
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Laninamivir
(4S,5R,6R)-5-acetamido-4-carbamimidamido-6-[(1R,2R)-3-hydroxy-2-methoxypropyl]-5,6-dihydro-4H-pyran-2-carboxylic acid
| Formula | C13H22N4O7 |
|---|---|
| Mol. mass | 346.33638 g/mol |
cas 203120-17-6,
Laninamivir (L174000) prodrug; a novel long-acting neuraminidase inhibitor.
laninamivir octanoate
 472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
Daiichi Sankyo (Originator)
R-118958 is a potent, long-acting neuraminidase inhibitor (LANI) approved and launched in 2010 in Japan as an inhalable formulation for the treatment of influenza A and influenza B in adults and pediatric patients. In 2013 the product was approved in Japan for the prevention of influenza A and influenza B.
| 5-(Acetylamino)-4-[(aminoiminomethyl)amino]-2,6-anhydro-3,4,5-trideoxy-7-O-methyl-D-glycero-D-galacto-non-2-enonic Acid 9-Octanoate |
| (2R,3R,4S)-3-Acetamido-4-guanidino-2-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-3,4-dihydro-2H-pyran-6-carboxylic Acid |
| (4S,5R,6R)-5-Acetamido-4-guanidino-6-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-5,6-dihydro-4H-pyran-2-carboxylic Acid |
| CS 8958 |
ATLANTA, Dec. 20, 2013 (GLOBE NEWSWIRE) — Biota Pharmaceuticals, Inc.
(Nasdaq:BOTA) (“Biota” or the “Company”) today reported that Daiichi Sankyo Company, Limited (“Daiichi Sankyo”) has been granted regulatory approval in Japan to manufacture and market Inavir(R) Dry Powder Inhaler 20mg (generic name laninamivir octanoate) for the prevention of influenza A and B. Inavir(R) was successfully developed and launched by Daiichi Sankyo in Japan for treatment of influenza A and B viruses in October, 2010. Biota is developing laninamivir octanoate outside of Japan for the treatment of influenza, and is currently conducting a large, multi-national Phase 2 trial of laninamivir octanoate in adults infected with influenza. In 2003, the Company and Daiichi Sankyo entered into a collaboration and license agreement to develop long-acting neuraminidase inhibitors, including laninamivir octanoate, and in March 2009, the parties entered into a commercialization agreement, pursuant to which Daiichi Sankyo obtained exclusive marketing rights to laninamivir octanoate in Japan.http://www.pharmalive.com/biota-flu-drug-okd-in-japan

Laninamivir (CS-8958) is a neuraminidase inhibitor which is being researched for the treatment and prophylaxis of Influenzavirus A and Influenzavirus B.[1] It is currently in Phase III clinical trials. [2]
Laninamivir was approved for influenza treatment in Japan in 2010 and is currently marketed under the name “Inavir” by Daiichi Sankyo. Biota Pharmaceuticals [3] and Daiichi Sankyo co-own Laninamivir. On 1st April 2011, BARDA awarded up to an estimated U$231m to Biota Pharmaceuticals (Formerly Biota Holdings Ltd) wholly owned subsidiary, Biota Scientific Management Pty Ltd, for the advanced development of Laninamivir in the US. [4]
patent
|
8-13-2010
|
DRUG FOR TREATMENT OF INFLUENZA
|
The recent flu scares – first H5N1 bird flu and then H1N1 swine flu – transformed Roche’s neuraminidase inhibitor Tamiflu (oseltamivir) into a household name, along with GSK’s Relenza (zanamivir). Both of these require twice-daily dosing, and the orally available oseltamivir is the first choice, but resistance is starting to appear.
A new neuraminidase inhibitor, laninamivir, is being developed by Daiichi Sankyo.5 When administered as the octanoate prodrug form, it appears that a single dose might be sufficient to treat influenza, weekly doses could be preventative, and it is active against extremely pathogenic H5N1 strains.
Laninamivir octanoateIn a double blind, randomised, placebo-controlled Phase I study in 76 healthy male volunteers, subjects were given inhaled single doses of 5, 10, 20, 40, 80 or 120mg of the prodrug, or twice-daily doses of 20 or 40mg for three days.6 No adverse events were observed, and while the prodrug disappeared from the plasma with a half-life of about two hours, the laninamivir itself was much more slowly eliminated, with a half-life of the order of three days, suggesting the potential for giving long-lasting activity against influenza.
In another Phase I trial, a total of 20 healthy subjects with renal function ranging from normal to severely impaired were given single inhaled 20mg doses of the prodrug.7 The degree of renal impairment did not affect the maximum concentration or the time to achieve it, but the half-life increased as renal function reduced. This indicates that the rate-limiting step for elimination is drug release rate to plasma from tissues rather than renal excretion. It was well tolerated, but systemic exposure increased with increasing renal impairment.
It has also been compared with oseltamivir in patients with influenza. A total of 186 children under 10 who had had febrile influenza symptoms for no longer than 36 hours were randomised to receive 20 or 40mg of laninamivir octanoate as a single inhalation or 2mg/kg oseltamivir orally twice a day for five days.8
The new drug gave a significant reduction, of 61 hours for the 40mg group and 66 for the 20mg group, in median time to illness alleviation compared with oseltamivir in those with oseltamivir-resistant H1N1 influenza A. However, there was no significant difference in the time to alleviation of illness with H3N2 influenza A, or influenza B.
The most common side-effects were gastrointestinal problems.
In a Phase III trial, a total of 1,003 adult patients with febrile influenza symptoms for no more than 36 hours were given similar doses to those in the trial in children.9 Median time to alleviation of illness was 73h for 40mg, 86h for 20mg, and 74h for oseltamivir, and the proportion of patients shedding virus at day 3 was significantly lower in the 40mg group than for those given oseltamivir.
- Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nasu H, Kubo S (January 2009).“CS-8958, a prodrug of the new neuraminidase inhibitor R-125489, shows long-acting anti-influenza virus activity”. Antimicrobial Agents and Chemotherapy53 (1): 186–92.doi:10.1128/AAC.00333-08. PMC2612152. PMID18955520.
- Hayden F (January 2009). “Developing new antiviral agents for influenza treatment: what does the future hold?”. Clinical Infectious Diseases. 48. Suppl 1 (S1): S3–13.doi:10.1086/591851. PMID19067613.
- http://www.biotapharma.com
- http://www.biotapharma.com/?page=1021001&subpage=1021019
5. T. Honda et al. Synthesis and in vivo influenza virus-inhibitory effect of ester prodrug of 4-guanidino-7-O-methyl-Neu5Ac2en, Bioorg Med Chem Lett 2009, 19(11): 2938
6. H. Ishizuka et al. J. Clin. Pharmacol. 2010, 50, 1319
7. H. Ishizuka et al. J. Clin. Pharmacol. 2010, epub ahead of print, doi 10.1177/0091270010361914
8. N. Sugaya and Y. Ohashi, Antimicrob. Ag. Chemother. 2010, 54, 2575
9 A. Watanabe et al. Clin. Inf. Dis. 2010, 51, 1167
A new route toward 2-acetamido-4-O-methyl-2-deoxy-D-mannopyranose from a Ferrier derivative of tri-O-acetyl-D-glucal, which contributes to aldolase-catalyzed synthesis of laninamivir (CS-8958)
Tetrahedron 2013, 39(37): 7931

Infection of poultry with H5N1 avian influenza virus has been expanding since 2003 in wide areas including Asia, Europe and Africa. Humans infected with this virus have been found not only in Asia but also in Middle East and Africa. If a new type of H5N1 influenza virus has appeared and its infection has started, it is believed that the infection will rapidly expand to cause a worldwide spread (i.e., influenza pandemic) because most people do not possess immunity against that virus and influenza viruses spread via droplet infection and airborne infection. More than half of human patients infected with H5N1 influenza virus have died so far. Thus, the virus is highly pathogenic. It is known that three influenza pandemics, the Spanish Flu, the Asian Flu and the Hong Kong Flu, occurred in the 20th century. In the Spanish Flu which caused the largest number of patients, it is estimated that about 20-40 million people died in the world and about 0.5 million people in Japan.
According to a report from Japanese Ministry of Health, Labour and Welfare made in November, 2005, if a new type influenza virus has spread, the number of patients who will consult medical doctors in Japan as a result of infection with that virus is estimated about 18-25 million. Further, when the pathogenicity of that new type influenza virus is severe, the number of inpatients is estimated about 0.2 million while the number of dead is estimated about 0.64 million. Therefore, not only health hazard but also significant influences upon social activities are feared.
Thus, a new type influenza can cause a highly severe disease. Early development of effective therapeutics is demanded.
Although it is reported that zanamivir (in particular, zanamivir hydrate) and oseltamivir (in particular, oseltamivir phosphate or oseltamivir carboxylate) which are influenza therapeutics with neuraminidase inhibitory activity show an inhibitory activity against H5N1 influenza virus, compounds with more excellent activity are desired (Non-Patent Document 1 or 2). Further, H5N1 influenza virus strains against which oseltamivir does not show any inhibitory activity (i.e., oseltamivir resistant virus strains) have been reported. Compounds which possess an inhibitory activity against these oseltamivir resistant H5N1 influenza virus strains are desired (Non-Patent Document 1 or 2).
Compounds represented by formula (I) are known to be useful as influenza therapeutics with neuraminidase inhibitory activity (Patent Documents 1 to 3). However, it has not been reported that these compounds have an inhibitory activity against H5N1 influenza virus. Further, the structures of the compounds represented by formula (I) resemble the structure of zanamivir but are completely different from the structure of oseltamivir.
Non-Patent Document 1: Nature, 2005, vol. 437, p. 1108
Non-Patent Document 2: N. Engl. J. Med., 2005, vol. 353, (25):2667-72
Patent Document 1: U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946)
Patent Document 2: U.S. Pat. No. 6,451,766 (Japanese Patent Publication No. Hei 10-109981)
Patent Document 3: U.S. Pat. No. 6,844,363 (Japanese Patent Publication No. 2002-012590)

………………………
Preparation Example 1 5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid

(1) Diphenylmethyl 5-acetamido-4-(N,N-bis-t-butyloxycarbonyl)guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoate (3.46 g, 4.1 mmol) disclosed in Example 35 (i) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was dissolved in methylene chloride (27 ml) and trifluoroacetic acid (14 ml). The resultant solution was stirred at room temperature overnight. The reaction solution was concentrated to dryness under reduced pressure, followed by three cycles of azeotropic distillation to dryness with toluene (5 ml). The resultant oily material was dissolved in ethyl acetate (10 ml). The solution was poured into a saturated aqueous solution of sodium hydrogencarbonate (50 ml). The pH of the resultant solution was adjusted to 8.5 by addition of 20% aqueous solution of sodium carbonate. Then, the solution was stirred at room temperature for 3 hr and its pH was adjusted to 5.0 with hydrochloric acid (3 ml), followed by stirring at room temperature for another 1 hr. The solution was further stirred for 1 hr while ice-cooling. Subsequently, precipitating crystals were suction filtered and vacuum dried for 10 hr at an external temperature of 50° C. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (0.97 g; yield 51%).
Infrared Absorption Spectrum (KBr) ν max cm−1: 3412, 2929, 2856, 1676, 1401, 1320, 1285, 1205, 1137, 722.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.88 (1H, d, J=2.5 Hz), 4.45 (3H, m), 4.27 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.15 (1H, m), 3.47 (21-1, m), 3.42 (3H, s), 2.37 (2H, t, J=7.4 Hz), 2.10 (3H, s), 1.31 (2H, m), 1.20-1.40 (8H, m), 0.85-0.95 (3H, m).
13C Nuclear Magnetic Resonance Spectrum (100 MHz, CD3OD) δ ppm: 176.5, 173.7, 164.7, 158.9, 146.7, 108.7, 80.1, 78.0, 69.3, 66.8, 61.4, 52.4, 35.1, 32.8, 30.2, 30.1, 26.0, 23.7, 22.8, 14.4.
(2) The subject compound was also obtained by the method described below.
5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 35 (ii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was subjected to reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque), 100 g] and eluted with methanol/water (0/1-1/1-2/1). Fractions containing the compound of interest were vacuum concentrated. The precipitating crystals were suction filtered and vacuum dried. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (1.2 g; yield 49%). The property data of the resultant compound were consistent with those of the compound obtained in (1) above.
Preparation Example 2 5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid

5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 28 (viii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was purified in an ion-exchange resin column [Dowex-50X; (i) water and (ii) 5% aqueous ammonium solution] and further purified by reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque); water]. Fractions containing the compound of interest were vacuum concentrated. The resultant solid was washed with methanol, filtered and dried to thereby yield the subject compound (1.43 g) as a colorless solid.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.64 (1H, d, J=2.0 Hz), 4.43 (2H, m), 4.22 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.00 (1H, m), 3.85 (1H, dd, J=10.0 Hz, 2.4 Hz), 3.68 (1H, dd, J=10.0 Hz, 5.5 Hz), 3.58 (1H, m), 3.43 (3H, s).
………………………….


…………………………..
Process W is known as a method for manufacturing a compound represented by the formula (Ia), which is embraced in a compound represented by the formula (I) or a pharmacologically acceptable salt thereof, (hereinafter also referred to as “compound (Ia)”; the same shall be applied with respect to other (Patent Document 1). In Process W, n-Hep represents a 1-heptyl group.


Process X is known as a method for manufacturing compound (Ib), which is embraced in compound (I) or a pharmacologically acceptable salt thereof (Patent Document 2). Compound (IVk) is a synthetic intermediate in Process W. In Process X, n-Hep represents a 1-heptyl group.

Process Y is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 1). The procedures from compound (IVc) to compound (IVe) and from compound (IVf) to compound (IVh) in Process Y are the same as in Process W.


Process Z is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 2). In Process Z, the procedure from compound (IVf) to compound (IVh) is the same as in Process W, and the procedure from compound (IVh) to compound (IIIa) is the same as in Process Y.
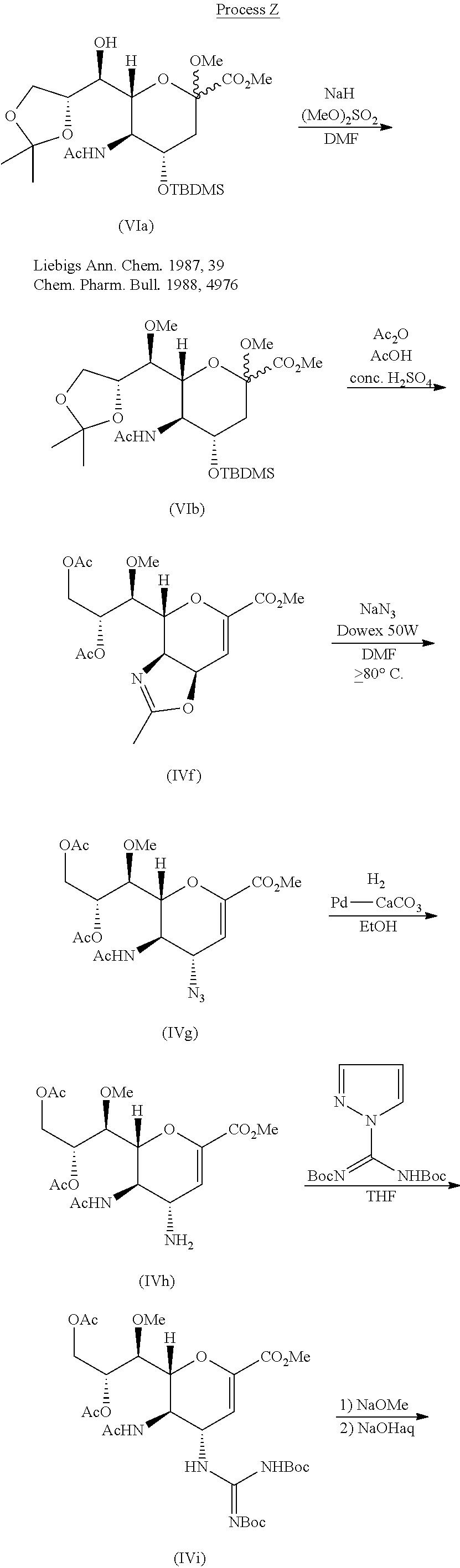

From the viewpoint of industrial production, the aforementioned Process W, Process Y, or Process Z could be improved in points such as the following:
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
update…………

:ACIE 10.1002/anie.201408138
Scheme zanamivir and Lanimiwei is based on N- acetylneuraminic acid as a starting material, the price is more expensive (ca.13000RMB / kg). Ma recently from Shanghai Institute of Organic Chemistry greatly researcher on ACIE published zanamivir, Lanimiwei and CS-8958 is more simple synthetic route. References: ACIE 10.1002 / anie.201408138

