
Home » Posts tagged 'GMP'
Tag Archives: GMP
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ozonization of Pharmaceutical Water and the Biocidal Products Regulation

With the new biocidal products regulation from 2013 in-situ generated ozone now also falls into the scope of this directive. Ozone generation systems with a biocide application (such as disinfection of pharma water) thus require an approval after the transitional period expires in the September 2017. The ozone registration group is active for this purpose. Read more about the Ozonization of Pharmaceutical Water and the Biocidal Products Regulation.
With the new biocidal products regulation from 2013 in-situ generated ozone now also falls into the scope of this regulation. Ozone generation systems with a biocide application (such as disinfection of pharma water) thus require an approval after the transitional period expires in the September 2017. We already reported about the impact of the new Biocidal Products Regulation – please see the GMP News “Pharmaceutical Water: Uncertainty caused by the New Biocidal Products Regulation” from 21 May 2014.
Admission will take place in two stages. In the first step, ozone is certified as an active ingredient and registered in the list of active substances authorised in the EU. In the second step, the ozone generation system is approved as a biocidal product. The major manufacturers of ozone generation systems have joined forces for this in the ozone registration group (ORG). It aims at relieving users of ozone systems from the registration procedure. That means the documents should be provided to the users. The access to the marketing authorisation dossier is supposed to be assured through a Letter of Access (LoA). One of the open questions seems to be resolved now: the question whether an authorisation document will be required for each ozone precurser (i.e. water, oxygen or air). As this seems to be unnecessary, only one authorisation document is currently being processed.
The question with regard to how reasonable it is to include ozone from pharmaceutical water systems in the biocidal products regulation cannot be clarified at this point. The same is true with regard to the question on who is supposed to control pharmaceutical companies and whether their ozone comes from approved ozone systems.
You can find more information on the page Ozone registration group.


GMP Matrix – EU GMP Guide – FDA cGMP Guide and ISO 9001 comparison
How can one find a certain GMP requirement in the EU GMP Guide, in the FDA cGMP Guide and in the ISO 9001 without searching for a long time? The Good Practice Guide developed by the ECA has become a standard in many companies and is aimed at providing this information. A 26 pages matrix provides information about where to find a GMP requirement e.g. on Validation, QC Lab testing etc in the three major Guidelines. The comprehensive booklet with 500 pages contains a full text version of all three guidelines. You can find the GMP Matrix here.
http://www.gmp-compliance.org/eca_handbuecher.html
Publications

ECA Good Practice Guide – “GMP Matrix”
“FDA cGMP, EU GMP and ISO 9001 Matrix for a pharmaceutical Quality System – A GMP Roadmap”. (Version 15 of April 2014)
The revised ECA Good Practice Guide is a comprehensive juxtaposition containing the requirements laid down in FDA’s cGMP Guide, the EU GMP Guide and ISO 9001. The updated Matrix now has 26 pages as well as further 500 pages for the following three regulations
- FDA cGMP Guide
- EU GMP Guide Part I, II, and III incl. all Annexes
- ISO 9001 Quality Management Systems
In addition, the Good Practice Guide contains a ISO 9001/ICH10 Matrix and the complete Part III to the EU GMP Guide.
Price*: € 149 Non ECA Members, € 99 ECA Members
What GMP 良好作業規範 Changes can we still expect for 2014?

What GMP Changes can we still expect for 2014?
Heraclitus once said: “There is nothing permanent except change”. This statement is even true for the rather conservative GMP environment. What can we still expect for 2014? The answer to that question can be found in a work plan of EMA’s GMP/GDP Inspectors Working Group.
What are the coming plans?
- Finalising the changes planned for the Chapters 3 and 5 of the EU GMP Guide
- Finalising the revision of Chapter 8 of the EU GMP guide (with regard to product shortage notifications and specific risk management concepts)
- Agreeing, in consultation with PIC/S, whether guidance is needed on biofilms concerning Annex 1 of the EU GMP Guide
- Finalising the revision of Annex 15 of the EU GMP Guide (comparison with the new EMA process validation guideline and inclusion of necessary changes in the light of ICH Q 8-10)
- Finalising the revision of Annex 16 of the EU GMP Guide
- Finalising the revision of Annex 17 of the EU GMP Guide
- Further measures regarding the EudraGMDP database
The finalisation of the revision of Chapter 6 (Quality Control) of the EU GMP Guide is already completed (April 2014). The revised chapter will apply as of October 2014.
The following topics are also addressed in the work paper:
- Inspections under the centralised system
- Mutual Recognition Agreements (MRAs)
- Harmonisation topics
- Collaboration with the EU Commission (the collaboration should enable by the end of 2014 the publication of the GDP guidelines for APIs and the risk assessment guidelines to establish GMP for excipients)
- Collaboration with other groups (i.e. Reverse Osmosis for the production of WFI and biological indicators for monitoring and the control of sterilisation are topics addressed together with the EDQM in Strasburg)
Please also see the complete “Work plan for GMP/GDP Inspectors Working Group for 2014“.
http://www.gmp-compliance.org/enews_04349_What-GMP-Changes-can-we-still-expect-for-2014%3F.html

|
email me amcrasto@gmail.com |
GMP Handbooks with all major GMP and GDP Guidelines

GMP Handbooks with all major GMP and GDP Guidelines
Everyone involved in the GMP/GDP environment needs to use the current GMP and GDP Guidelines for reference. The ECA offers a range of booklets with all major Guidelines such as the EU GMP Guide (with all current Annexes), the new EU GDP Guideline, the FDA cGMP Guide and many more. You can order the GMP booklets here.http://www.gmp-compliance.org/eca_handbuecher.html
GMP Publications

….NEW….
….NEW….
ECA Good Practice Guide on Validation
(1st Edition of October 2012)
This document is intended to provide support to both regulators and industry. On one hand, the guide contains the main elements of the new approach (“what to do”). On the other hand, it also serves as a supporting guide for the implementation (“how to do”). The guide contains 163 pages divided in 5 chapters and 4 annexes. The topics covered are among others:•Risk based qualification and validation legacy products
•Statistics
•Case study about process validation in biopharmaceutical manufacturing
•Case study about continuous process verification
•Paperback in the handy format 14,8 x 21 cm
Price*: € 149 Non ECA Members, € 99 ECA Members
Booksellers receive a 15% discount – please ask for a COUPON CODE before ordering!
http://www.gmp-compliance.org/eca_handbuecher.htm
If you want to use the major GMP Guidelines on your smartphone or tablet we recommend to use the free of charge GMP Web App developed by the ECA Academy
http://www.gmp-compliance.org/eca_app.html
The new GMP WebApp from ECA
——————————————————————————–

ECA is pleased to announce a major development: now you can have all GMP information on your smartphone or Tablet PC (e.g. iPad) – with the new free of charge ECA GMP WebApp.
The unique new WebApp provides a number of GMP features. The App, which works on all smartphones (Apple and Android), is a useful tool for all professionals in the GMP environment. To open it, just go to app.gmp-compliance.org in your browser and the WebApp opens immediately.
To use the App in a convenient way you need to add the ECA icon to the Home screen (see below).
GMP News
From ECA‘s weekly GMP Newsletter you are used to get the latest trends in the GMP environment. Now you can have these news at hand and keep track of all GMP developments any time. You will always find the latest GMP News on your App.
Major GMP Guidelines
The App allows you to access the major GMP Guidelines very easily. Whenever a revised GMP Guide is published the document is available without any update of the App. So you can always check the relevant Guidelines in seconds.
GMP Search
If you are looking for additional GMP information, the „Search“ function is very helpful. Just enter a keyword and select a specific database – or just search in all databases. The GMP Database contains hundreds of GMP articles and more than 1.000 GMP Guidelines. You do not need to search on different websites for the information. The GMP Database provides the links to the most relevant information.
GMP Courses & Conferences
On the ECA website you can scroll a list with all currently offered courses and conferences. The new WebApp does provide that list as well. Simply go on „GMP Courses & Conferences“ to access the complete ECA course and conference programme any time. If you just want to get a list with courses and conferences in a certain area, simply use the „GMP Search“ function decsribed before. And… by the way… if you found the programme you were just looking for… you can even register by using the App.
GMP Guideline Manager
Access to more than 1.200 GMP Guidelines This function is an exclusive service for ECA Members (Company Members will get access for all employees*). After login you will have access to all GMP Guidelines from EU/EMA, FDA, ICH, PIC/S, ICH, APIC, IPEC and WHO. To log in simply use your user name and password from your ECA Membership account. ECA Members have access to two so called Webtrees. One Guideline Tree is structured according to GMP topics. The second Guideline Tree is structured according to authorities. By using the Guideline Trees you can easily access the Guideline of interest.
* Employees of all sites in the country in which the company signed up for the membership.
What GMP Changes can we still expect for 2014?
What GMP Changes can we still expect for 2014?

Heraclitus once said: “There is nothing permanent except change”. This statement is even true for the rather conservative GMP environment. What can we still expect for 2014? The answer to that question can be found in a work plan of EMA’s GMP/GDP Inspectors Working Group.
What are the coming plans?
- Finalising the changes planned for the Chapters 3 and 5 of the EU GMP Guide
- Finalising the revision of Chapter 8 of the EU GMP guide (with regard to product shortage notifications and specific risk management concepts)
- Agreeing, in consultation with PIC/S, whether guidance is needed on biofilms concerning Annex 1 of the EU GMP Guide
- Finalising the revision of Annex 15 of the EU GMP Guide (comparison with the new EMA process validation guideline and inclusion of necessary changes in the light of ICH Q 8-10)
- Finalising the revision of Annex 16 of the EU GMP Guide
- Finalising the revision of Annex 17 of the EU GMP Guide
- Further measures regarding the EudraGMDP database
The finalisation of the revision of Chapter 6 (Quality Control) of the EU GMP Guide is already completed (April 2014). The revised chapter will apply as of October 2014.
The following topics are also addressed in the work paper:
- Inspections under the centralised system
- Mutual Recognition Agreements (MRAs)
- Harmonisation topics
- Collaboration with the EU Commission (the collaboration should enable by the end of 2014 the publication of the GDP guidelines for APIs and the risk assessment guidelines to establish GMP for excipients)
- Collaboration with other groups (i.e. Reverse Osmosis for the production of WFI and biological indicators for monitoring and the control of sterilisation are topics addressed together with the EDQM in Strasburg)
Please also see the complete “Work plan for GMP/GDP Inspectors Working Group for 2014“.

India under Pressure: GMP Conformity Not Guaranteed in many APIs Facilities

|
India under Pressure: GMP Conformity Not Guaranteed in many APIs Facilities |
| The pressure on India is getting bigger because of GMP deficiencies found during inspections. An article of the news agency Reuters summarised impressive information on the topic. Read more here about the Reuters article. |
read here
|
India under Pressure: GMP Conformity Not Guaranteed in many APIs Facilities |
|
The pressure on India is getting bigger because of GMP deficiencies found during inspections. A wide range of FDA Warning Letters and FDA Import Alerts are given special attention. The GMP deviations observed are extreme and partly alarming with regard to the potential risks for the patients. Even elementary GMP requirements have been neglected. Production areas were often found in an uncontrolled status (risk of cross contamination) that medicinal products had to be recalled. The Warning Letter for Wockhardt is one of the most prominent examples. According to an article published in RAPS Online, FDA inspectors wrote in this letter about the location: “a wide range of disturbing allegations, including bathrooms that allowed for the collection of standing urine on floors, products contaminated with glass and unknown “black particles,” staff that repeatedly lied to FDA on multiple occasions, and manufacturing lines that were kept hidden from investigators.” At the same time, this shows that effective and extensive GMP monitoring in India is inexistent. An article of the news agency Reuters summarised impressive information on the topic. According to it, 1,500 inspectors are responsible for 10,000 factories. In one out of every 22 samples, lack of quality has been observed. These data come from a study already performed two years ago. Reuters refers to industry analysts who say that companies which cannot deliver into the USA because of an Import Alert might continue their production and sell their products to other countries which are not aware of those GMP deficiencies. This is a terrifying scenario which is – according to experts’ statements – current practice in India. Such serious problems can only be explained because of totally insufficient monitoring of medicinal products in India. A GMP inspector in India told Reuters: “I took salaries for 30 years without doing anything. I visited some of the plants … not with the intention of taking any action, but just out of curiosity.” Reuters quotes an employee of India’s health ministry who says that only the US FDA complains about Indian factories. “(…) other countries have no problems with our drugs. They have never raised any objections or have found fault,” This statement can be easily refuted. The EudraGMDP database currently lists 38 facilities in India which have been classified as “GMP non-compliant” because of negative GMP inspections. The lack of adequate GMP supervision (GMP Inspections) by Indian Authorities also raises questions on the EU procedure to require Written (GMP) Confirmations from agencies around the world. In order to import an API from a manufacturer located outside the EU the legal provisions require that a Written Confirmation has to be issued by the exporting countries (only some countries like e.g. Switzerland, USA have been found to have equivalent GMP Inspection systems and do not need to issue Written Confirmations). India has published many Written Confirmations for API manufacturers in India. However, some of the companies who own a Written Confirmation received FDA Warning Letters or EU GMP non-compliance statements only some months after the Written Confirmations were issued. This questions the value of the Written Confirmations also for all other facilities in India not inspected recently by EU or FDA inspectors. The information published by Reuters, RAPS Online and other well recognized media sources may require to initiate additional actions by industry and regulators in the EU in order to safeguard APIs imported from India. |
EMA publishes New Process Validation Guideline
![]()
EMA publishes New Process Validation Guideline
After the publication of the Annex 15 draft at the beginning of February 2014, the EMA made a move towards the revision of its process validation guideline. The final document was published on 27 February 2014. For a long time now, the EMA had already announced this revision in a concept paper. What’s new? click here
After the publication of the Annex 15 draft at the beginning of February 2014, the EMA made a move towards the revision of its process validation guideline. The final document was published on 27 February 2014. For a long time now, the EMA had already announced this revision in a concept paper. The objective of the revision was to integrate modern GMP aspects:
- Integration of the ICH Q8, Q9 and Q10 Guidelines
- Incorporation of Process Analytical Technology (PAT), Quality by Design (QbD) and Real-Time Release Testing (RTRT).
- Extension with regard to an “enhanced approach” and integration of “continuous process verification”
- Integration of the Annexes to the current Note for Guidance
- Harmonisation with the current FDA Guidance on Process Validation
The deadline for comments on the draft for the revision of the process validation guideline ended in October 2012 already. Now, elements in accordance with the Annex 15 have also flowed into the final document. In the following, you will read a short evaluation of the document with regard to the original draft from March 2012, the (still) applicable Note for Guidance on Process Validation and FDA’s Guidance on Process Validation. The GMP relevant aspects of the documents will also be addressed.
The original 7-page long Note for Guidance on Process Validation has more than doubled and now contains 15 pages. Even the original revision draft had only 11 pages. The change in the title to “Guideline on process validation for finished products- information and data to be provided in regulatory submissions” is noticeable. The title itself gives indication about the content of the document, namely marketing authorisation matters.
Like in the draft, the document is composed of 8 numerated chapters, a summary, definitions, references, an Annex I (Process validation scheme) and an Annex II (Standard/non-standard processes) which is a new part compared to the draft. A sub section on “Design space verification” has been newly added to the chapter on process validation.
There haven’t been big changes to the draft document released in 2012. Only the chapter “Design space verification” is brand new, all other parts have been mostly updated. The chapter on ongoing process validation has been removed. Compared to the draft, indications about standard/ non-standard processes are now available in the Annex II – like in the currently applicable Note for Guidance.
What are the changes to the currently applicable Note for Guidance on Process Validation?
Compared to the current Note for Guidance, the revision remains in its final version pretty difficult to read and rather general. This is a marketing authorisation document, which is clearly addressed in the title and only applies to finished dosage forms of chemical medicinal products for human and veterinary use but not for old ones, which are already authorised and on the market. The introduction of a validation life cycle and the integration of continued process verification (CPV) are completely new although this approach is already acquainted from ICH Q8. The “traditional approach” remains accepted. Like in the Annex 15 draft the hybrid approach remains here in the final document “nebulous”. The idea to integrate modern elements from ICH Q8, Q10 (and Q11) into the document is clearly noticeable. Yet, far less concrete references are made to ICH Q9.
A stronger overlap of the FDA Guidance would have been desirable. FDA’s Guidance also deals with APIs and biologicals, and the process validation life cycle runs like a thread through the whole FDA document. FDA’s Guidance also contains GMP aspects. The FDA Guidance explicitly addresses old products which should be integrated to stage 3 of the life cycle. Yet, there is another big difference. The revised document doesn’t highlight statistical methods like the FDA Guidance.
Before the finalisation, a comparison with the Annex 15 has been made which is a nice thing. This explains the long period between the publication of the draft (March 2012) and that of the finalisation (February 2014).
What is significant for the GMP world? On the one hand almost nothing, on the other hand quite a lot: one may wonder why? Direct references to the Annex 15 can be found with regard to the “ongoing process verification” and “concurrent validation”, which is almost nothing looking at the whole document. Moreover, validation in general is required to be executed according to the GMP regarding “continuous process verification” and “change control”; these are the essential parts of the document, and (almost) the complete document should therefore be seen from a GMP perspective.
The new EMA guideline on process validation will apply by the end of August 2014.

Japanese Pharmacopoeia and Japanese GMP Regulations available online
Japanese Pharmacopoeia and Japanese GMP Regulations available online
On Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) website, you can download documents on GMP as well as on marketing authorisations for medicinal products. An English version of the Japanese Pharmacopoeia (JP) is also available. You will find the direct links in the News.
On Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) website, you can find in the section “Regulations and Procedures” under the heading “GMP” requirements regarding the inspection of manufacturers of medicinal products and APIs who want to introduce their products into Japan.
Now, a document was supplemented in January 2014 which describes which documents have to be submitted to the Japanese Agency within a pre-approval inspection and/ or a periodical post-approval inspection.
Go to the PMDA webpage to get more information.
There, you can also access the current Japanese Pharmacopoeia Sixteenth Edition in English.
Source: PMDA, Japan
Indian Regulators promote two levels of GMP
GMP deviations and even data falsification have been identified in a number of companies in India. How is it possible that interpretation of FDA and EU authorities on one side and the Indian authority on the other side come to a completely different picture? Read more in our GMP News
GMP deviations and even data falsification have been identified in a number of companies in India. The FDA has issued numerous Warning Letters, the EU has published GMP Non Compliance Reports in its EudraGMDP database and EDQM has withdrawn various CEPs because of GMP inspection findings.
In an article published by Regulatory Focus on 28 January 2014 the question has been raised whether Indian companies have a chronic data falsification problem. The article lists 7 companies in India which have received a Warning Letter in the past months – all of them because of GMP deviations and because of “actually or potentially tampering with their data”. In addition to the 7 companies the Ranbaxy case is a story of its own. Not only one facility was found to manipulate data but several sites of the company are involved. For this reason the US FDA has issued a consent decree of permanent injunction against Ranbaxy. All manufactured products in the facilities concerned are now subject to an FDA import alert. In a press release the FDA states: “Because this company continued to violate current good manufacturing practice regulations and falsify information on drug applications, the FDA took these actions in an effort to protect consumers.” Dara Corrigan, FDA associate commissioner for regulatory affairs goes on: “The FDA continues to be committed to protecting consumers from potentially unsafe products that may be offered on the market.” On January 23, 2014 the FDA added an additional facility of Ranbaxy to the existing consent decree.
So far, the Indian Authority did not initiate the same measures like US and European counterparts. This also questions the supervision system in India. If inspections have been performed by Indian Inspectors at the concerned facilities why did they fail to make the same findings? The Drug Controller General of India, Mr. G.N. Singh, gave an interesting interpretation: According to an interview published by live mint & Wall Street Journal he said: “…it must be stated that every country has different measures and we cannot judge Ranbaxy by standards set up by the American drug regulator“. When Mr Singh was asked about the problems identified at three Ranbaxy plants he stated: “Some of those were found to be true and my office had told Ranbaxy to take corrective measures. Similar procedures will be followed in this case as well. But I do not think this is a situation which will warrant withdrawal of drugs from the domestic market. Our biggest objective is to maintain good quality of medicines and we are doing that. There are no drugs in the Indian market that are not up to the standards stated under the Drugs and Cosmetics Act.” In a final statement in the interview he also mentioned that he is “not worried about issues of quality.” In another interview with the Business Standard Press Mr Singh made an alarming statement for all customers of medicinal products and APIs in Europe and the US. “If I follow US standards, I will have to shut almost all drug facilities“. If this is the truth EU and US customers are in big trouble because products not complying to EU/US GMP standard (e.g. ICH Q7 GMP for APIs) would need to be taken from the market immediately.
This all looks like it will not fit together. How is it possible that interpretation of FDA and EU authorities on one side and the Indian authority on the other side come to a completely different picture? It can only mean that dual standards exist. This would result in two quality levels, an international and a domestic quality level. Such a policy possibly causes questions by Indian patients who have to accept a different and probably lower quality standard.
It does not look like the Indian Regulators will re-think the GMP inspection approach and the quality standard in their country. Instead of acting in his own country the Drug Controller General of India announced inspections in the US and the EU.
But what are the international implications of this strategy? European Regulators need to react as they require from the Indian Authority to issue Written Confirmations of GMP compliance. Without a Written Confirmation APIs can not enter EU market. Currently more than 200 Written Confirmations have been issued by Indian Authority. If the inspections which have been performed as a prerequisite for issuing a Written confirmation were not based on the international standard ICH Q7 (GMP for APIs) the Written Confirmations are no longer valid documents. This issue might be raised by an EU court if a substandard API in a medicinal product will cause a health risk to patients in Europe.
RAMOSETRON

Ramosetron (INN),(1-methylindol-3-yl)-[(5R)-4,5,6,7-tetrahydro-3H-benzimidazol-5-yl]methanone, 132036-88-5 cas no
| C17H17N3O | |
| 279.33 g/mol |
(1-methyl-1H-indol-3-yl)[(5R)-4,5,6,7-tetrahydro-1H-benzimidazol-5-yl]methanone
YM060
- Nasea
- Nor-YM 060
- Ramosetron
- UNII-7ZRO0SC54Y
…………………………………………………………………………………..
HYDROCHLORIDE SALT
hyrochloride salt, cas no 132907-72-3

(−)-(R)-5-[(1-methyl-1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole monohydrochloride (yield 78.8%, 99.5% e.e.). FAB-MS (m/z): 280 [M+H+]
1H NMR (DMSO-d6, 30° C.): δ ppm (TMS internal standard): 1.82-1.95 (1H, m), 2.12-2.22 (1H, m), 2.66-2.94 (4H, m), 3.63-3.72 (1H, m), 3.88 (3H, s), 7.24 (1H, t, J=8.0 Hz), 7.30 (1H, t, J=8.0 Hz), 7.56 (1H, d, J=8.0 Hz), 8.22 (1H, d, J=8.0 Hz), 8.53 (1H, s), 8.90 (1H, s), 14.42 (1H, br)
…………………………………………………………………………………….
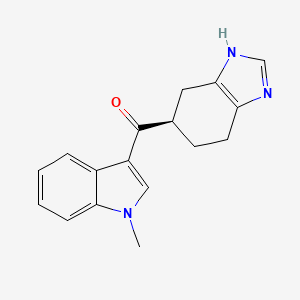
Ramosetron (INN) is a serotonin 5-HT3 receptor antagonist for the treatment of nausea and vomiting.[1] Ramosetron is also indicated for a treatment of “diarrhea-predominant irritable bowel syndrome in males”.[2] In India it is marketed under the brand name of“IBset”.
It is only licensed for use in Japan and selected Southeast Asian countries. In Japan it is sold under the tradename Iribo (イリボー). [3] Elsewhere it is commonly sold under the tradename Nasea and in India as Nozia (300 mcg/ml Inj. & 100 mcg Tab.) [4]
- Fujii Y, Saitoh Y, Tanaka H, Toyooka H (February 2000). “Ramosetron for preventing postoperative nausea and vomiting in women undergoing gynecological surgery”.Anesth. Analg. 90 (2): 472–5. doi:10.1097/00000539-200002000-00043.PMID 10648342.
- http://www.astellas.com/en/corporate/news/detail/astellas-launches-irribow-for.html
- Summary in Japanese. Retrieved on September 4, 2012.
- Abridged prescribing information – Nasea (MIMS Philippines). Retrieved on June 13, 2008.
- Synthesis and 5-HT3 antagonistic activities of 4,5,6, 7-tetrahydrobenzimidazole derivatives
200th ACS Natl Meet (August 26-31, Washington DC) 1990, Abst MEDI 39
|
1-27-2010
|
Process for producing ramosetron or its salt
|
|
|
11-20-1996
|
Intrabuccally dissolving compressed moldings and production process thereof
|
|
|
3-6-1996
|
5-substituted tetrahydrobenzimidazole compounds
|
|
|
11-15-1995
|
Intrabuccally disintegrating preparation and production thereof
|
|
|
9-7-1994
|
Tetrahydrobenzimidazole derivatives and pharmaceutical compositions containing same
|
|
|
6-24-1994
|
NEW USE OF 5-HT3 RECEPTOR ANTAGONISTS
|
AU 9048890; EP 0381422; JP 1991223278; US 5344927
| CN1696128A | Nov 2, 2004 | Nov 16, 2005 | 天津康鸿医药科技发展有限公司 | New method for synthesizing Ramosetron Hydrochloride |
| CN1765896A | Oct 28, 2004 | May 3, 2006 | 北京博尔达生物技术开发有限公司 | Novel preparation method of ramosetron hydrochloride |
| US5496942 * | 14 Feb 1994 | 5 Mar 1996 | Yamanouchi Pharmaceutical Co., Ltd. | 5-substituted tetrahydrobenzimidazole compounds |
| US5677326 * | 30 Sep 1994 | 14 Oct 1997 | Tokyo Tanabe Company Limited | Indoline compound and 5-HT.sub.3 receptor antagonist containing the same as active ingredient |
| US7358270 | 28 Jan 2005 | 15 Apr 2008 | Astellas Pharma Inc. | Treating agent for irritable bowel syndrome |
| US7683090 | 18 Oct 2006 | 23 Mar 2010 | Astellas Pharma Inc. | Treating agent for irritable bowel syndrome |
| US7794748 | 27 Aug 2004 | 14 Sep 2010 | Yamanouchi Pharmaceutical Co., Ltd. | Stable oral solid drug composition |
WO 2010024306
WO 2013005760
WO 2013100701
WO 2011001954
The chemical name of ramosetron is (−)-(R)-5-[(1-methyl-1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole, and it has the structure represented by the formula (II).

It is known that ramosetron or a salt thereof has a potent 5-HT3 receptor antagonism (Patent Reference 1, Non-patent references 1 and 2), and it is on the market as a preventive or therapeutic agent for digestive symptoms (nausea, emesis) caused by administration of an anti-malignant tumor agent (cisplatin or the like). In addition, a possibility has been reported that ramosetron or a salt thereof may be useful as an agent for treating diarrheal-type irritable bowel syndrome or an agent for improving diarrheal symptoms of irritable bowel syndrome (Patent Reference 1), and its clinical trials are now in progress as an agent for treating diarrheal-type irritable bowel syndrome or an agent for improving diarrheal symptoms of irritable bowel syndrome.
As a process for producing ramosetron or a salt thereof, the following production methods are known.
Patent Reference 1 describes a production method shown by the following Production method A, namely a method for producing a tetrahydrobenzimidazole derivative (V) by allowing a heterocyclic compound (III) to react with a carboxylic acid represented by a formula (IV) or its reactive derivative.
(Production Method A)

(In the formula, X2 is a single bond and binds to a carbon atom on the heterocyclic ring represented by Het.)
As an illustrative production method of ramosetron, Patent Reference 1 describes a production method (Production method A-1) in which racemic ramosetron are obtained by using 1-methyl-1H-indole as the compound (III), and N,N-diethyl-4,5,6,7-tetrahydrobenzimidazole-5-carboxamide or N-[(4,5,6,7-tetrahydrobenzimidazol-5-yl)carbonyl]pyrrolidine, which are acid amides, as the reactive derivative of compound (IV), and allowing them to undergo treatment with phosphorus oxychloride (Vilsmeyer reaction), and then their optical resolution is carried out by fractional crystallization using (+)-dibenzoyltartaric acid.
In addition, the Patent Reference 1 exemplifies an acid halide as one of the reactive derivatives of the compound (IV), and also describes another production method of the compound (V) (Production method A-2) in which the heterocyclic compound (III) is condensed with an acid halide of the compound (IV) by the Friedel-Crafts acylation reaction using a Lewis acid as the catalyst. However, illustrative production example of ramosetron by the Friedel-Crafts acylation reaction is not described therein.
Also, a method similar to the Production example A-1 is described in Non-patent References 1 and 2 as a production method of ramosetron.
In addition, Non-patent Reference 3 describes a method for producing ramosetron labeled with 11C, represented by a Production method B. However, it discloses only the methylation step, and does not disclose a production method of nor-YM060 as the starting material.
(Production Method B)

(In the formula, nor-YM060 means (R)-5-[(1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole which was provided by the present applicant, DMF means dimethylformamide.)
- Non-patent Reference 1: Chemical & Pharmaceutical Bulletin, 1996, vol. 44, no. 9, p. 1707-1716
- Non-patent Reference 2: Drugs of the Future, 1992, vol. 17, no. 1, p. 28-29
- Non-patent Reference 3: Applied Radiation and Isotopes, 1995, vol. 46, no. 9, p. 907-910
- Patent Reference 1: JP-B-6-25153

LIU Qing-wen, XU Hao, TIAN Hua, ZHENG Liang-yu, ZHANG Suo-qin
Chemoenzymatic Synthesis of Ramosetron Hydrochloride
2012 Vol. 28 (1): 70-72 [Abstract] ( 1143 ) [HTML 1KB] [PDF 206KB] ( 1052 )
doi:http://www.cjcu.jlu.edu.cn/hxyj/EN/abstract/abstract13356.shtml
…………………………………………………………………………..

The Vilsmeier-type reaction of 1-methylindole (I) with 5 – (1-pyrrolidinocarbonyl) -4,5,6,7-1 H-tetrahydrobenzimidazole hydrochloride (II) and phosphorous oxychloride in 1,2-dichloroethane gives (-5? -. [(1-methyl-3-indolyl) carbonyl] -4,5,6,7-tetrahydro-1H-benzimidazol e (III) Optical resolution of (III) with (+)-dibenzoyltartaric acid (DIBTA) in DMF -H2O, followed by exchange of the salt affords YM060.
………………………………………………….
Ondansetron: 1,2,3 ,9-Tetrahydro-9-methyl-3-[(2-methyl1-H-imidazole-1-yl)methyl]-4H-carbazol-4-one

Granisetron: Endo-1-methyl-N-(9-methyl-9-azabicyclo[3.3.1]non-3-yl)-1H-indazole-3-carboxamide

Tropisetron: Endo-1H-indole-3-carbocylic acid8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester

Dolasetron: 1H-Indole-3 -carboxylic acid (2a, 6a, 8a, 9up)-octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl Ester

Azasetron: (±)-N-Azabicyclo[2.2.2]oct-3-yl-6-chloro-3,4-dihydro-4-methyl-3-oxo-1,4-benzoxazine-8-carboxamide

Alosetron: 2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl- 1H-imidazol-4-yl)methyl]-1H-pyrido[4,3-b]indol-1-one

Ramosetron

