
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

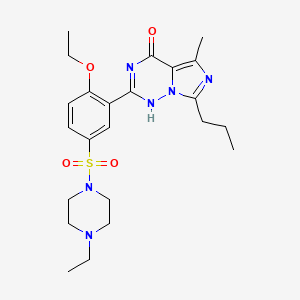
VARDENAFIL
224785-90-4 CAS NO
Vardenafil hydrochloride (CAS NO.224785-91-5)
| Formula | C23H32N6O4S |
|---|---|
| Mol. mass | 488.604 g/mol |
4-[2-Ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-9-methyl-7-propyl-3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one
Vivanza, Vardenafil (INN), Levitra (TN), STK642629, , LEVITRA
Vardenafil (INN) is a PDE5 inhibitor used for treating erectile dysfunction that is sold under the trade names Levitra (Bayer AG, GSK, and SP) andStaxyn.
Vardenafil was co-marketed by Bayer Pharmaceuticals, GlaxoSmithKline, and Schering-Plough under the trade name Levitra. As of 2005, the co-promotion rights of GSK on Levitra have been returned to Bayer in many markets outside the U.S. In Italy, Bayer sells vardenafil as Levitra and GSK sells it as Vivanza. Thus, because of European Union trade rules, parallel imports might result in Vivanza sold next to Levitra in the EU.
Vardenafil (Levitra) is an oral therapy for the treatment of erectile dysfunction. It is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). Penile erection is a hemodynamic process initiated by the relaxation of smooth muscle in the corpus cavernosum and its associated arterioles. During sexual stimulation, nitric oxide is released from nerve endings and endothelial cells in the corpus cavernosum. Nitric oxide activates the enzyme guanylate cyclase resulting in increased synthesis of cyclic guanosine monophosphate (cGMP) in the smooth muscle cells of the corpus cavernosum. The cGMP in turn triggers smooth muscle relaxation, allowing increased blood flow into the penis, resulting in erection. The tissue concentration of cGMP is regulated by both the rates of synthesis and degradation via phosphodiesterases (PDEs). The most abundant PDE in the human corpus cavernosum is the cGMPspecific phosphodiesterase type 5 (PDE5); therefore, the inhibition of PDE5 enhances erectile function by increasing the amount of cGMP.
An orally disintegrating form, marketed as Staxyn, has been gaining approvals in countries such as the United States[1] and Canada.[2]
Vardenafil’s indications and contra-indications are the same as with other PDE5 inhibitors; it is closely related in function to sildenafil citrate (Viagra) and tadalafil (Cialis). The difference between the vardenafil molecule and sildenafil citrate is a nitrogen atom’s position and the change of sildenafil’spiperazine ring methyl group to an ethyl group. Tadalafil is structurally different from both sildenafil and vardenafil. Vardenafil’s relatively short effective time is comparable to but somewhat longer than sildenafil’s.
Beyond its indications for erectile dysfunction, vardenafil may be effective in the treatment of premature ejaculation, where it may significantly increase the time from vaginal penetration to ejaculation.[3]
The common, adverse drug reactions (side-effects) are the same as with other PDE5 inhibitors. The frequent vardenafil-specific side-effect is nausea; the infrequent side-effects are abdominal pain, back pain, photosensitivity, abnormal vision, eye pain, facial edema, hypotension, palpitation,tachycardia, arthralgia, myalgia, rash, itch, and priapism.

One possibly serious, but rare, side-effect with vardenafil is heart attack. Also, in rare cases, vardenafil use may cause priapism, a very painful emergency condition that can cause impotence if left untreated.[4]
On 18 October 2007, the U.S. Food and Drug Administration (FDA) announced that a warning about possible deafness (sudden hearing loss) would be added to the drug labels of Vardenafil, and other PDE5 inhibitors.[5]
Vardenafil, as with all PDE5 inhibitors, should not be used by men taking nitrate medications, because combining them with vardenafil might provoke potentially life-threatening hypotension (low blood pressure).
Further, Vardenafil causing lengthening of the QT interval. Therefore it should not be taken by men taking other medications that affect the QT interval (such as amiodarone).

Levitra 20mg Oral Tablet
It is available in 2.5 mg, 5 mg, 10 mg, and 20 mg doses in round orange tablets. The normal starting dose is 10 mg (roughly equivalent to 50 mg of sildenafil). Vardenafil should be taken 1 to 2 hours prior to sexual activity, with a maximum dose frequency of once per day. In some territories, such as the UK, only certain doses may be available.
Vardenafil is also available under the name Staxyn as a tablet which dissolves on the tongue rather than being swallowed in the form of a pill.
STAXYN is an oral therapy for the treatment of erectile dysfunction. This monohydrochloride salt of vardenafil is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific PDE5.
Vardenafil HCl is designated chemically as piperazine, 1-[[3-(1,4-dihydro-5-methyl-4-oxo-7-propylimidazo[5,1f][1,2,4]triazin-2-yl)-4-ethoxyphenyl]sulfonyl]-4-ethyl-, monohydrochloride and has the following structural formula:
 |
Vardenafil HCl is a nearly colorless, solid substance with a molecular weight of 579.1 g/mol and a solubility of 0.11 mg/mL in water.
TRIHYDRATE, HCL SALT


US2002137930A

vardenafil hydrochloride is piperazine, 1-[[3-(1,4-dihydro-5-methyl-4-oxo-7-propylimidazo[5,1-f][1,2,4]triazin-2-yl)-4-ethoxyphenyl]sulfonyl]-4-ethyl-, mono -hydrochloride and can be structurally represented by Formula I.

The monohydrochloride salt of vardenafil is a selective inhibitor of cyclic guaosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). It is commercially available in products sold under the brand name LEVITRA formulated as 2.5 mg, 5 mg, 10 mg, 20 mg film-coated tablets.
U.S. Pat. No. 6,362,178 B1 discloses vardenafil, its related compounds and processes for their preparation. The patent describes a process in which vardenafil is obtained by recrystallization in ether in Example 19. Vardenafil produced as per Example 19 is hereinafter referred as “crystalline Form I” of vardenafil. The patent also describes processes for the preparation of its monohydrochloride and dihydrochloride salts, which are formed in a combination of ether and dichloromethane. The patent also describes a process for the preparation of vardenafil monohydrochloride trihydrate.
U.S. Patent Application Publication No. 2005/0203298 also describes a process for the preparation of vardenafil, and its monohydrochloride trihydrate.
Chemical synthesis of vardenafil has mostly been directed to the preparation of the trihydrate of monohydrochloride of vardenafil.
In WO 99/24433, sulphonamide-substituted imidazotriazinones are described as potent inhibitors of either one or more of the cyclic guanosine 3′,5′-monophosphate-metabolizing phosphodiesterases (cGMP PDEs). According to the nomenclature of Beavo and Reifsnyder (Trends in Pharmacol. Sci. 11, 150-155, 1990), these cGMP PDEs are the phosphodiesterase isoenzymes PDE-I, PDE-II and PDE-V.
According to WO 99/24433, the sulphonamide-substituted imidazotriazinones described therein are prepared from corresponding 2-ethoxyphenyl-substituted imidazotriazinones by reaction with chlorosulphonic acid and subsequent reaction with an appropriate amine, as is illustrated by the following scheme (R1 to R6 here have the meanings indicated in WO 99/24433):
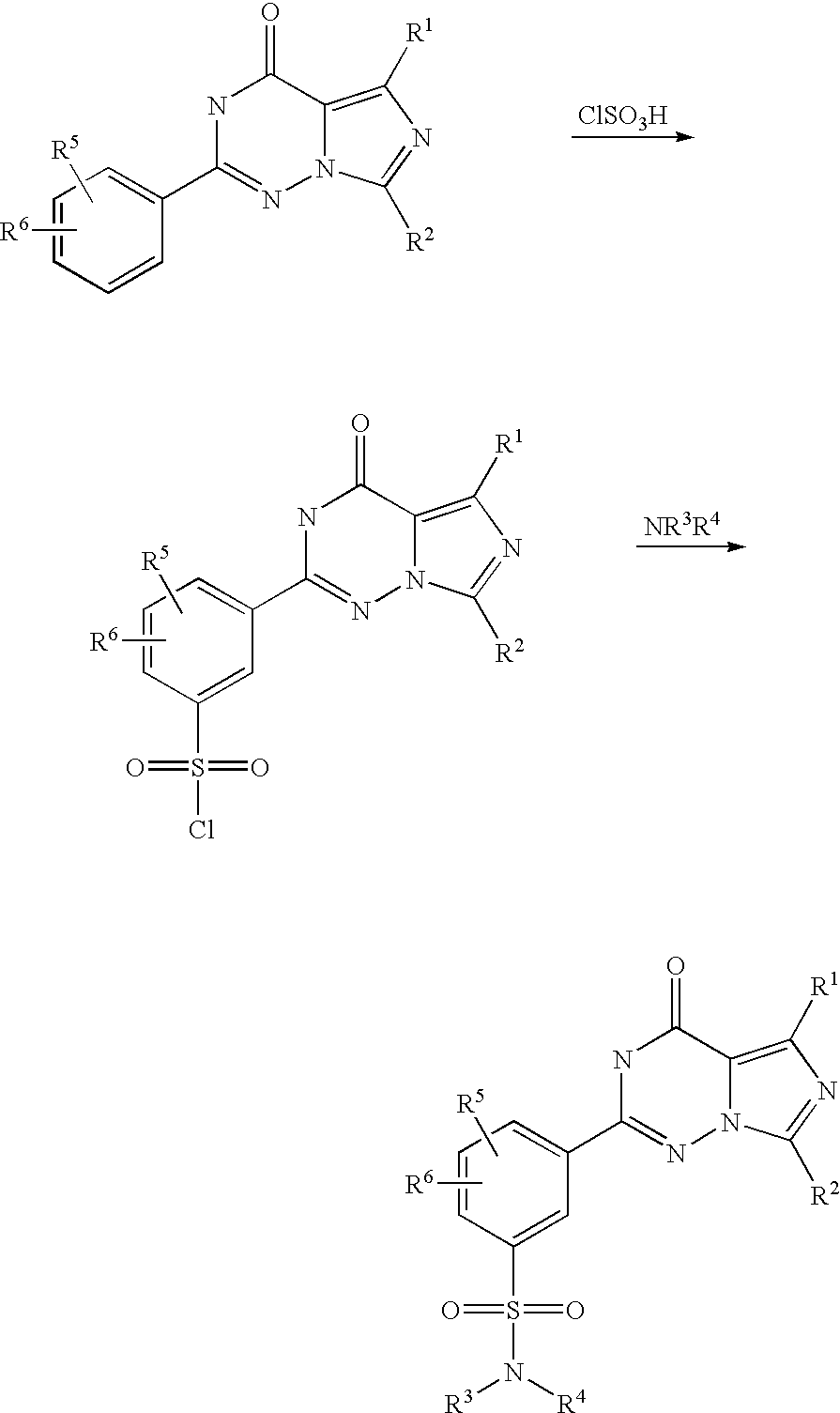
In this process, highly reactive chlorosulphonic acid has to be used as a reagent. Moreover, the imidazotriazinonesulphonyl chlorides formed as intermediates are sensitive to hydrolysis, which, in particular in the conversion of this preparation process to the industrial scale, can lead to not inconsiderable yield variations.
It was therefore the object of the present invention to make available a process for the preparation of sulphonamide-substituted imidazotriazinones in which the disadvantages of the above process known from the prior art are avoided.
This object is achieved according to the present invention by a process as in claim 1. In particular, in the process according to the invention as in claim 1 the use of chlorosulphonic acid is avoided by introduction of the sulphonic acid via a reaction with sulphuric acid and subsequent reaction with thionyl chloride. Moreover, the reaction with thionyl chloride and the subsequent reaction with an amine is carried out in a one-pot process, so that the imidazotriazinonesulphonyl chloride intermediate, which is sensitive to hydrolysis, does not need to be isolated. By means of this, yield variations on account of partial hydrolysis of this intermediate can be excluded. As a result of these advantages, the process according to the invention is much simpler to carry out on the industrial scale than the process described in WO 99/24433.
………………….
SYNTHESIS
2-butyrylamino-propionic acid
EXAMPLE 1A 2-Butyrylaminopropionic acid

22.27 g (250 mmol) of D,L-alanine and 55.66 g (550 mmol) of triethylamine are dissolved in 250 ml of dichloromethane, and the solution is cooled to 0° C. 59.75 g (550 mmol) of trimethylsilyl chloride are added dropwise, and the solution is stirred for 1 hour at room temperature and for 1 hour at 40° C. After cooling to −10° C., 26.64 g (250 mmol) of butyryl chloride are added dropwise, and the resulting mixture is stirred for 2 hours at −10° C. and for one hour at room temperature.
With ice-cooling, 125 ml of water are added dropwise and the reaction mixture is stirred at room temperature for 15 minutes. The aqueous phase is evaporated to dryness, the residue is titrated with acetone and the mother liquor is filtered off with suction. The solvent is removed and the residue is chromatographed. The resulting product is dissolved in 3N aqueous sodium hydroxide solution and the resulting solution is evaporated to dryness. The residue is taken up in conc. HCl and once more evaporated to dryness. The residue is stirred with acetone, precipitated solid is filtered off with suction and the solvent is removed under reduced pressure. This gives 28.2 g (71%) of a viscous oil which crystallizes after some time.
200 MHz 1H-NMR (DMSO-d6): 0.84, t, 3H; 1.22, d, 3H; 1.50, hex, 2H; 2.07, t, 2H; 4.20, quin., 1H; 8.09, d, 1H.
EXAMPLE 3A 2-Ethoxybenzonitrile

25 g (210 mmol) of 2-hydroxybenzonitrile are refluxed with 87 g of potassium carbonate and 34.3 g (314.8 mmol) of ethyl bromide in 500 ml of acetone overnight. The solid is filtered off, the solvent is removed under reduced pressure and the residue is distilled under reduced pressure. This gives 30.0 g (97%) of a colourless liquid.
200 MHz 1H-NMR (DMSO-d6): 1.48, t, 3H; 4.15, quart., 2H; 6.99, dt, 2H; 7.51, dt, 2H.
EXAMPLE 4A 2-Ethoxybenzamidine hydrochloride

21.4 g (400 mmol) of ammonium chloride are suspended in 375 ml of toluene, and the suspension is cooled to 0° C. 200 ml of a 2M solution of trimethylaluminium in hexane are added dropwise, and the mixture is stirred at room temperature until the evolution of gas has ceased. After addition of 29.44 g (200 mmol) of 2-ethoxybenzonitrile, the reaction mixture is stirred at 80° C. (bath) overnight.
With ice-cooling, the cooled reaction mixture is added to a suspension of 100 g of silica gel and 950 ml of chloroform, and the mixture is stirred at room temperature for 30 minutes. The mixture is filtered off with suction, and the filter residue is washed with the same amount of methanol. The mother liquor is concentrated, the resulting residue is stirred with a mixture of dichloromethane and methanol (9:1), the solid is filtered off with suction and the mother liquor is concentrated. This gives 30.4 g (76%) of a colourless solid.
200 MHz 1H-NMR (DMSO-d6): 1.36, t, 3H; 4.12, quart., 2H; 7.10, t, 1H; 7.21, d, 1H; 7.52, m, 2H; 9.30, s, broad, 4H.
EXAMPLE 10A 2-(2-Ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

7.16 g (45 mmol) of 2-butyrylamino-propionic acid and 10.67 g of pyridine are dissolved in 45 ml of THF and, after addition of a spatula tip of DMAP, heated to reflux. 12.29 g (90 mmol) of ethyl oxalyl chloride are slowly added dropwise, and the reaction mixture is refluxed for 3 hours. The mixture is poured into ice-water and extracted three times with ethyl acetate and the organic phase is dried over sodium sulphate and concentrated using a rotary evaporator. The residue is taken up in 15 ml of ethanol and refluxed with 2.15 g of sodium bicarbonate for 2.5 hours. The cooled solution is filtered.
With ice-cooling, 2.25 g (45 mmol) of hydrazine hydrate are added dropwise to a solution of 9.03 g (45 mmol) of 2-ethoxybenzamidine hydrochloride in 45 ml of ethanol, and the resulting suspension is stirred at room temperature for another 10 minutes. The ethanolic solution described above is added to this reaction mixture, and the mixture is stirred at a bath temperature of 70° C. for 4 hours. After filtration, the mixture is concentrated, the residue is partitioned between dichloromethane and water, the organic phase is dried over sodium sulphate and the solvent is removed under reduced pressure.
This residue is dissolved in 60 ml of 1,2-dichloroethane and, after addition of 7.5 ml of phosphorus oxychloride, refluxed for 2 hours. The mixture is diluted with dichloromethane and neutralized by addition of sodium bicarbonate solution and solid sodium bicarbonate. The organic phase is dried and the solvent is removed under reduced pressure. Chromatography using ethyl acetate and crystallization afford 4.00 g (28%) of a colourless solid, Rf=0.42 (dichloromethane/methanol=95:5)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.56, t, 3H; 1.89, hex, 2H; 2.67, s, 3H; 3.00, t, 2H; 4.26, quart., 2H; 7.05, m, 2H; 7.50, dt, 1H; 8.17, dd, 1H; 10.00, s, 1H.
EXAMPLE 15A 4-Ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride

At 0° C., 2.00 g (6.4 mmol) of 2-(2-ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are slowly added to 3.83 ml of chlorosulphonic acid. At room temperature, the reaction mixture is stirred ovemight, and then poured into ice-water and extracted with dichloromethane. This gives 2.40 g (91%) of a colourless foam.
200 MHz 1H-NMR (CDCl3): 1.03, t, 3H; 1.61, t, 2H; 1.92, hex, 2H; 2.67, s, 3H; 3.10, t, 2H; 4.42, quart., 2H; 7.27, t, 1H; 8.20, dd, 1H; 8.67, d, 1H; 10.18, s, 1H.
Example 19 2-[2-Ethoxy-5-(4-ethyl-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

470 mg (1.14 mmol) of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride are dissolved in 20 ml of dichloromethane and cooled to 0° C. 390 mg (3.42 mmol) of N-ethylpiperazine are added, and the reaction mixture is stirred at room temperature overnight. The mixture is diluted with dichloromethane, the organic phase is washed twice with water and dried over sodium sulphate and the solvent is removed under reduced pressure. Crystallization from ether gives 370 mg (66%) of a colourless solid.
400 MHz 1H-NMR (CDCl3): 1.01, t, 3H; 1.59, t, 3H; 1.88, hex, 2H; 2.42, quart., 2H; 2.56, m, 4H; 2.63, s, 3H; 3.00, t, 2H; 3.10, m, 4H; 4.33, quart., 2H, 7.17, d, 1H; 7.88, dd, 1H; 8.44, d, 1H; 9.75, s, 1H.
…………………….
EXAMPLE 7 Preparation of the Trihydrate of Vardenafil Monohydrochloride
14 g of vardenafil hydrochloride was taken into a round bottom flask followed by the addition of 70 ml water and the pH of the reaction mass was adjusted using sodium hydroxide to 11 at 30° C. 280 ml of dichloromethane was added to the above reaction mass and the layers were separated. The organic layer was dried over sodium sulfate and the organic layer was transferred into a round bottom flask and subjected to heating for distillation at 40° C. for 1.5 hours. The solid material was transferred into a round bottom flask and 36 ml of a mixture of acetone and water in 12:1 ratio was added with stirring, then 2.2 ml of 36% aqueous hydrochloric acid was added with stirring. The reaction mass was heated to a temperature of about 45° C. and the undissolved particles were removed by filtration. The filtrate was taken into a round bottom flask and cooled to 5° C., maintained for 45 minutes at 3 to 5° C. followed by the filtration of the solid which was then subjected to suction drying and finally dried at 40° C. to yield 9.0 g of the trihydrate of vardenafil monohydrochloride.
……………………..
STARTING COMPOUNDS
Example I Preparation of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo-[5,1-f][2,4]triazin-4-oneIa) Preparation of 2-butyrylaminopropionic acid
A solution of 100 kg of D,L-alanine in aqueous sodium hydroxide solution is reacted in the cold with 119 kg of butyryl chloride. After addition of butyl acetate, the mixture is acidified with hydrochloric acid, the organic phase is separated off and the aqueous phase is re-extracted. The organic phase is dried by azeotropic distillation. The crystallizate is isolated, washed with butyl acetate and dried.
Yield: 132.6 kg (68%)
1H-NMR: δ=0.8 (t, 3H), 1.25 (d, 3H), 1.5 (m, 2H), 2.1 (t, 2H), 4.2 (q, 1H), 8.1 (d, NH), 12.0-12.7 (s, COOH)
MS: 336 (2M+NH4, 40), 319 (2M+H, 15), 177 (M+NH4, 100), 160 (M+H, 20)
Ib) Preparation of 2-ethoxybenzonitrile

260 kg of thionyl chloride are added at 85-95° C. to a suspension of 250 kg of 2-ethoxybenzamide in toluene under metering control. The reaction mixture is stirred in the presence of heat. Thionyl chloride and toluene are then distilled off in vacuo. The product is employed in the subsequent stage as a crude product.
Yield: 228.5 kg (crude product)
1H-NMR: δ=1.45 (t, 3H), 4.15 (q, 2H), 7.0 (m, 2H, phenyl), 7.5 (m, 2H, phenyl)
MS: 312 (2M+N4, 35), 165 (M+NH4, 100), 147 (5)
Ic) Preparation of 2-ethoxy-N-hydroxybenzamidine

111 kg of 2-ethoxybenzonitrile (crude product) from Example Ib are heated under reflux with 164 1 of triethylamine and 73 kg of hydroxylamine hydrochloride in isopropanol. The reaction mixture is treated with water and cooled. The crystallizate is isolated, washed and employed in the subsequent stage as a moist product.
Yield: 92.6 kg (moist product)
1H-NMR: δ=1.35 (t, 3H), 4.1 (q, 2H), 5.6 (s, 2H), 6.9-7.4 (4H, phenyl), 9.4 (s, 1H, OH)
MS: 361 (2M+H, 30), 198 (M+N, 30), 181 (M+H, 100)
Id) Preparation of 2-ethoxybenzamidine hydrochloride

135 kg of 2-ethoxy-N-hydroxybenzamidine (moist product) from Example Ic are hydrogenated at 50-60° C. in acetic acid using palladium on carbon as a catalyst. For the work-up, the hydrogenation reaction is freed from the catalyst, treated with hydrochloric acid and concentrated. Residual acetic acid and water are removed by azeotropic distillation with toluene. The crystallizate is isolated and dried in vacuo.
Yield: 136.4 kg
H-NMR: 1.35 (t, 3H), 4.15 (q, 2H), 7.1-7.7 (m, 4H, phenyl), 9.1-9.4 (2×s, 3H), 10.5-10.7 (s, 1H)
MS: 329 (2M+H, 10), 165 (M+H, 100)
Ie) Preparation of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]-triazin-4-one

231 kg of 2-butyrylaminopropionic acid from Example Ia are treated in tetrahydrofuran with 341 kg of pyridine, catalytic amounts of 4-N,N-dimethylaminopyridine and 392 kg of ethyl chloroxalate and stirred with heating under reflux. The reaction mixture is taken up in ethyl acetate, washed with water and the ethyl acetate phase is concentrated. The distillation residue is taken up in methanol and reacted with the following solution.
192 kg of 2-ethoxybenzamidine hydrochloride from Example Id are treated in methanol with 47.5 kg of hydrazine hydrate and the mixture is stirred at room temperature. The solution is combined with the solution of 2-butyrylamino-1-ethoxycarbonylpropenyl ethyl oxalate prepared above. The reaction mixture thus obtained is stirred with heating under reflux. Methanol is removed by distillation and replaced by acetic acid.
Option A:
138.6 kg of phosphorus oxychloride are added and stirred in the presence of heat.
Acetic acid is distilled off in vacuo. The residue is treated with water and dichloromethane or optionally methyl isobutyl ketone and rendered neutral using sodium hydroxide solution. The organic phase is concentrated, and the residue is dissolved in acetone and crystallized with cooling. The crystallizate is isolated, washed and dried.
Option B:
At least 190 kg of acetyl chloride are added and stirred in the presence of heat. Acetic acid is distilled off in vacuo. The distillation residue is treated with acetone and water, and the product is crystallized by rendering neutral with sodium hydroxide solution. The product is isolated, washed and dried.
Yield: 90-160 kg
1H-NMR: δ=1.0 (t, 3H), 1.6 (t, 3H), 1.9 (m, 2H), 2.8 (s, 3H), 3.3 (t, 2H), 4.3 (q, 2H), 7.0-8.2 (Ar, 4H), 10.3 (CONH, 1H)
MS: 313 (M+H, 100), 149 (25), 151 (40), 121 (15)
HPLC: Kromasil C-18 phase, neutral phosphate buffer, acetonitrile, 233 nm, linear gradient of 30% acetonitrile ->80% acetonitrile (30 min.): 99 area % (Rt 19.1)
PREPARATION EXAMPLES Example 1a 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydroimidazo[5,1-fl-][1,2,4]triazin-2-yl)benzenesulphonic acid

194 kg of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one from Example Ie are reacted with 504 kg of concentrated sulphuric acid. The reaction mixture is added to water, cooled, and the crystallizate is isolated and dried in vacuo.
Yield: 195.2 kg
1H-NMR: δ=0.95 (t, 3H), 1.3 (t, 3H), 1.8 (m, 2H), 2.6 (s, 3H), 3.05 (t, 2H), 4.1 (q, 2H), 7.15 (Ar, 1H), 7.75 (m, 2H), 12.3 (SO2OH)
MS: 393 (M+H, 100), 365 (25), 151 (40)
HPLC: X-Terra C-18 phase, aqueous phosphoric acid, acetonitrile, 242 nm, linear gradient of 10% acetonitrile ->90% acetonitrile (20 min.):
98 area % (R, 9.2)
Example 1b) 2-[2-ethoxy-5-(4-ethlylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

22.5 kg of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]-triazin-2-yl)benzenesulphonic acid from Example 1a are reacted with 74 kg of thionyl chloride and catalytic amounts of dimethylformamide until the evolution of gas has ended. Xylene is repeatedly added to the reaction mixture and thionyl chloride is distilled off. 15.1 kg of N-ethylpiperazine are added to the suspension and it is stirred. After the addition of water, it is adjusted to pH 1 using hydrochloric acid, and the phases are separated. The aqueous phase is treated with acetone and rendered neutral by addition of sodium hydroxide solution. The mixture is cooled, and the crystallizate is isolated, washed and dried in vacuo.
Yield: 26.1 kg
1H-NMR: δ=1.0 (2×t, 6H), 1.6 (t, 3H), 1.9 (m, 2H), 2.45 (q, 2H), 2.55 (m, 4H), 2.65 (s, 3H), 3.0 (t, 2H), 3.1 (m, 4H), 4.35 (q, 2H), 7.15 (Ar, 1H), 7.9 (Ar, 1H), 8.4 (Ar, 1H), 9.8 (CONH)
MS: 489 (M+H, 100), 345 (10), 313, (10), 285 (10), 113 (20)
HPLC: X-Terra C-18 phase, neutral phosphate buffer, acetonitrile, 242 nm, linear gradient of 20% acetonitrile ->75% acetonitrile (20 min.): 98 area % (Rt 16.3)
1 c) 2-[2-ethoxy-5-(4-ethylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-fl][1,2,4]triazin-4-one hydrochloride trihydrate

22.5 kg of 2-[2-ethoxy-5-(4-ethylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one from Example 1b are dissolved in 5.1 kg of concentrated hydrochloric acid and acetone/water (12:1 v/v) in the presence of heat. The clear solution is filtered hot and crystallized by cooling and seeding. The crystallizate is isolated, washed and dried in vacuo at about 30° C. and about 300 mbar.
Yield: 25.4 kg
M.p. (DSC): 192° C.
HPLC: X-Terra C-18 phase, neutral phosphate buffer, acetonitrile, 242 nm, linear gradient of 20% acetonitrile ->75% acetonitrile (20 min.): 99 area % (Rt 16.3)
- http://www.pharmpro.com/News/Feeds/2010/06/pharmaceutical-companies-bayer-new-erectile-dysfunction-treatment-staxyn-approve/
- http://www.newswire.ca/en/story/832217/staxyn-new-innovation-in-erectile-dysfunction-helps-younger-men-rise-to-the-occasion
- A Aversa et al. “Effects of vardenafil administration on intravaginal ejaculatory latency time in men with lifelong premature ejaculation”. Retrieved 2010-12-14.
- Schools of Pharmacy (Glen L. Stimmel, Pharm.D., and Mary A. Gutierrez, Pharm.D.) and Medicine (Glen L. Stimmel, Pharm.D.), University of Southern California, Los Angeles, California. “Counseling Patients About Sexual Issues: Drug-Induced Priapism”. Medscape. Retrieved 2010-12-06.
- “FDA Announces Revisions to Labels for Cialis, Levitra and Viagra”. Food and Drug Administration. 2007-10-18. Retrieved 2009-08-06.
- Official Levitra website
- PubChem Information
-
2-1-2013Synthesis of quinoline derivatives: discovery of a potent and selective phosphodiesterase 5 inhibitor for the treatment of Alzheimer’s disease.European journal of medicinal chemistry
PATENTS
| US6362178 * | Oct 31, 1998 | Mar 26, 2002 | Bayer Aktiengesellschaft | 2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors |
| US20050203298 * | May 5, 2005 | Sep 15, 2005 | Bayer Healthcare Aktiengesellschaft | Process for the preparation of sulphonamide-substituted imidazotriazinones |
| US20060111354 * | Jul 3, 2003 | May 25, 2006 | Peter Serno | Medicaments containing vardenafil hydrochloride trihydrate |
| WO2004006894A1 * | Jul 3, 2003 | Jan 22, 2004 | Bayer Healthcare Ag | Medicaments containing vardenafil hydrochloride trihydrate |
|
11-4-2011
|
ROFLUMILAST FOR THE TREATMENT OF DIABETES MELLITUS
|
|
|
9-14-2011
|
Roflumilast for the Treatment of Diabetes Mellitus
|
|
|
8-5-2011
|
N-BUTYRAMIDE, THE PREPARATION METHOD AND USE THEREOF
|
|
|
3-4-2011
|
Fatty Acid Oxidation Inhibitors Treating Hyperglycemia and Related Disorders
|
|
|
1-14-2011
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
|
|
|
9-17-2010
|
SUBSTITUTED PDE5 INHIBITORS
|
|
|
7-16-2010
|
Combination treatment for diabetes mellitus
|
|
|
4-28-2010
|
2-Phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
|
|
4-14-2010
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
|
|
|
2-5-2010
|
Heterocyclic Compounds And Uses Thereof In The Treatment Of Sexual Disorders
|
|
12-25-2009
|
Therapeutic Compositions Comprising a Specific Endothelin Receptor Antagonist and a PDE5 Inhibitor
|
|
|
11-27-2009
|
Substituted PDE5 inhibitors
|
|
|
9-4-2009
|
Uses of 2-Phenyl-Substituted Imidazotriazinone Derivatives for Treating Pulmonary Hypertension
|
|
|
8-28-2009
|
Roflumilast for the Treatment of Pulmonary Hypertension
|
|
|
8-7-2009
|
Use of Phosphodiesterase Inhibitor as a Component of Implantable Medical Devices
|
|
|
6-26-2009
|
Method for healing a wound using a phosphodiesterase type five inhibitor
|
|
|
3-20-2009
|
Pde5 inhibitor compositions and methods for immunotherapy
|
|
|
3-6-2009
|
Pde5 inhibitor compositions and methods for treating cardiac indications
|
|
|
10-31-2008
|
Formulations with Controlled Release of Active Ingredient
|
|
|
8-15-2008
|
HIGHLY SELECTIVE and LONG-ACTING PDE5 MODULATORS
|
|
8-8-2008
|
Formulations With Controlled Release Of Active Ingredient
|
|
|
4-11-2008
|
Use of 2-alkoxyphenyl-substituted imidazotriazinones
|
|
|
1-2-2008
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors, for treatment of hypertension
|
|
|
12-28-2007
|
Novel Uses of 2-Phenyl-Substituted Imidazotriazinone Derivatives
|
|
|
10-3-2007
|
Use of 2-alkoxyphenyl-substituted imidazotriazinones
|
|
|
11-24-2006
|
Methods for synthesizing imidazotriazinones
|
|
|
10-18-2006
|
2-Phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
|
|
2-15-2006
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
|
|
|
8-17-2005
|
Use of 2-alkoxyphenol-substituted imidazotriazinones
|
|
|
5-11-2005
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
|
1-21-2005
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
|
|
|
8-18-2004
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
|
|
|
8-6-2004
|
Novel use of 2-phenyl-substituted imidazotriazinones
|
|
|
7-32-2003
|
Daily treatment for erectile dysfunction using a PDE5 inhibitor
|
|
|
5-21-2003
|
2-phenyl substituted imidatriazinones as phosphodiesterase inhibitors
|
|
|
3-27-2002
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
|
|
12-21-2001
|
Daily treatment for erectile dysfunction using a PDE5 inhibitor
|
|
|
5-21-1999
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
|


Thanks for sharing this blog its helps me a lot.
LikeLike