to a mere 16,384.
Fang joined Eisai in 1998 as it selected eribulin for further development, and worked to develop a production process for a route that produced it from three fragments. He again strove to exploit recrystallisation and use the NHK reaction, although making it reliable enough for manufacturing was challenging. ‘There was an appreciation for the somewhat sensitive nature of the reaction, particularly the asymmetric variant,’ he recalls.
The Eisai researchers therefore studied the NHK procedure as they applied it to redesigning the synthesis for part of the eribulin molecule they refer to as the C14–C26 fragment. Featuring just one ring, this fragment isn’t the most structurally complex of the three, but is still very difficult to make. That’s because it is a long chain with several stereocentres, whose stereochemistry is hard to link together.
Fang’s team initially broke this section down into two sub-fragments, C14–C19 and C20–C26, using asymmetric NHK reactions on each, learning about the reaction’s parameters as they did so.5 They then used what they’d found out to devise NHK reactions linking the two sub-fragments and attaching the two fragments on either side, which included closing the eribulin macrocycle. ‘We gained knowledge through our studies on the C19–C20 NHK coupling and were ultimately able to utilise that knowledge to try to execute an asymmetric NHK reaction in fixed equipment on multi-kilogram scale and construct the C19–C20, C26–C27, and C13–C14 bonds,’ Fang explains.
 Synthesis of eribulin relies heavily on Nozaki–Hiyama–Kishi (NHK) coupling reactions to make key C–C bonds
Synthesis of eribulin relies heavily on Nozaki–Hiyama–Kishi (NHK) coupling reactions to make key C–C bonds
Halaven was approved in the US in 2010 to treat breast cancer and earned ¥2.89 billion in sales (£159 million) in 2014. The commercial route initially took 62 steps across a convergent synthesis bringing together three fragments, with a longest linear sequence of 30 steps. Fang’s team has since added seven steps to the C14–C26 fragment route, which counterintuitively cuts costs and waste by 80% by eliminating chromatography.6 ‘I am hopeful that we can find the lessons applicable in future work,’ Fang says.
Cheaper synthesis would appear welcome, given that Halaven’s price tag has been criticised. In the UK it currently costs £2,000 per 21 day treatment cycle according to data from the British National Formularyand the country’s National Institute for Health and Clinical Excellence (Nice). As a result, Nice refused to cover the drug, and in January 2015 the remaining funding in England looked set to be closed off with Halaven being taken off the Cancer Drugs Fund (CDF)’s list. But Eisai was told in March that the drug would stay on the list, pending reconsideration, after an appeal against the decision.
In defence, Fang claims that Halaven is actually one of the most affordable breast cancer treatments on the CDF. ‘Eisai was given no opportunity to lower the price of Halaven before NHS England announced that the treatment would be removed from the fund, despite this being something we were, and still are, very willing to do,’ he adds.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009124237A1 * | Apr 3, 2009 | Oct 8, 2009 | Eisai R&D Management Co., Ltd. | Halichondrin b analogs |
| US6214865 * | Jun 16, 1999 | Apr 10, 2001 | Eisai Co., Ltd. | Macrocyclic analogs and methods of their use and preparation |
| Reference | ||
|---|---|---|
| 1 | * | DONG, C.-G. ET AL.: “New Syntheses of E7389 C 14-C35 and Halichondrin C 14- C38 Building Blocks: Reductive Cyclization and Oxy-Michael Cyclization Approaches“, J. AM. CHEM. SOC., vol. 131, 2009, pages 15642 – 15646, XP002629056 |
| 2 | * | See also references of EP2831082A4 |
| 3 | * | ZHENG, W. ET AL.: “Macrocyclic ketone analogues of halichondrin B“, BIOORG. MED. CHEM. LETT., vol. 14, 2004, pages 5551 – 5554, XP004598592 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015000070A1 * | May 30, 2014 | Jan 8, 2015 | Alphora Research Inc. | Synthetic process for preparation of macrocyclic c1-keto analogs of halichondrin b and intermediates useful therein including intermediates containing -so2-(p-tolyl) groups |
| WO2015066729A1 * | Nov 4, 2014 | May 7, 2015 | Eisai R&D Management Co., Ltd. | Macrocyclization reactions and intermediates useful in the synthesis of analogs of halichondrin b |
| WO2015131286A1 * | Mar 6, 2015 | Sep 11, 2015 | Alphora Research Inc. | Crystalline derivatives of (s)-1-((2r,3r,4s,5s)-5-allyl-3-methoxy-4-(tosylmethyl)tetrahydrofuran-2-yl)-3-aminopropan-2-ol |
| CN103483352A * | Oct 18, 2013 | Jan 1, 2014 | 李友香 | Medicinal bulk drug for resisting tumors |
| US9062020 | Dec 24, 2012 | Jun 23, 2015 | Alphora Research Inc. | 2-((2S,3S,4R,5R)-5-((S)-3-amino-2-hydroxyprop-1-yl)-4-methoxy-3-(phenylsulfonylmethyl)tetrahydrofuran-2-yl)acetaldehyde derivatives and process for their preparation |
| US9174956 | Dec 14, 2012 | Nov 3, 2015 | Alphora Research Inc. | Process for preparation of 3-((2S,5S)-4-methylene-5-(3-oxopropyl)tetrahydrofuran-2-yl)propanol derivatives and intermediates useful thereof |
| US9181152 | Nov 29, 2012 | Nov 10, 2015 | Alphora Research Inc. | Process for preparation of (3R)-2,4-di-leaving group-3-methylbut-1-ene |
| WO2012129100A1 * | Mar 16, 2012 | Sep 27, 2012 | Eisai R&D Management Co., Ltd. | Methods and compositions for predicting response to eribulin |
| WO2012166899A2 * | May 31, 2012 | Dec 6, 2012 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of thyroid and kidney cancer subjects to lenvatinib compounds |
| CA2828946A1 * | Apr 16, 2012 | Oct 26, 2012 | Eisai R&D Management Co., Ltd. | Therapeutic agent for tumor |
| US7982060 * | Jun 3, 2005 | Jul 19, 2011 | Eisai R&D Management Co., Ltd. | Intermediates for the preparation of analogs of Halichondrin B |
References
- ^“FDA approves new treatment option for late-stage breast cancer” (Press release). USFDA. 2010-11-15. Retrieved November 15, 2010.
- ^Notice of Decision for HALAVEN
- ^http://www.clinicaltrials.gov/ct2/results?term=eribulin+OR+E7389
- ^ Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsky BM, Palme MH, Habgood GJ, Singer LA, Dipietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ, Littlefield BA (February 2001). “In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B”. Cancer Res.61 (3): 1013–21. PMID11221827.
- ^ Yu MJ, Kishi Y, Littlefield BA (2005). “Discovery of E7389, a fully synthetic macrocyclic ketone analogue of halichondrin B”. In Newman DJ, Kingston DGI, Cragg, GM. Anticancer agents from natural products. Washington, DC: Taylor & Francis. ISBN0-8493-1863-7.
- ^ Hirata Y, Uemura D (1986). “Halichondrins – antitumor polyether macrolides from a marine sponge”. Pure Appl. Chem.58 (5): 701–710. doi:10.1351/pac198658050701.
- ^ Bai RL, Paull KD, Herald CL, Malspeis L, Pettit GR, Hamel E (August 1991). “Halichondrin B and homohalichondrin B, marine natural products binding in the vinca domain of tubulin. Discovery of tubulin-based mechanism of action by analysis of differential cytotoxicity data”. J. Biol. Chem.266 (24): 15882–9. PMID1874739.
- Jordan MA, Kamath K, Manna T, Okouneva T, Miller HP, Davis C, Littlefield BA, Wilson L (July 2005). “The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth”. Mol. Cancer Ther.4 (7): 1086–95. doi:10.1158/1535-7163.MCT-04-0345. PMID16020666.
- Okouneva T, Azarenko O, Wilson L, Littlefield BA, Jordan MA (July 2008). “Inhibition of Centromere Dynamics by Eribulin (E7389) during Mitotic Metaphase”. Mol. Cancer Ther.7 (7): 2003–11. doi:10.1158/1535-7163.MCT-08-0095. PMC2562299. PMID18645010.
- Smith JA, Wilson L, Azarenko O, Zhu X, Lewis BM, Littlefield BA, Jordan MA (February 2010). “Eribulin Binds at Microtubule Ends to a Single Site on Tubulin to Suppress Dynamic Instability”. Biochemistry49 (6): 1331–7. doi:10.1021/bi901810u. PMC2846717. PMID20030375.
- Kuznetsov G, Towle MJ, Cheng H, Kawamura T, TenDyke K, Liu D, Kishi Y, Yu MJ, Littlefield BA (August 2004). “Induction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin B macrocyclic ketone analog E7389”. Cancer Res.64 (16): 5760–6. doi:10.1158/0008-5472.CAN-04-1169. PMID15313917.
- ^ Towle MJ, Salvato KA, Wels BF, Aalfs KK, Zheng W, Seletsky BM, Zhu X, Lewis BM, Kishi Y, Yu MJ, Littlefield BA (January 2011). “Eribulin induces irreversible mitotic blockade: implications of cell-based pharmacodynamics for in vivo efficacy under intermittent dosing conditions”. Cancer Res.71 (2): 496–505. doi:10.1158/0008-5472.CAN-10-1874. PMID21127197.
- ^ Kim DS, Dong CG, Kim JT, Guo H, Huang J, Tiseni PS, Kishi Y (November 2009). “New syntheses of E7389 C14-C35 and halichondrin C14-C38 building blocks: double-inversion approach”. J. Am. Chem. Soc.131 (43): 15636–41. doi:10.1021/ja9058475. PMID19807076.
SEE https://wordpress.com/post/newdrugapprovals.org/3955
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(3-Amino-2-hydroxypropyl)hexacosahydro-3-methoxy- 26-methyl-20,27-bis(methylene)11,15-18,21-24,28-triepoxy- 7,9-ethano-12,15-methano-9H,15H-furo(3,2-i)furo(2′,3′-5,6) pyrano(4,3-b)(1,4)dioxacyclopentacosin-5-(4H)-one
|
|
| Clinical data | |
| Trade names | Halaven |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a611007 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 253128-41-5 |
| ATC code | L01XX41 (WHO) |
| PubChem | CID 17755248 |
| ChemSpider | 21396142 |
| UNII | LR24G6354G |
| ChEMBL | CHEMBL1237028 |
| Chemical data | |
| Formula | C40H59NO11 |
| Molar mass | 729.90 g/mol |
////////Halaven, ERIBULIN, anticancer drug , Eisai Co. E7389, ER-086526, US NCI designation, NSC-707389. breast cancer, liposarcoma, halichrondrin B analog, B1939, E7389, ER-086526, 441045-17-6, FDA 2010, 253128-41-5 , ERIBULIN MESYLATE, Antineoplastic, エリブリンメシル酸塩
CC1CC2CCC3C(=C)CC(O3)CCC45CC6C(O4)C7C(O6)C(O5)C8C(O7)CCC(O8)CC(=O)CC9C(CC(C1=C)O2)OC(C9OC)CC(CN)O.CS(=O)(=O)O
C[C@@H]1C[C@@H]2CC[C@H]3C(=C)C[C@@H](O3)CC[C@]45C[C@@H]6[C@H](O4)[C@H]7[C@@H](O6)[C@@H](O5)[C@@H]8[C@@H](O7)CC[C@@H](O8)CC(=O)C[C@H]9[C@H](C[C@H](C1=C)O2)O[C@@H]([C@@H]9OC)C[C@@H](CN)O.CS(=O)(=O)O
C[C@@H]1C[C@@H]2CC[C@H]3C(=C)C[C@@H](O3)CC[C@]45C[C@@H]6[C@H](O4)[C@H]7[C@@H](O6)[C@@H](O5)[C@@H]8[C@@H](O7)CC[C@@H](O8)CC(=O)C[C@H]9[C@H](C[C@H](C1=C)O2)O[C@@H]([C@@H]9OC)C[C@@H](CN)O.CS(=O)(=O)O
CREDIT
http://www.chm.bris.ac.uk/motm/eribulin/eribulinh.htm

253128-41-5 CAS

