
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
![]()

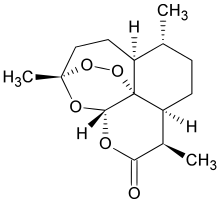
New Patent, WO 2016110874, Artemisinin , IPCA Laboratories Ltd
FOR Cancer; Parasitic infection; Plasmodium falciparum infection; Viral infection
KUMAR, Ashok; (IN).
SINGH, Dharmendra; (IN).
MAURYA, Ghanshyam; (IN).
WAKCHAURE, Yogesh; (IN)

Dr. Ashok Kumar, President – Research and Development (Chemical) at IPCA LABORATORIES LTD
IPCA LABORATORIES LIMITED [IN/IN]; 48, Kandivli Industrial Estate, Charkop, Kandivali (West), Mumbai 400067 (IN)

Novel process for preparing artemisinin or its derivatives such as dihydroartemisinin, artemether, arteether and artesunate. Also claims novel intermediates of artemesinin such as artemisinic acid or dihydroartemisinic acid. Discloses the use of artemisinin or its derivatives, for treating malaria, cancer, viral and parasitic infections.
In July 2016, Newport Premium™ reported that IPCA was capable of producing commercial quantities of artemether, arteether and artesunate; and holds an inactive US DMF for artemether since February 2009. In July 2016, IPCA’s website lists artemether, arteether and artesunate under its products and also lists artemether and artesunate as having EDMF and WHO certificates. The assignee also has Canada HPFB certificate for artemether.
The Central Drug Research Institute (CDRI) in collaboration with IPCA is developing CDRI-97/78 (1,2,4 trioxane derivative), a synthetic artemisinin substitute for treating drug resistant Plasmodium falciparum infection. In July 2016, CDRI-97/78 was reported to be in phase 1 clinical development. IPCA in collaboration with CDRI was also investigating CDRI-99/411, a synthetic artemisinin substitute for treating malaria; but its development had been presumed to have been discontinued; however, this application’s publication would suggest otherwise.

Writeup
Artemisinin is an active phytoconstituent of Chinese medicinal herb Artemisia annua, useful for the treatment of malaria. Generally, artemisinin/artemisinic acid is obtained by extraction of the plant, Artemisia annua. The plant Artemisia annua was first mentioned in an ancient Chinese medicine book written on silk in the West Han Dynasty at around 200 B.C. The plant’s anti-malarial application was first described in a Chinese pharmacopeia, titled “Chinese Handbook of Prescriptions for Emergency Treatments,” written at around 340 A.D.
Artemisinin being poorly bioavailable limits its effectiveness. Therefore semisynthetic derivatives of artemisinin such as artesunate, dihydroartemisinin, artelinate, artemether, arteether have been developed to improve the bioavailability of Artemisinin.
Artemisinin and its derivatives – dihydroartemisinin, artemether, arteether, and artesunate being a class of antimalarials compounds used for the treatment of uncomplicated, severe complicated/cerebral and multi drug resistant malaria. Additionally, there are research findings that artemisinin and its derivatives show anti-parasite, anti-cancer, and anti-viral activities.


Dihydroartemisinin Artesunate
The content of Artemisinin in the plant Artemisia annua varies significantly according to the climate and region/geographical area where it is cultivated. Further, the extraction methods provide artemisinin or artemisinic acid from the plant in very poor yields and therefore not sufficient to accommodate the ever-growing need for this important drug. Consequently, widespread use of these valuable drugs has been hampered due to the low availability of this natural product. Therefore, research has focused on the syntheses of this valuable drug in a larger scale to meet the increasing global demand and accordingly ample literature is available on the synthesis of artemisinin or its derivatives, but no commercial success being reported / known till date.
Artemisinin can be prepared synthetically from its precursors such as artemisinic acid or dihydroartemisinic acid according to literature methods known to skilled artisans. For example, dihydroartemisinic acid can be converted to artemisinin by a combination of photooxidation and air-oxidation processes as described in U.S. Patent No. 4,992,561.
Amorphadiene is an early starting material for synthesis of Artemisinic acid or dihydroartemisinic acid, which is an important intermediate for producing Artemisinin commercially, and WO2006128126 reported a preparation method as mentioned in scheme- 1.

acid
In accordance with the scheme 1, the amorphadiene is treated with di(cyclohexyl)borane ( δΗι ΒΗ followed by reaction with H2O2 in presence of NaOH to obtain the amorph-4-ene 12-ol which is further oxidized to dihydroartemisinic acid using CrCb/ifcSC^. The formation of amorph-4-ene 12-ol is taking place via epoxidation of the exocyclic double bond. However, the reported yields of this synthesis are very low, making it unviable to produce artemisinic acid at a cheaper cost than natural extraction, for commercial use.

Amorpha -4, 11-diene
A similar method is published in, WO2009088404, for synthesis of dihydroartemisinic acid through preparation of amorph-4-ene-12-ol via epoxide formation, albeit, predominantly at exo position by reacting the amorpha-4,11-diene with H2O2 in presence of porphyrin catalyst (TDCPPMnCl). During reaction, epoxidation also occurred at endo position leading to formation of Amorphadiene- 4,5- epoxide that remain as impurity. The formed exo epoxide (amorphadiene – 11, 12 – epoxide) is further reduced to get amorph- 4-ene 12-ol and then converted to dihydroartemisinic acid and finally converted into artemisinin.

Amorphadiene-11,12-epoxide
This process involves expensive & industry unfriendly reagents. Moreover, desired stereo isomers were obtained only in poor yields, because several purification steps were needed to get desired stereo isomers leading to escalated production/operational costs.
Therefore there remains a need in the art to improve the yield of Dihydroartemisinic acid, which could potentially reduce the cost of production of Artemisinin and/or its derivatives. Consequently it is the need of the hour to provide a synthetic and economically viable process to meet the growing worldwide demand by improving the process for Artemisinin and/or its derivatives to obtain them in substantially higher yields with good purity by plant friendly operations like crystallization/extractions rather than column chromatography/other cost constraint procedures.
Therefore, the object of the invention is to prepare Artemisinic acid of formula-II, Dihydroartemisinic acid of formula-IIa, Artemisinin and its derivatives through Amorphadiene- 4,5- epoxide.


DHAA methyl ester
Scheme 2
Method 4 (From compound of formula IV (R = CI)):
In the 4-neck round bottom flask was charged Diphenyl sulfoxide (23.8 g), NaHC03 (32.96 g) and DMSO (80 ml) at 30°C. Further a solution of compound of formula IV (R = CI) (10 g) in DMSO (20 ml) was charged to the reaction mass at 30°C followed by heating and maintaining the temperature for 40 hours at 80°C till completion. DMSO was distilled out under vacuum. The reaction mass was cooled followed by charging water
(100 ml) and toluene (100 ml) to the reaction mass with stirring for 30 minutes at 28°C. The layers were separated out and aqueous layer was back extracted with toluene (2 X 100 ml). The organic layer was washed with water (100 ml) and saturated brine solution (100 ml). Solvent was distilled out under vacuum at 50°C, and the crude mass degassed under vacuum at 50-55°C. IPA (40 ml) was charged to the mass. Simultaneous addition of hydrazine hydrate (65% in aqueous solution) (3.8 g) and hydrogen peroxide (50% in aqueous solution) (2.5 ml) was done at 30-32°C over a period of 3.25 hours. After completion, reaction mass was cooled up to 5-10°C and water (100ml) was added to the reaction mass. The pH of the reaction mass was adjusted to 3.8 with dilute 8% aqueous HC1 (24 ml) at 10°C. Ethyl acetate (60 ml) was added to the reaction mass at 10°C and stirred for 15 minutes at 15-20°C. The layers were separated. Aqueous layer was back extracted with ethyl acetate (2 X 20 ml). The combined organic layer was washed with 10%) sodium metabisulfite solution (50 ml), water (50 ml) and saturated brine solution (50 ml). The organic layer was distilled out under vacuum at 45°C and the obtained crude mass was degassed at 50-55°C. To this was added DME (40 ml), Biphenyl (0.9 g) and Li-metal (1.63 g) and the reaction mass was maintained for 10 hours at 80-85°C till reaction completion. The reaction mass was cooled up to 0-5°C followed by drop wise addition of water within one hour, and the reaction stirred for two hours at 20-25°C. Toluene (35 ml) was charged with stirring and layers were separated. The aqueous layer was washed with toluene (35 ml) and the combined toluene layer was washed with water (20 ml). The combined aqueous layer was again washed with toluene (20 ml). The aqueous layer was cooled to 10-15°C and pH adjusted to 3.5-4 with dilute 16% aqueous HC1. MDC (50 ml) was charged and stirred 30 minutes at 20-25°C followed by separation of layers. The aqueous layer extracted with MDC (25 ml) and the combined MDC layer was washed with water (50 ml), then with saturated NaCl solution (25 ml). The solvent was distilled out under vacuum at 40-45°C and the crude mass (Purity: 70-80%>) was degassed at 65-70°C. The crude product (10 g) was dissolved in ethyl acetate (200 ml). 10%> aqueous NaOH (100 ml) was charged to the reaction mass and stirred one hour at 20°C followed by layer separation. Again 10%> aqueous NaOH (100ml) was added to the organic layer, stirred for 30 minutes and layers were separated out. The pH of the combined NaOH solution wash was adjusted to 4.0 with dilute 16%> aqueous HC1 at 5-10°C under stirring. Ethyl acetate (850 ml) was charged to aqueous acidic mass, stirred 30 minutes and layers were separated out. The aqueous layer was back extracted with ethyl acetate (2 X 30 ml) and the combined organic layer was washed with water (100 ml) and saturated brine (50 ml). The organic layer was dried over sodium chloride, solvent was distilled out under vacuum and the purified mass was degassed under vacuum at 50-55°C to obtain Dihydroartemisinic acid (Purity: 90-95%).
b) Methyl ester of Dihydroartemisinic acid:
To a clear solution of Dihydroartemisinic acid (40 g) dissolved in MDC (120 ml) was added thionyl chloride (SOCh) (14.85 ml) at 10±2°C and reaction mass was heated to reflux temperature 40±2°C. After the completion of reaction, solvent was distilled out and excess SOCh was removed under reduced pressure. The resulting concentrated mass of acid chloride was dissolved in MDC (200 ml). In another RBF was taken triethylamine (30.6 ml) and methanol (120 ml). To this solution was added above acid chloride solution at 30±2°C and maintained till completion of reaction. To the reaction mass was added water (400 ml) and organic layer was separated. The aqueous layer was washed with MDC and mixed with main organic layer and the combined organic layer was back washed with water till neutral pH. Then organic layer was concentrated to give methyl ester of Dihydroartemisinic acid as a brown color oily mass.
Weight: 41.88 gm
Yield = 98%
c) Artemisinin:
Methyl ester of dihydroartemisinic acid (67.7 g) was dissolved in methanol (338 ml). To this solution was added Sodium molybdate (29.5 g), 50% hydrogen peroxide (147.3 g) was added at 30±2°C and reaction was maintained for 3-4 hours. After completion of reaction was added water (300 ml) and MDC (300 ml) to the reaction mass. The organic layer was separated and aqueous layer washed with MDC (100 ml). The combined organic layer was concentrated to 475 ml containing hydroperoxide intermediate and directly used for next stage reaction. In another RBF containing MDC (475 ml) was added benzene sulfonic acid (1.27 g) and Indion resin (6.7 g). This heterogeneous solution was saturated with oxygen by passing O2 gas for 10 min at 0±2°C. To this was added previous stage hydroperoxide solution at same temperature with continuous 02 gas purging within 30-40 minutes. The oxygen gas was passed at same temp for 4 hours and temperature raised to 15±2°C with continued passing of oxygen for 5 hours. The
mixture was stirred at 25-30°C for 8-10 hours followed by filtration of resin. The filtrate was washed with water (200 ml X 3) and the combined aqueous layer back washed with MDC (50 ml). The combined organic layer was concentrated to give crude Artemisinin. Weight: 54 gm
Yield= 70.7%
Purification of Artemisinin:
Crude Artemisinin (10 g) was dissolved in ethyl acetate (25 ml) at 45-50°C. The solution was cooled to 30-35°C followed by addition of n-Hexane (100 ml). The material was isolated, stirred for 2 hours, filtered and vacuum dried at 45°C.
Weight: 4 gm
Yield: 40%

THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India
////////New Patent, WO 2016110874, Artemisinin , IPCA Laboratories Ltd, malaria, Cancer, Parasitic infection, Plasmodium falciparum infection, Viral infection, artemether artemisinin, artemotil, artenimol, artesunate,

