
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


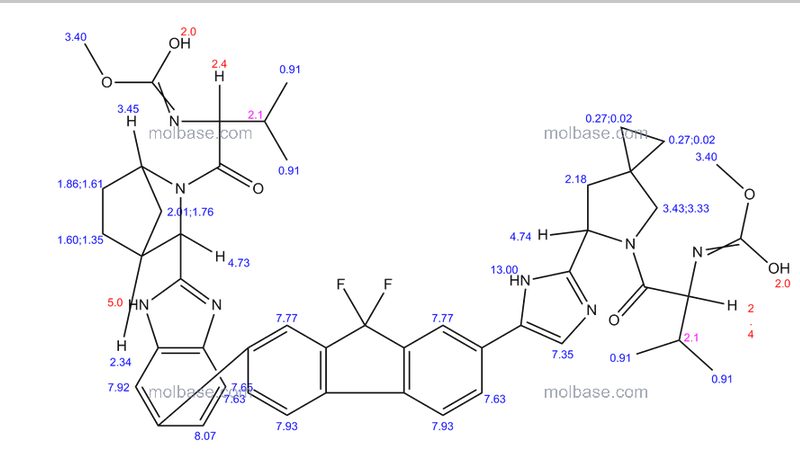
Carbamic acid, N-((1S)-1-(((6S)-6-(5-(9,9-difluoro-7-(2-((1R,3S,4S)-2-((2S)-2-((methoxycarbonyl)amino)-3-methyl-1-oxobutyl)-2-azabicyclo(2.2.1)hept-3-yl)-1H-benzimidazol-6-yl)-9H-fluoren-2-yl)-1H-imidazol-2-yl)-5-azaspiro(2.4)hept-5-yl)carbonyl)-2-me
Chemical Formula:C52H60F2N8O7
Molecular Weight:947.08


The structure of ledipasvir was unambiguously confirmed by 1 H, 13C and 19F NMR spectroscopy, UV spectroscopy, IR spectroscopy, high resolution mass spectrometry, elemental analysis and X-ray crystallography. LDV-AS is a white to tinted (off-white, tan, yellow, orange, or pink), slightly hygroscopic crystalline solid. It shows pH dependent solubility in aqueous media: it is slightly soluble in pH 2.3 buffer but practically insoluble in pH 4-7.5 buffers. It is freely soluble in ethanol and DMSO and slightly soluble in acetone. Ledipasvir is chiral and possesses 6 stereogenic centres and enantiomeric purity is controlled in starting material specifications. Three crystalline forms are known and ledipasvir acetone solvate is the designated commercial form. The first step for finished product manufacture involves the dissolution of ledipasvir in ethanol followed by spray-drying and thus precise control of morphology and particle size is not considered important. Ledipasvir is a chemical substance not previously authorised as a medicinal product in the European Union. Furthermore, it is not a salt, complex, derivative or isomer, (nor mixture of isomers), of a previously authorised substance. Whilst it contains some structural features in common with daclastavir, it is metabolically stable and the applicant presented data indicating that there are no common active metabolites. Therefore, the therapeutic moieties are not the same. Ledipasvir thus meets the definition of a New Active Substance according to the Notice to Applicants (NtA), Vol 2A, Chapter 1, Annex 3.
The mode of action of ledipasvir has not been directly established but indirect evidence is consistent with the compound targeting the NS5A molecule. In vitro resistance selection and cross-resistance studies, and the lack of HCV enzyme or kinase inhibition was taken to support the conclusion that ledipasvir targets NS5A as its mode of action. Ledipasvir has shown antiviral activity against HCV genotypes 1a and 1b with mean EC50 values of 0.031 and 0.004 nM, respectively. Antiviral activity determined as EC50 against genotypes 2 to 6 ranged from 0.15 to 530 nM. Ledipasvir showed no relevant antiviral activity at the highest concentration tested, or the highest concentration without cytotoxicity, against other virus such as bovine viral diarrhea virus (BVDV), RSV, HBV, HIV-1, HRV, influenza A and B, and a panel of flaviviruses (including West Nile virus, yellow fever virus, dengue virus, and banzai virus). Cytotoxicity of ledipasvir was characterised by CC50 of 4029 to >50000 nM using different cell lines (1b-Rluc-2, Huh-luc, 1a-HRlucp, Hep G2, SL3, Huh7, Hep-2, AD-38 and MT4 cells). Ledipasvir at 10 µM showed significant binding to 3 ion channels and 1 receptor in a radioligand binding assay screen against a panel of 68 mammalian ion channels and receptors. The IC50s of ledipasvir were 0.210 and 3.47 μM against sodium channel site 2 and calcium channel L-type (dihydropyridine), respectively. A 50% inhibition of androgen receptor was noted at 10 μM. Ledipasvir activity against 442 kinases was assessed using a quantitative polymerase chain reaction (qPCR)-based competition assay. Results showed weak competition for binding of 2 kinases, Bruton’s tyrosine kinase (BTK) and homeodomain-interacting protein kinase 1 (HIPK1) at 0.1 and 1 μM, respectively. Taking into account the high protein binding, >99.5%, of ledipasvir the large margin between unbound maximum clinical plasma levels (0.8 nM) and potential ion channel/receptor inhibition indicates limited clinical relevance.
Ledipasvir (formerly GS-5885) is a drug for the treatment of hepatitis C that was developed by Gilead Sciences.[1] After completingPhase III clinical trials, on February 10, 2014 Gilead filed for U.S. approval of a ledipasvir/sofosbuvirfixed-dose combination tablet for genotype 1 hepatitis C.[2][3] The ledipasvir/sofosbuvir combination is a direct-acting antiviral agent that interferes with HCV replication and can be used to treat patients with genotypes 1a or 1b without PEG-interferon or ribavirin.
Ledipasvir is an inhibitor of the hepatitis C virusNS5A protein.
Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of the nucleotide analog inhibitor sofosbuvir, ledipasvir, and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[4][5] The sofosbuvir/ledipasvir coformulation is being tested with and without ribavirin. In February 2014 Gilead has filed for United StatesFood and Drug Administration (FDA) approval of ledipasvir/sofosbuvir oral treatment, without interferon and ribavirin.[6]
On October 10, 2014 the FDA approved the combination product ledipasvir 90 mg/sofosbuvir 400 mg called Harvoni.[7]


https://www.google.co.in/patents/WO2013184698A1
CLIP
SYN


https://www.google.co.in/patents/WO2013184702A1









PATENT
https://www.google.co.in/patents/US8088368
Example ED Preparation of Intermediate 5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-methyl ester

4-Methylene-pyrrolidine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester
Example ED′


2,7-Dibromo-9,9-difluoro-9H-fluorene
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-[2-(7-bromo-9,9-difluoro-9H-fluoren-2-yl)-2-oxo-ethyl]ester
6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carboxylic acid benzyl ester
(1-{6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester
(1-{3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
https://www.google.co.in/patents/US8088368

2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester: A mixture of 2-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carboxylic acid tert-butyl ester (324 mg, 0.627 mmol), 3-[6-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (1.1 eq., 304 mg), [1,1′ bis(diphenylphosphino)ferrocene]dichloropalladium(II)(3%, 15 mg), tetrakis(triphenylphosphine)palladium (3%, 22 mg) and potassium carbonate (3.3 eq., 285 mg) in 10 mL DME and 3 mL water was heated to 90° C. under Argon for 3 hours. The reaction mixture was cooled and diluted with ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was dried (MgSO4), concentrated and purified by flash column chromatography (silica gel, 20 to 100% ethyl acetate/hexane) to give 2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester (361 mg, yield 77%). LCMS-ESI−: calc’d for C43H46F2N6O4: 748.86. Found: 749.2 (M+H+).
PATENTS
SEE
WO 2010132601
WO 2013040492
WO 2013059630
WO 2013059638
CLIP
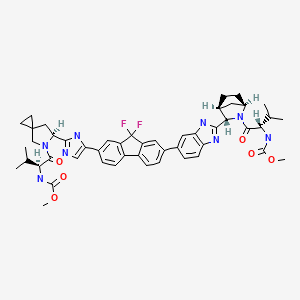
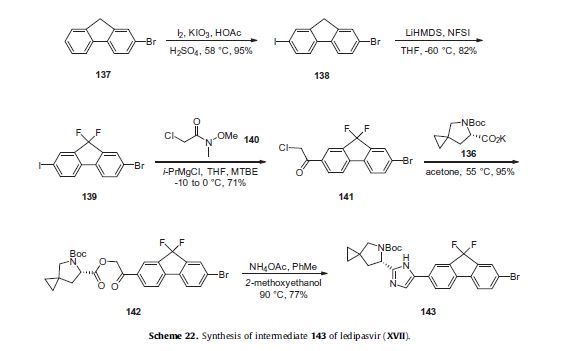
Ledipasvir (Harvoni) Ledipasvir is a potent NS5A inhibitor that is approved for use in combination with sofosbuvir, a nucleotide inhibitor of viral polymerase, for the treatment of chronic hepatitis C virus genotype 1 infection.14,130,131 This combination was discovered and developed at Gilead Sciences and is marketed as the fixed combination with brand name of Harvoni. The synthesis of ledipasvir has been reported in the literature132 and the routes shown in Schemes 22–24 below represent the most efficient and largest scale sequence reported in the patent literature.133,134 The synthesis of the spirocyclopropane proline intermediate 136 is described in Scheme 21. Bis-iodination of cyclopropane-1,1-diyldimethanol (131) in the presence of triphenylphosphine gave diiodide 132 in 70% yield. N-Boc-glycine ethyl ester (133) was then treated with sodium hydride followed by diiodide 132 to give the protected proline analog 134 in 61% yield. Saponification of the ester followed by a classical resolution with (1S,2R)-amino-indanol gave enantomerically pure salt 135. Liberation of the free acid with 1 M HCl followed by treatment with potassium tert-butoxide provided enantiopure potassium salt 136 in high yield. The synthesis of the difluoro-fluorene Suzuki coupling intermediate 143 is described in Scheme 22. Iodination of 2-bromofluorene (137) produced aryl iodide 138 in 95% yield, which was then treated with lithium hexamethyldisilazide and N-fluorobenzenesulfonimide (NFSI) to give the difluoro intermediate 139 in 82% yield. Formation of the Grignard reagent of 139 through reaction with isopropylmagnesium chloride followed by condensation with Weinreb amide 140 gave chloroketone 141 in 71% yield. The potassium salt of the cyclopropyl proline intermediate 136 (described in Scheme 21) was coupled with 141 to give keto ester 142 in high yield. Heating 142 with ammonium acetate resulted in formation of the imidazole ring in intermediate 143 in 77% yield. The completion of the synthesis of ledipasvir is described in Scheme 23. Commercially available (1R,3S,4S)-N-Boc-2-azabicyclo [2.2.1]heptane-3-carboxylic acid (144) was coupled to 4-bromo- 1,2-benzenediamine (145) using EDC/HOBt to give a mixture ofamides 146a/146b in 72% yield. Heating mixture 146a/146b with acetic acid affected cyclization to benzimidazole 147 in 94% yield. Palladium mediated coupling of bromide 147 to bis(pinacolato)diboron gave intermediate148 which was then coupled in the same reaction vessel to bromide 143 generated in Scheme 22. This was followed by formation of the oxalate salt to give the protected central core of ledipasvir (149) in good overall yield. Removal of the amine protecting groups gave diamine 150 which was coupled to two equivalents of Moc-valine (151) via EDC/HOBt to give ledipasvir XVII in 73% yield. 19. Lobeglitazone sulfate



130. Gentile, I.; Buonomo, A. R.; Borgia, F.; Castaldo, G.; Borgia, G. Expert Opin.Invest. Drugs 2014, 23, 561.
131. Smith, M. A.; Chan, J.; Mohammad, R. A. Ann. Pharmacother. 2015, 49, 343.132. Link, J. O.; Taylor, J. G.; Xu, L.; Mitchell, M.; Guo, H.; Liu, H.; Kato, D.;Kirschberg, T.; Sun, J.; Squires, N.; Parrish, J.; Keller, T.; Yang, Z. Y.; Yang, C.;Matles, M.; Wang, Y.; Wang, K.; Cheng, G.; Tian, Y.; Mogalian, E.; Mondou, E.;Cornpropst, M.; Perry, J.; Desai, M. C. J. Med. Chem. 2014, 57, 2033.
133. Guo, H.; Kato, D.; Kirschberg, T. A.; Liu, H.; Link, J. O.; Mitchell, M. L.; Parrish, J.P.; Squires, N.; Sun, J.; Taylor, J.; Bacon, E. M.; Canales, E.; Cho, A.; Cottell, J. J.;Desai, M. C.; Halcomb, R. L.; Krygowski, E. S.; Lazerwith, S. E.; Liu, Q.;Mackman, R.; Pyun, H. J.; Saugier, J. H.; Trenkle, J. D.; Tse, W. C.; Vivian, R. W.;Schroeder, S. D.; Watkins, W. J.; Xu, L.; Yang, Z. Y.; Kellar, T.; Sheng, X.; Clarke,M. O. N. H.; Chou, C. H.; Graupe, M.; Jin, H.; McFadden, R.; Mish, M. R.;Metobo, S. E.; Phillips, B. W.; Venkataramani, C. WO Patent 2010132601A1,2010.
134. Scott, R. W.; Vitale, J. P.; Matthews, K. S.; Teresk, M. G.; Formella, A.; Evans, J.W. US Patent 2013324740A1, 2013.
135. Jin, S. M.; Park, C. Y.; Cho, Y. M.; Ku, B. J.; Ahn, C. W.; Cha, B.-S.; Min, K. W.;Sung, Y. A.; Baik, S. H.; Lee, K. W.; Yoon, K.-H.; Lee, M.-K.; Park, S. W. Diab.Obes. Metab. 2015, 17, 599.
136. Lee, H. W.; Ahn, J. B.; Kang, S. K.; Ahn, S. K.; Ha, D.-C. Org. Process Res. Dev.2007, 11, 190.
137. Lee, H. W.; Kim, B. Y.; Ahn, J. B.; Kang, S. K.; Lee, J. H.; Shin, J. S.; Ahn, S. K.; Lee,S. J.; Yoon, S. S. Eur. J. Med. Chem. 2005,
PAPER
The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection
http://pubs.acs.org/doi/abs/10.1021/jm401499g?prevSearch=LEDIPASVIR&searchHistoryKey=
http://pubs.acs.org/doi/pdf/10.1021/jm401499g
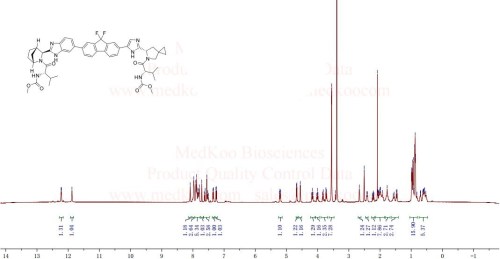
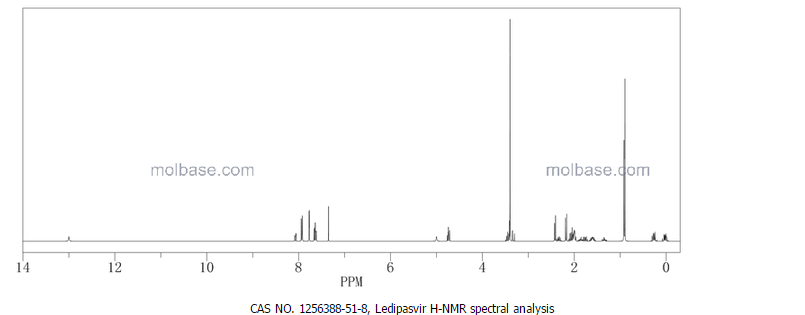
1H-NMR: 300 MHz, (dmso-d6) δ: 8.20-7.99 (m, 8H), 7.73 (s, 2H), 7.37 – 7.27
(m, 2H), 5.25 (dd, J = 7.2 Hz, 1H), 4.78 (s, 1H) 4.54 (s, 1H), 4.16 (m, 1H), 4.02 (m,
1H), 3.87 (m,1H), 3.74 (m, 1H), 3.55 (s, 3H), 3.53 (s, 3H), 2.75 (m, 1H), 2.25 (m,
2H), 2.09 – 2.04 (m, 2H), 1.88 – 1.79 (m, 2H), 1.54 (m, 1H), 0.94 – 0.77 (m, 15H)
0.63 (m, 4H) ppm.
19F-NMR: 282 MHz, (dmso-d6) δ: -109.1 ppm [-74.8 ppm TFA].
HRMS (ESI-TOF) m/z: [M + H]+
calc’d for C49H55F2N8O6: 889.4207; Found: 889.4214.
methyl [(2S)-1-{(6S)-6-[5-(9,9-difluoro-7-{2-[(1R,3S,4S)-2-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-azabicyclo[2.2.1]hept-3-yl]-1H-benzimidazol-6-yl}-9H-fluoren-2-yl)-1H-imidazol-2-
yl]-5-azaspiro[2.4]hept-5-yl}-3-methyl-1-oxobutan-2-yl]carbamate (39 NOS IS LEDISPAVIR
PATENT
Synthesis of 25
25
B. Synthesis of 26 and 27
25 26 27
[0186] To a flask was charged 25 (20.00 g, 0.083 mol), 4-bromo-l,2-benzenediamine (16.74 g, 0.089 mol, 1.08 equiv.), hydroxybenzotriazole (HOBt) (13.96 g, 0.091 mol, 1.1 equiv.), and l-ethyl-3-(3-dimethylaminopropyl) carbodiimide HC1 (EDC.HC1) (17.48 g, 0.091 mol, 1.1 equiv.). The flask was cooled in an ice bath, and was charged with N,N- dimethylacetamide (DMAc, 80 mL). The reaction was allowed to cool to ca. 10 °C with stirring. N-methylmorpholine (NMM) (27.34 mL, 0.249 mol, 3 equiv.) was added over 5 minutes keeping the internal temperature below 20 °C. The reaction was stirred at rt for 20 h. Upon reaction completion, the reaction mixture was added to MTBE (200 mL) and water (600 mL) in a separatory funnel and was gently shaken. The layers were allowed to separate, and the aqueous layer was removed. The aqueous layer was extracted twice with MTBE (50 mL), and the organic extracts were combined. The combined organic extracts were then extracted with water (500 mL), forming a mixture that did not separate well. The mixture was filtered over an appropriate solid support and the layers were separated. The organic phase was concentrated under vacuum, and the resulting residue was dissolved in diisopropyl ether (100 mL). The solution was cooled to ca. 5 °C with stirring. Acetic acid (5.22 mL, 0.091 mol, 1.1 equiv.) was added slowly keeping the internal temperature below 10 °C, and the resulting suspension was stirred 2 h at 5 °C. The thick suspension was then filtered, and the solid was rinsed with diisopropyl ether (100 mL), followed by heptane (100 mL). The cake was dried under vacuum to give the product as a light-beige solid as a mixture of regioisomers 26 and 27 (28.19 g, 72%, >99% AN). 1H NMR (400 MHz, DMSO) mixture of 26 & 27 (data is for the two rotamers of the major regioisomer): δ 9.25 (s, 0.5H), 9.13 (s, 0.5H), 7.08 (d, J= 8.3 Hz, 0.5H); 7.06 (d, J= 8.2 Hz, 0.5H), 6.92 (d, J= 2.2 Hz, 0.5H), 6.89 (d, J= 2.1 Hz, 0.5H), 6.71 (dd, J= 8.4, 2.2, 0.5H), 6.66 (dd, J= 8.4, 2.2, 0.5H), 5.10 (br s, 1H), 5.05 (br s, 1H), 4.15 (br s, 0.5H), 4.10 (br s, 0.5H), 3.76 (s, 1H), 2.64 (br s, 1H), 1.96- 1.88 (m, 1H), 1.77-1.67 (m, 1H), 1.67-1.19 (m, 4H), 1.41 (s, 4.5H), 1.33 (s, 4.5H). MS-ESI+: [M + H]+ calcd for Ci8H25Br03N3, 410.1, 412.1; found, 410.0, 412.0
[0187] The disclosure provides in some embodiments the use of other coupling reagents. These include but are not limited to N,N”-dicyclohexylcarbodiimide (DCC), NJV- diisopropylcarbodiimide (DIC), 6-chloro-2,4-dimethoxy-s-triazine (CDMT), O- benzotriazole-N^N^A^-tetramethyl-uronium-hexafluoro-phosphate (HBTU), and 2-(7-Aza- 1H- benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU).
[0188] The amine base also can be varied or omitted completely. For instance the amine is selected from tertiary amines (R3N), 2,6-lutidine, pyridine, dicyclohexylmethylamme, and N- methylmorpholine (NMM).
[0189] Suitable solvent alternatives are selected from DMF, NMP, dialkyl and cyclic ethers R20, THF, 2-MeTHF, DCM, DCE, toluene, EtOAc, IP Ac, acetone, MIBK, and MEK.
[0190] Suitable temperatures for the reaction range from about -20 °C to 80 °C.
NMR PREDICT

1H/13C NMR PREDICT


COSY
Links
1)Link, John O.et al; The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection; Journal of Medicinal Chemistry (2013), Ahead of Print.DOI:10.1021/jm401499g
2)Ray, Adrian S. et al; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., WO2013040492
3) Delaney, William E. et al ; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., wo2012087596
4) Delaney, William E., IV et al; Preparation of quinoline derivatives and analogs for use in the treatment of hepatitis C virus infection in combination with ribavirin; PCT Int. Appl., wo2011156757
5) Guo, Hongyan et al; Preparation of biaryls, arylheteroaryls, heteroaryls, biarylacetylenes and related compounds end-capped with amino acid or peptide derivatives as antiviral agents; PCT Int. Appl., WO2010132601
6)Phase III (Sofosbuvir + Ledipasvir) ION-1 study: (Clinical Trial number: NCT01701401):
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection
7) Phase III (Sofosbuvir + Ledipasvir) ION-2 study: (Clinical Trial number: NCT01768286)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/GS-5885 Fixed-Dose Combination ± Ribavirin for 12 and 24 Weeks in Treatment-Experienced Subjects With Chronic Genotype 1 HCV Infection
8) Phase III (Sofosbuvir + Ledipasvir) ION-3 study: (Clinical trial number: NCT01851330)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection

References
- “Ledipasvir” (PDF). United States Adopted Name.
- “Ledipasvir-submitted-to-FDA”.
- “GS-5885”. Gilead Sciences.
- ELECTRON: 100% Suppression of Viral Load through 4 Weeks’ Post-treatment for Sofosbuvir + Ledipasvir (GS-5885) + Ribavirin for 12 Weeks in Treatment-naïve and -experienced Hepatitis C Virus GT 1 Patients. Gane, Edward et al. 20th Conference on Retroviruses and Opportunistic Infections. March 3–6, 2013. Abstract 41LB.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, Liz. HIVandHepatitis.com. 4 March 2013.
- “Gilead Files for U.S. Approval of Ledipasvir/Sofosbuvir Fixed-Dose Combination Tablet for Genotype 1 Hepatitis C”. Gilead Sciences. 10 February 2014.
- “U.S. Food and Drug Administration Approves Gilead’s Harvoni® (Ledipasvir/Sofosbuvir), the First Once-Daily Single Tablet Regimen for the Treatment of Genotype 1 Chronic Hepatitis C”. 10 October 2014. Retrieved 10 October 2014.
- Afdhal, N; Zeuzem, S; Kwo, P; Chojkier, M; Gitlin, N; Puoti, M; Romero-Gomez, M; Zarski, J. P.; Agarwal, K; Buggisch, P; Foster, G. R.; Bräu, N; Buti, M; Jacobson, I. M.; Subramanian, G. M.; Ding, X; Mo, H; Yang, J. C.; Pang, P. S.; Symonds, W. T.; McHutchison, J. G.; Muir, A. J.; Mangia, A; Marcellin, P; Ion-1, Investigators (2014). “Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection”. New England Journal of Medicine 370 (20): 1889–98. doi:10.1056/NEJMoa1402454. PMID 24725239.
- http://www.gilead.com/~/media/Files/pdfs/medicines/liver-disease/harvoni/harvoni_pi.pdf
- http://www.hepatitisc.uw.edu/page/treatment/drugs/ledipasvir-sofosbuvir
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Methyl N-[(2S)-1-[(6S)-6-[5-[9,9-Difluoro-7-[2-[(1S,2S,4R)-3-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]-3-azabicyclo[2.2.1]heptan-2-yl]-3H-benzimidazol-5-yl]fluoren-2-yl]-1H-imidazol-2-yl]-5-azaspiro[2.4]heptan-5-yl]-3-methyl-1-oxobutan-2-yl]carbamate
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 76% |
| Protein binding | >99% |
| Metabolism | No cytochromemetabolism |
| Biological half-life | 47 hrs |
| Identifiers | |
| CAS Registry Number | 1256388-51-8 |
| ATC code | None |
| ChemSpider | 29271894 |
| ChEBI | CHEBI:85089 |
| Chemical data | |
| Formula | C49H54F2N8O6 |
| Molecular mass | 889.00 g/mol |
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT
////////////////GS-5885, LEDIPASVIR
PATENT 1
Patent application WO2010132601A1 (primary patent) discloses the base compound of ledipasvir. The application claims a general structural formula (Markush) of new amide compounds useful for treating disorders associated with HCV. This patent, if granted, serves as a blocking patent preventing competitors from making the product. The claims are very broad, using a Markush structure of antiviral agents. As per the WIPO ISR, claims 1-19 are novel and inventive. However, according to the ISR, all remaining claims (claims 20 to 173), covering a large number of compounds, lack both novelty and inventive step, due to lack of support from the patent specification and in the light of prior art. Prosecution at the USPTO Three patents have been granted in the United States: US8088368B2, claiming the base compound by general structural formula; US8273341B2 (a division of US8088368B2), claiming a method of inhibiting HCV; and US8575118B2 (a continuation of US8273341B2 and a division of US8088368B2), claiming specific amide compounds not covered in the other two related patents. The examination report of US8088368B2 reveals that the application was allowed after the applicant cancelled and amended claims on Markush substuents. The examination report of US8273341B2 reveals that the application was allowed after the applicant amended a claim ‘A method of treating HCV’ to ‘A method of inhibiting HCV´. The examination report of US8575118B2 reveals that the application was allowed after the applicant cancelled claims already covered by the related patents, and limited claims to four specific compounds. Patent 1 has been filed in various jurisdictions: The patent has been granted by the ARIPO, in South Africa, and the United States. The patent (or a related patent) is pending in Argentina, Australia, Canada, China, as well as China, Hong Kong SAR, the EAPO, the EPO, Israel, India, Japan, New Zealand, Singapore, and Ukraine. Legal status is not available for Colombia, Ecuador, Mexico, Peru, Uruguay, and Viet Nam. 13 Litigation / Opposition on Patent 1 In December 2013, Gilead Sciences filed apatent infringement lawsuit against Abbott Laboratories and AbbVie Inc., in the United States District Court for the District of Delaware (case Number: 1:13cv02034). The case involves Gilead Sciences patents US8088368B2, US8273341B2, and US8575118B2.
PATENT 2 Patent application WO2013184698A1 is a product and process patent, claiming new crystalline solvate forms of ledipasvir useful for treating a subject suffering from HCV infection. The application also claims processes of manufacture of such amorphous and crystalline forms with specific X-ray diffraction peaks, and compositions and combinations comprising them. The application has just recently been published and no written opinion on patentability is available at this stage. As per the available information (details available in the Annex): The patent is pending at the EPO and the United States. There are no litigation or opposition procedures reported.
PATENT 3 Patent application WO2013184702A1 is a process patent, claiming processes for the preparation of ledipasvir. The disclosure also provides compounds that are synthetic intermediates to compounds of ledipasvir. The claims are moderately narrow covering crystalline and amorphous forms of ledipasvir with specific X-ray diffraction peaks. The application has just recently been published and no written opinion on patentability is available at this stage. As per the available information (details available in the Annex): The patent is pending at the EPO and the United States. There are no litigation or opposition procedures reported.
PATENT 4 Patent application WO2012087596A1 is a formulation patent, claiming various formulations comprising a combination of ledipasvir with GS-9256, or tegobuvir or with other compounds. The application also claims methods of treatment with the said combinations for reducing viral load in a person infected with HCV. 14 As per the WIPO ISR, the application is novel but not inventive in comparison to the closest prior art retrieved during the search. The combinations claimed in the instant application are not disclosed in the prior art, thus the combinations are novel. However, the prior art discloses various combinations, therefore, the problem to be solved through the invention should be new combinations with fewer side effects. Further, no experimental data of synergism has been provided to support double, triple, or quadruple combinations. Thus, according to the ISR, the instant invention cannot be regarded as inventive. As per the available information (details available in the Annex): The patent has been granted in Argentina. The patent is pending in Australia, Canada, the EPO, and the United States. Legal status is not available for Japan and Uruguay. There are no litigation or opposition procedures reported.
PATENT 5 Patent application WO2013040492A2 is a formulation and method of use patent, claiming compositions and a method of using the combination for the treatment of HCV. Drug combinations are used, and the compositions include sofosbuvir, PSI-7851 and ledipasvir. Since the application claims a group of compounds of Markush structure, it gives the claims a broad scope. As per the WIPO ISR the application is novel but lacks the inventive step in light of prior art. The invention lacks an inventive step as it would be obvious to a person skilled in the art to combine the diastereoisomer of the present invention, disclosed in the prior art, with other antiviral agents to provide an alternative HCV therapy. As per the available information (details available in the Annex): The patent is pending in Australia, Canada, the EPO, and the United States. There are no litigation or opposition procedures reported. This patent is listed in the sofosbuvir report as Patent No. 7
http://www.who.int/phi/implementation/ip_trade/ledipasvir_report_2014-09-02.pdf
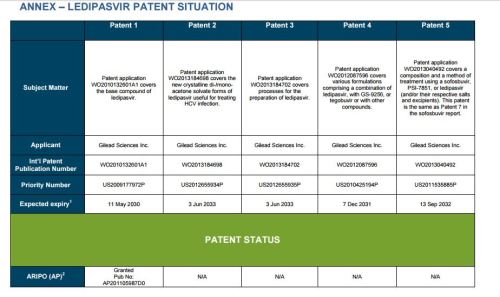
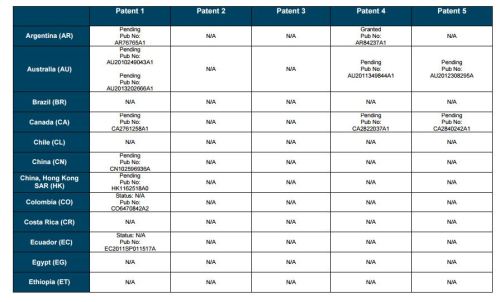
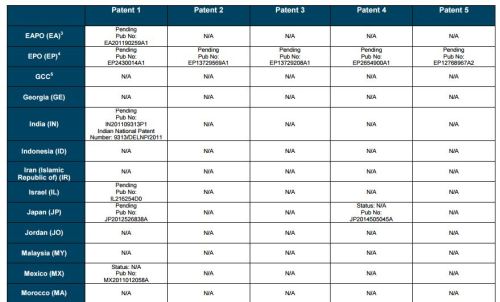
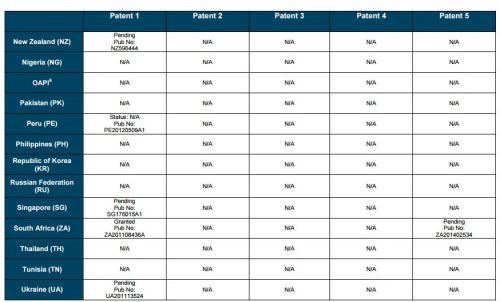

SUMMARY The search revealed patents filed with respect to ledipasvir by the Sponsor as well as a nonSponsor. The ledipasvir Sponsor patent collection comprises 5 different patents (patent families) with 47 family members published in 23 jurisdictions. The majority of these patent applications are still pending in the respective patent offices (see Patents 1 to 5 in the Annex). Patent 1 is the primary patent, claiming the base compound through a Markush claim, along with various substituents. Where granted, this patent can prevent competitors from making ledipasvir. Patents 2 and 3 claim processes to make ledipasvir and thus if granted will require competitors to design around these patents and use other production processes. The chemical product itself is not protected. Patents 4 and 5 claim combinations of different HCV drugs with ledipasvir, and their formulations. There is competition in the field by AbbVie, Inc., which filed formulation patents. Note: The search also revealed two patents that are relevant for all seven reports. Patent applications WO2013059630A1 and WO2013059638A1 inter alia claim the use of combinations of unnamed direct-acting antiviral agents for treating HCV, where the treatment does not include administration of interferon or ribavirin, and the treatment lasts between 8-12 weeks. The description and the dataset for these two patents can be found in the Working Paper on ombitasvir (Patents No 3 and 4). These patents are in litigation. Detailed information can be found in the Working Paper on sofosbuvir under Patent No 2.
World Drug Tracker: LEDIPASVIR
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
LEDIPASVIR
Biological Activity of Ledipasvir
Ledipasvir(GS5885) is an inhibitor of the hepatitis C virus NS5A protein. Ledipasvir is an experimental drug for the treatment of hepatitis C.
IC50 Value: 141 nM (EC50, JFH1/3a-NS5A hybrid replicon) [1]
Target: HCV NS5A
in vitro: Against JFH1/3a-NS5A, DCV was more potent (EC(50) = 0.52 nM) than GS-5885 (EC(50) = 141 nM). DCV sensitivity was increased against JFH1/3a-NS5A-M28V (EC50 = 0.006 nM), A30V (EC(50) = 0.012 nM), and E92A (EC(50) = 0.004 nM) while the NS5A-A30K and -Y93H variants exhibited reduced sensitivity to DCV (EC50 values of 23 nM and 1120 nM, respectively) and to GS-5885 (EC50 values of 1770 nM and 4300 nM, respectively) [1].
in vivo: GS-5885 was well tolerated and resulted in median maximal reductions in HCV RNA ranging from 2.3 log(10) IU/ml (1 mg QD) to 3.3 log(10) IU/ml (10 mg QD in genotype 1b and 30 mg QD). E(max) modeling indicated GS-5885 30 mg was associated with>95% of maximal antiviral response to HCV genotype 1a. HCV RNA reductions were generally more sustained among patients with genotype 1b vs. 1a. Three of 60 patients had a reduced response and harbored NS5A-resistant virus at baseline. NS5A sequencing identified residues 30 and 31 in genotype 1a, and 93 in genotype 1b as the predominant sites of mutation following GS-5885 dosing. Plasma pharmacokinetics was consistent with QD dosing [2].
Toxicity:
Clinical trial: Combination Therapy for Chronic Hepatitis C Infection. Phase 2
Clinical Information of Ledipasvir
| Product Name | Sponsor Only | Condition | Start Date | End Date | Phase | Last Change Date |
|---|---|---|---|---|---|---|
| Ledipasvir | Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-12 | 31-DEC-14 | Phase 3 | 12-SEP-13 |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-13 | 31-JAN-15 | Phase 3b | 11-NOV-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-MAY-13 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-DEC-10 | 30-APR-14 | Phase 2b | 28-AUG-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-JUN-13 | Phase 2 | 22-AUG-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-APR-13 | Phase 2b | 03-OCT-12 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-13 | 31-JAN-15 | Phase 3 | 11-NOV-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-MAY-13 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-OCT-12 | 31-DEC-14 | Phase 3 | 12-SEP-13 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-APR-13 | Phase 2 | 03-OCT-12 | |
| Gilead Sciences Inc | Hepatitis C virus infection | 31-JUL-11 | 30-JUN-13 | Phase 2b | 22-AUG-13 |
update………..
![]()

WO 2016145990, Ledipasvir, New patent, SHANGHAI FOREFRONT PHARMACEUTICAL CO., LTD
(WO2016145990) METHOD OF PREPARATION FOR LEDIPASVIR AND DERIVATIVE THEREOF, AND INTERMEDIATE COMPOUND FOR PREPARATION OF LEDIPASVIR
SHANGHAI FOREFRONT PHARMCEUTICAL CO., LTD [CN/CN]; Room 1306, No.781 Cailun Road China (Shanghai) Pilot Free Trade Zone, Pudong New Area Shanghai 201203 (CN)
HUANG, Chengjun; (CN).
FU, Gang; (CN).
FU, Shaojun; (CN).
WEI, Zhewen; (CN).
LI, Wei; (CN).
ZHANG, Xixuan; (CN)
chinese machine translation please bear………..
SMILES COC(=O)N[C@@H](C(C)C)C(=O)N1CC2(CC2)C[C@H]1c3ncc([nH]3)c4ccc5c6ccc(cc6C(F)(F)c5c4)c7ccc8nc([nH]c8c7)[C@@H]9[C@H]%10CC[C@H](C%10)N9C(=O)[C@@H](NC(=O)OC)C(C)C


Thank you for the good writeup. It in fact was a amusement account it.
Look advanced to more added agreeable from you!
However, how can we communicate?
Hello, i think that i saw you visited my website
thus i came to “return the favor”.I\’m attempting to find things
to enhance my web site!I suppose its ok to use some of your ideas!\\
LikeLike