
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Lumacaftor
3-[6-[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropylcarboxamido]-3-methylpyridin-2-yl]benzoic acid
3-{6-{[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino}-3-methylpyridin-2-yl}benzoic acid
VRT-826809
VX-809
US patents: US8124781, US8461342
Indication:Cystic fibrosis
Developmental status:Phase III (US, UK, EU)
Developer:Vertex
| Vertex Pharmaceuticals |
| Company | Vertex Pharmaceuticals Inc. |
| Description | Small molecule cystic fibrosis transmembrane conductance regulator (CFTR) corrector |
| Molecular Target | Cystic fibrosis transmembrane conductance regulator (CFTR) |
| Mechanism of Action | CFTR stabilizer |
| Latest Stage of Development | Phase III |
| Indication | Cystic fibrosis (CF) |
| cas | 936727-05-8 |
http://www.ama-assn.org/resources/doc/usan/lumacaftor.pdf for all data
see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.html
Lumacaftor (USAN, codenamed VX-809) is an experimental drug for the treatment of cystic fibrosis being developed by Vertex Pharmaceuticals. The drug is designed to be effective in patients that have the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), the defective protein that causes the disease. F508del, meaning that the amino acid phenylalanine in position 508 is missing, is found in about 60% of cystic fibrosis patients in Europe,[1] and in about 90% of persons with some mutation in the CFTR gene.
A corrector molecule, one of two new classes of ion channel modulators. The corrector modulators enhance the number of channels of the CFTR protein at the cell surface. in combination with ivacaftor in homozygous F508del pts
Results from a Phase II clinical trial indicate that patients with the most common form of genetic mutation causing cystic fibrosis—homozygous F508del—had a mean increase of 7.4% in lung function (FEV1) on a combination of lumacaftor and ivacaftor.[2]
VX-809 is an investigational corrector compound in a phase II clinical trial for oral the treatment of cystic fibrosis. The trial will evaluate single and multiple doses of VX-809 in healthy volunteers. This compound has resulted from a collaboration with the Cystic Fibrosis Foundation Therapeutics, Inc. (CFFT) . In 2010, orphan drug designation was assigned in the E.U. and the U.S. for the treatment of CF.
VX-809 may act to restore the function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, the defective cell membrane protein responsible for the progression of CF. VX-809 and other corrector compounds were designed to increase the amount of DF508-CFTR on the surface of cells lining the airway, which may result in an increase in chloride transport across the cell surface in patients with the DF508-CFTR mutation.
On January 11, 2013, the combination regimen of Lumacaftor (VX-809) and Kalydeco (Ivacaftor) was awarded by U.S. FDA with Breakthrough Therapy Designation as part of the agency’s efforts to accelerate the development and approval of drugs for serious and life-threatening disease.Breakthrough Therapy Designation for the combination regimen of VX-809 with ivacaftor was based on the Phase II combination data announced in 2012. Vertex Pharmaceuticals will report results from two Phase III trials (NCT01807949 (TRANSPORT) and NCT01807923 (TRAFFIC)) of the combination of Kalydeco + VX-809 in the middle of 2014. Positive data from TRAFFIC and TRANSPORT could open up a market with peak sales of approximately $6 billion, estimate analysts.

- 1 Merk; Schubert-Zsilavecz. Pharmazeutische Zeitung (in German) 156 (37): 24–27.
- 2 Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery medicine 15 (81): 127–133. PMID 23449115

see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.htm
…………………………
PATENT
http://www.google.com/patents/EP2639222A1?cl=en
-
CFTR correctors useful in the treatment of cystic fibrosis. Such compounds include 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (hereinafter “Compound 1”) which has the structure below:

-
Compound 1 and pharmaceutically acceptable compositions thereof are useful for treating or lessening the severity of a variety of CFTR mediated diseases.

-
Scheme 1. Synthesis of the acid chloride moiety.


Scheme 2. Synthesis of the amine moiety.

Scheme 3. Formation of an acid salt of 3-(6-(1-(2,2-difluorobcnzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid.

-
Synthesis of 3-(6-(1-(2,2-difluorobenzord[d][1,3]dioxol-5-yl) cyclopropancearboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl.
-
[0238]Acid Chloride Moiety
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-methanol (Compound 18).

-
Commercially available 2,2-difluoro-1,3-benzodioxole-5-carboxylic acid (1.0 eq) is slurried in toluene (10 vol). Vitride® (2 eq) is added via addition funnel at a rate to maintain the temperature at 15-25 °C. At the end of addition the temperature is increased to 40 °C for 2 h then 10% (w/w) aq. NaOH (4.0 eq) is carefully added via addition funnel maintaining the temperature at 40-50 °C. After stirring for an additional 30 minutes, the layers are allowed to separate at 40 °C. The organic phase is cooled to 20 °C then washed with water (2 x 1.5 vol), dried (Na2SO4), filtered, and concentrated to afford crude Compound 18 that is used directly in the next step.
-
Synthesis of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (Compound 19).

-
Compound 18 (1.0 eq) is dissolved in MTBE (5 vol). A catalytic amount of DMAP (1 mol %) is added and SOCl2 (1.2 eq) is added via addition funnel. The SOCl2 is added at a rate to maintain the temperature in the reactor at 15-25 °C. The temperature is increased to 30 °C for 1 hour then cooled to 20 °C then water (4 vol) is added via addition funnel maintaining the temperature at less than 30 °C. After stirring for an additional 30 minutes, the layers are allowed to separate. The organic layer is stirred and 10% (w/v) aq. NaOH (4.4 vol) is added. After stirring for 15 to 20 minutes, the layers are allowed to separate. The organic phase is then dried (Na2SO4), filtered, and concentrated to afford crude Compound 19 that is used directly in the next step.
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (compound 20).

-
A solution of Compound 19 (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude compound 20 (95%) that is used directly in the next step.
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile (compound 21).

-
A mixture of compound 20 (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) is heated at 70 °C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford compound 21.
-
Synthesis of 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid (compound 22).

-
Compound 21 is hydrolyzed using 6 M NaOH (8 equiv) in ethanol (5 vol) at 80 °C overnight. The mixture is cooled to room temperature and ethanol is evaporated under vacuum. The residue is taken into water and MTBE, 1 M HCl was added and the layers are separated. The MTBE layer was then treated with dicyclohexylamine (0.97 equiv). The slurry is cooled to 0 °C, filtered and washed with heptane to give the corresponding DCHA salt. The salt is taken into MTBE and 10% citric acid and stirred until all solids dissolve. The layers are separated and the MTBE layer was washed with water and brine. Solvent swap to heptane followed by filtration gives compound 22 after drying in a vacuum oven at 50 °C overnight.
-
Synthesis of 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonyl chloride (compound 7).

-
Compound 22 (1.2 cq) is slurried in toluene (2.5 vol) and the mixture heated to 60 °C. SOCl2 (1.4 eq) is added via addition funnel. The toluene and SOCl2 are distilled from the reaction mixture after 30 minutes. Additional toluene (2.5 vol) is added and distilled again.
-
Synthesis of 14C-(2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (compound 23).

-
A solution of Compound 19 (1 eq) in DMSO (1.25 vol) is added to a slurry of Na14 CN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude compound 23 that is purified by chromatography.
-
Synthesis of 14C-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile (compound 24).

-
A mixture of compound 23 (1.0 eq) and 1,2-dibromoethane (1.8 eq) in THF (3 vol) is cooled to -10 °C via external chiller. 1 M LHMDS in THF (2.5 eq) is added via an addition funnel and at a rate to maintain the temperature in the reactor below 10 °C. One hour after addition is complete, 20% w/v aq. citric acid (13 vol) is added via addition funnel maintaining the temperature in the reactor below 20 C. The external chiller is turned off and after stirring for 30 min the layers are separated. The organic layer is filtered and concentrated to afford crude compound 24 that is purified by chromatography.
-
Synthesis of 14C-1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid (compound 25).

-
Compound 24 is hydrolyzed using 6 M NaOH (8 equiv) in ethanol (5 vol) at 80 °C overnight. The mixture is cooled to room temperature and ethanol is evaporated under vacuum. The residue is taken into water and MTBE. 1 M HCl is added to the mixture and the organic layer is filtered and concentrated to afford compound 25.
-
Synthesis of 14C-1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonyl chloride (compound 26).

-
A mixture of Compound 25, 4-dimethylaminopyridine, and thionyl chloride (SOCl2) in CH2Cl2 is stirred to produce compound 26, which may be further reacted with compound 6 without isolation.
-
Amine Moiety
-
Synthesis of tert-butyl-3-(3-methylpyridin-2-yl)benzoate (compound 4).

-
2-Bromo-3-methylpyridine (1.0 eq) is dissolved in toluene (12 vol). K2CO3 (4.8 eq) is added followed by water (3.5 vol) and the mixture heated to 65 °C under a stream of N2 for 1 hour. 3-(t-Butoxycarbonyl)phenylboronic acid (1.05 eq) and Pd(dppf)Cl2-CH2Cl2 (0.015 eq) are then added and the mixture is heated to 80 °C. After 2 hours, the heat is turned off, water is added (3.5 vol) and the layers are allowed to separate. The organic phase is then washed with water (3.5 vol) and extracted with 10% aqueous methanesulfonic acid (2 eq MsOH, 7.7 vol). The aqueous phase is made basic with 50% aqueous NaOH (2 eq) and extracted with EtOAc (8 vol). The organic layer is concentrated to afford crude compound 4 (82%) that is used directly in the next step.
-
Synthesis of 2-(3-(tert-butoxycarbonyl)phenyl)-3-methylpyridine-1-oxide (compound 5).

-
Compound 4 (1.0 eq) is dissolved in EtOAc (6 vol). Water (0. 3 vol) is added followed by urea-hydrogen peroxide (3 cq). The phthalic anhydride (3 cq) is added portion-wise as a solid to maintain the temperature in the reactor below 45 °C. After completion of phthalic anhydride addition, the mixture is heated to 45 °C. After stirring for an additional 4 hours, the heat is turned off. 10% w/w aqueous Na2SO3 (1.5 eq) is added via addition funnel. After completion of Na2SO3 addition, the mixture is stirred for an additional 30 minutes and the layers separated. The organic layer is stirred and 10% w/w aq. Na2CO3 (2 eq) is added. After stirring for 30 minutes, the layers are allowed to separate. The organic phase is washed 13% w/v aq NaCl. The organic phase is then filtered and concentrated to afford crude compound 5 (95%) that is used directly in the next step.
-
Synthesis of tert-butyl-3-(6-amino-3-methylpyridin-2-yl)benzoate (compound 6).

-
A solution of compound 5 (1 eq) and pyridine (4 eq) in MeCN (8 vol) is heated to 70 °C. A solution of methanesulfonic anhydride (1.5 eq) in MeCN (2 vol) is added over 50 min via addition funnel maintaining the temperature at less than 75 °C. The mixture is stirred for an additional 0.5 hours after complete addition. The mixture is then allowed to cool to ambient. Ethanolamine (10 eq) is added via addition funnel. After stirring for 2 hours, water (6 vol) is added and the mixture is cooled to 10 °C. After stirring for NLT 3 hours, the solid is collected by filtration and washed with water (3 vol), 2:1 MeCN/water (3 vol), and MeCN (2×1.5 vol). The solid is dried to constant weight (<1% difference) in a vacuum oven at 50 °C with a slight N2 bleed to afford compound 6 as a red-yellow solid (53% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (compound 8).

-
Compound 7 is dissolved in toluene (2.5 vol based on acid chloride) and added via addition funnel to a mixture of compound 6 (1 eq), dimethylaminopyridine (DMAP, 0.02 eq), and triethylamine (3.0 cq) in toluene (4 vol based on compound 6). After 2 hours, water (4 vol based on compound 6) is added to the reaction mixture. After stirring for 30 minutes, the layers are separated. The organic phase is then filtered and concentrated to afford a thick oil of compound 8 (quantitative crude yield). MeCN (3 vol based on crude product) is added and distilled until crystallization occurs. Water (2 vol based on crude product) is added and the mixture stirred for 2 h. The solid is collected by filtration, washed with 1:1 (by volume) MeCN/water (2 x 1 vol based on crude product), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford 3-(6-(1-(2,2-difluorobenzo[d] [1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate as a brown solid.
-
Syntheisis of Syntheisis of 3-(6-(1-(2,2-difluorobenzo[d] [1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCL salt (compound 9).

-
To a slurry of compound 8 (1.0 eq) in MeCN (3.0 vol) is added water (0.83 vol) followed by concentrated aqueous HCl (0.83 vol). The mixture is heated to 45 ± 5 °C. After stirring for 24 to 48 hours the reaction is complete and the mixture is allowed to cool to ambient. Water (1.33 vol) is added and the mixture stirred. The solid is collected by filtration, washed with water (2 x 0.3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford compound 9 as an off-white solid.















-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1).

-
A slurry of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) is stirred at ambient temperature. A sample is taken after stirring for 24 hours. The sample is filtered and the solid washed with water (2 x). The solid sample is submitted for DSC analysis. When DSC analysis indicates complete conversion to Compound 1, the solid is collected by filtration, washed with water (2 x 1.0 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid (98% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) using water and base.


-
To a slurry of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) stirred at ambient temperature is added 50% w/w aq. NaOH (2.5 eq). The mixture is stirred for NLT 15 min or until a homogeneous solution. Concentrated HCl (4 eq) is added to crystallize Compound 1. The mixture is heated to 60 °C or 90 °C if needed to reduce the level of the t-butylbenzoate ester. The mixture is heated until HPLC analysis indicates NMT 0.8% (AUC) t-butylbenzoate ester. The mixture is then cooled to ambient and the solid is collected by filtration, washed with water (3 x 3.4 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid (97% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) directly from benzoate.

-
A solution of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 °C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 °C and the mixture stirred. The mixture is then heated to 70 ± 10 °C until the level of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid.



-
1HNMR spectra of Compound 1 are shown in Figures 9-11 (Figures 9 and 10 depict Compound 1 in Form I in a 50 mg/mL, 0.5 methyl cellulose-polysorbate 80 suspension, and Figure 11 depicts Compound 1 as an HCl salt).
-
Table 3 below recites additional analytical data for Compound 1.
-
Table 3.
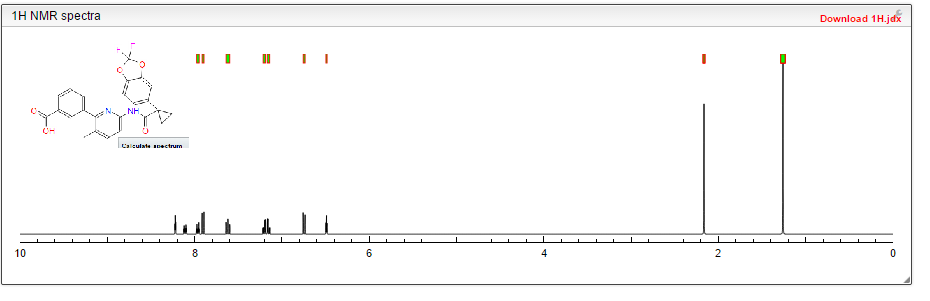
Cmpd. No. LC/MS M+1 LC/RTmin NMR 1 453.3 1.93 H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)



| WO2002096421A1 * | May 22, 2002 | Dec 5, 2002 | Neurogen Corp | 5-substituted-2-arylpyridines as crf1 modulators |
| WO2004072038A1 * | Feb 10, 2004 | Aug 26, 2004 | Vertex Pharma | Processes for the preparation of n-heteroaryl-n-aryl-amines by reacting an n-aryl carbamic acid ester with a halo-heteroaryl and analogous processes |
| WO2007056341A1 | Nov 8, 2006 | May 18, 2007 | Vertex Pharma | Heterocyclic modulators of atp-binding cassette transporters |
| see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.htm |
References
David Andrew Siesel;Processes for producing cycloalkylcarboxamido-pyridine benzoic acids,US patent number US8124781 B2 ;Also published as CA2707494A1, CN101910134A, EP2231606A2, EP2231606B1, EP2639222A1, EP2639223A1, EP2639224A1, US8592602, US20090176989, US20120190856, WO2009076142A2, WO2009076142A3;Filing date:Dec 4, 2008;Original Assignee:Vertex Pharmaceuticals Incorporated
David Andrew Siesel;Processes for producing cycloalkylcarboxamido-pyridine benzoic acids,US patent number US8461342 B2 ;Also published as US20100036130, US20120203006, US20130274477, WO2010138484A2, WO2010138484A3;Original Assignee:Vertex Pharmaceuticals Incorporated
Van Goor, Fredrick F. et al;Pharmaceutical compositions in the treatment of CFTR-mediated diseases such as cystic fibrosis;PCT Int. Appl., WO2011133956
Van Goor, Fredrick F. et al;Pharmaceutical compositions in the treatment of CFTR-mediated diseases such as cystic fibrosis.PCT Int. Appl., WO2011133951
Van Goor, Fredrick F. et al;Pharmaceutical compositions for treatment of CFTR-mediated diseases;PCT Int. Appl., WO2011133953
Verwijs, Marinus Jacobus et al;Preparation and pharmaceutical compositions of Lumacaftor for the treatment of cystic fibrosis and other diseases associated with CFTR mutations;PCT Int. Appl., WO2011127241
Keshavarz-Shokri, Ali et al;Preparation of Lumacaftor for therapeutical use;PCT Int. Appl., WO2011127290
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;U.S. Pat. Appl. Publ., US20100036130
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;PCT Int. Appl., WO2010138484
Young, Christopher;Dosage units of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid;PCT Int. Appl., WO2010037066
Hadida-Ruah, Sara et al;paration of N-pyridinyl carboxamide derivatives as modulators of ATP-binding cassette transporters;U.S. Pat. Appl. Publ., 20080019915
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;PCT Int. Appl., WO2009076142
Hadida Ruah, Sara et al;Preparation of N-pyridinyl carboxamide derivatives as modulators of ATP-binding cassette transporters;PCT Int. Appl., WO2007056341
video on cystic fibrosis
second video
Update on 26 mar 2015
LUMACAFTOR
VX 809
| 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic Acid | |
| CAS No.: | 936727-05-8 |
|---|---|
| Synonyms: |
|
| Formula: | C24H18F2N2O5 |
| Exact Mass: | 452.11800 |
SMILLES…. Cc1ccc(nc1c2cccc(c2)C(=O)O)NC(=O)C3(CC3)c4ccc5c(c4)OC(O5)(F)F
NMR…………….http://file.selleckchem.com/downloads/nmr/S156503-VX-809-HNMR-Selleck.pdf
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) directly from benzoate.
-
A solution of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 °C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 °C and the mixture stirred. The mixture is then heated to 70 ± 10 °C until the level of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid.
-
1HNMR spectra of Compound 1 are shown in Figures 9-11 (Figures 9 and 10 depict Compound 1 in Form I in a 50 mg/mL, 0.5 methyl cellulose-polysorbate 80 suspension, and Figure 11 depicts Compound 1 as an HCl salt).
-
Table 3 below recites additional analytical data for Compound 1.
-
Table 3.
Cmpd. No. LC/MS M+1 LC/RTmin NMR 1 453.3 1.93 H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)

1H NMR PREDICT
![3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid NMR spectra analysis, Chemical CAS NO. 936727-05-8 NMR spectral analysis, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-29/001/530/195/936727-05-8-1h.png)
13C NMR PREDICT
![3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid NMR spectra analysis, Chemical CAS NO. 936727-05-8 NMR spectral analysis, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-29/001/530/195/936727-05-8-13c.png) CAS NO. 936727-05-8, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectral analysisCOSY PREDICT
CAS NO. 936727-05-8, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectral analysisCOSY PREDICT


13C NMR PREDICT




| WO2002096421A1 * | May 22, 2002 | Dec 5, 2002 | Neurogen Corp | 5-substituted-2-arylpyridines as crf1 modulators |
| WO2004072038A1 * | Feb 10, 2004 | Aug 26, 2004 | Vertex Pharma | Processes for the preparation of n-heteroaryl-n-aryl-amines by reacting an n-aryl carbamic acid ester with a halo-heteroaryl and analogous processes |
| WO2007056341A1 | Nov 8, 2006 | May 18, 2007 | Vertex Pharma | Heterocyclic modulators of atp-binding cassette transporters |

http://www.google.co.in/patents/US8124781



Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1)

Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1) Using Water and Base

Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1) Directly from Benzoate

Compound 1
Compound 1 is used as the starting point for the other solid state forms and can be prepared by coupling an acid chloride moiety with an amine moiety according to Schemes 1-4.
Scheme 1. Synthesis of the acid chloride moiety.

1. NaCN
2. H20

socio

Scheme 1 depicts the preparation of l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride, which is used in Scheme 3 to make the amide linkage of Compound 1.
The starting material, 2,2-difluorobenzo[d][l,3]dioxole-5-carboxylic acid, is commercially available from Saltigo (an affiliate of the Lanxess Corporation). Reduction of the carboxylc acid moiety in 2,2-difluorobenzo[d][l ,3]dioxole-5-carboxylic acid to the primary alcohol, followed by conversion to the corresponding chloride using thionyl chloride (SOCl2), provides 5-(chloromethyl)-2,2-difluorobenzo[d][l,3]dioxole, which is subsequently converted to 2-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)acetonitrile using sodium cyanide. Treatment of 2-(2,2- difluorobenzo[d][l,3]dioxol-5-yl)acetonitrile with base and l-bromo-2-chloroethane provides 1- (2,2-difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarbonitrile. The nitrile moiety in l-(2,2- difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarbonitrile is converted to a carboxylic acid using base to give l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarboxylic acid, which is converted to the desired acid chloride using thionyl chloride.
Scheme 2. Alternative synthesis of the acid chloride moiety.

Touene, H20, 70 °C3 N HC1,
DMSO,
75 °C

Scheme 2 depicts an alternative synthesis of the requisite acid chloride. 5- bromomethyl-2,2-difluoro-l,3-benzodioxole is coupled with ethyl cyanoacetate in the presence of a palladium catalyst to form the corresponding alpha cyano ethyl ester. Saponification of the ester moiety to the carboxylic acid gives the cyanoethyl compound. Alkylation of the cyanoethyl compound with l-bromo-2-chloro ethane in the presence of base gives the cyanocyclopropyl compound. Treatment of the cyanocyclopropyl compound with base gives the carboxylate salt, which is converted to the carboxylic acid by treatment with acid. Conversion of the carboxylic acid to the acid chloride is then accomplished using a chlorinating agent such as thionyl chloride or the like.
Scheme 3. Synthesis of the amine moiety.

ptBu urea-hydrogen peroxide hthalic anhydride EtOAc, water

Scheme 3 depicts the preparation of the requisite tert-butyl 3-(6-amino-3- methylpyridin-2-yl)benzoate, which is coupled with l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride in Scheme 3 to give Compound 1. Palladium-catalyzed coupling of 2-bromo-3-methylpyridine with 3-(tert-butoxycarbonyl)phenylboronic acid gives tert-butyl 3-(3-methylpyridin-2-yl)benzoate, which is subsequently converted to the desired compound. Scheme 4. Formation of an acid salt of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid.

Scheme 4 depicts the coupling of l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride with tert-butyl 3-(6-amino-3-methylpyridin-2-yl)benzoate using triethyl amine and 4-dimethylaminopyridine to initially provide the tert-butyl ester of Compound 1.
……………………..
WO2010037066
http://www.google.im/patents/WO2010037066A2?cl=en
Syntheisis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCL salt.

HCl
To a slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in MeCN (3.0 vol) is added water (0.83 vol) followed by concentrated aqueous HCl (0.83 vol). The mixture is heated to 45 ± 5 0C. After stirring for 24 to 48 hours the reaction is complete and the mixture is allowed to cool to ambient. Water (1.33 vol) is added and the mixture stirred. The solid is collected by filtration, washed with water (2 x 0.3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford 3-(6-(l-(2,2- difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2- yl)benzoic acid • HCl as an off-white solid.
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I).

HCl

Compound 1 in Form I
A slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) is stirred at ambient temperature. A sample is taken after stirring for 24 hours. The sample is filtered and the solid washed with water (2 x). The solid sample is submitted for DSC analysis. When DSC analysis indicates complete conversion to Compound 1, the solid is collected by filtration, washed with water (2 x 1.0 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 as an off-white solid (98% yield).
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I) using water and base.


Compound 1 in Form I
To a slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) stirred at ambient temperature is added 50% w/w aq. NaOH (2.5 eq). The mixture is stirred for NLT 15 min or until a homogeneous solution. Concentrated HCl (4 eq) is added to crystallize Compound 1. The mixture is heated to 60 0C or 90 0C if needed to reduce the level of the t-butylbenzoate ester. The mixture is heated until HPLC analysis indicates NMT 0.8% (AUC) t-butylbenzoate ester. The mixture is then cooled to ambient and the solid is collected by filtration, washed with water (3 x 3.4 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 as an off-white solid (97% yield).
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I) directly from benzoate.


Compound 1 in Form I
A solution of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 0C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 0C and the mixture stirred. The mixture is then heated to 70 ± 10 0C until the level of 3- (6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin- 2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 in Form I as an off-white solid.


[…] syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/ […]
LikeLike