Ranbaxy to introduce malarial treatment Synriam in African nations
Ranbaxy Laboratories has obtained regulatory approval to introduce India’s first new chemical entity (NCE) Synriam (arterolane maleate 150mg and piperaquine phosphate 750mg drug) in seven African countries.
read at
http://www.pharmaceutical-technology.com/news/newsmalarial-treatment-synriam-4471331?WT.mc_id=DN_News
Synriam is a new age therapy recommended to treat uncomplicated Plasmodium falciparum malaria in adults. It was launched in India in April 2012.
The product was also launched in Uganda and is set to be introduced in Nigeria, Senegal, Cameroon, Guinea, Kenya and Ivory Coast by the end of January 2015.

Arterolane
cas 664338-39-0, UNII-3N1TN351VB, OZ277, RBX-11160, NCGC00274173-01
Molecular Formula: C22H36N2O4
Molecular Weight: 392.53224
cis-adamantane-2-spiro-3’-8’-[[[(2’-amino-2’ methylpropyl) amino] carbonyl] methyl] 1’,2’,4’-trioxaspiro [4.5] decane
cis-adamantane-2-spiro-3′-8′-[[[(2′- amino-2′-methylpropyl)amino]carbonyl]-methyl]- 1 ‘,2′,4′-trioxaspiro[4.5]decane
Arterolane, also known as OZ277 or RBx 11160,is a substance being tested for antimalarial activity[1] by Ranbaxy Laboratories.[2] It was discovered by US and European scientists who were coordinated by the Medicines for Malaria Venture (MMV).[3] Its molecular structure is uncommon for pharmacological compounds in that it has both an ozonide group and an adamantane substituent.[4]
Phase III clinical trials of arterolane, in combination with piperaquine, began in India in 2009.[5] When clinical trial results were disappointing, the MMV withdrew support[2] and Ranbaxy continued developing the drug combination on its own.

Ranbaxy launched India’s first new drug, SynriamTM, treating Plasmodium falciparummalaria in adults. The drug provides quick relief from most malaria-related symptoms, including fever, and has a high cure rate of over 95 %.
Just one tablet per day is required, for three days, instead of two to four tablets, twice daily, for three or more days with other medicines. The drug is independent of dietary restrictions for fatty foods or milk.
Ranbaxy developed Synriam as a fixed-dose combination of arterolane maleate and piperaquine phosphate, where arterolane is the new chemical entity (NCE) that was developed as an alternative to artemisinin. It is the first recently developed antimalarial not based on artemisinin, one of the most effective treatments for malaria, which has shown problems with resistance in recent years. Arterolane was discovered by a collaborative drug discovery project funded by the Medicines for Malaria Venture. Since SynriamTM has a synthetic source, unlike artemisinin-based drugs, production can be scaled up whenever required and a consistent supply can be maintained at a low cost.

The new drug, has been approved by the Drug Controller General of India (DCGI) for marketing in India and conforms to the recommendations of the World Health Organization (WHO) for using combination therapy in malaria. Ranbaxy is also working to make it available in African, Asian and South American markets where Malaria is rampant. SynriamTM trials are ongoing for Plasmodium vivax malaria and a paediatric formulation.
Derek Lowe of the famous In the Pipeline blog had written about arterolane in 2009. At the time it was in Phase III trial, which I assumed were the trials that Ranbaxy was conducting. But it turned out that arterolane was developed by a collaboration between researchers in the US, the UK, Switzerland and Australia who were funded by the World Health Organization and Medicines for Malaria Venture (a Swiss non-profit).
They published this work in Nature in 2004 and further SAR (Structure Activity Relationship) studies in J Med Chem in 2010. So Ranbaxy did not develop the drug from scratch? But the press release quotes Arun Sawhney, CEO and Managing Director of Ranbaxy which misleads people to think so: “It is indeed gratifying to see that Ranbaxy’s scientists have been able to gift our great nation its first new drug, to treat malaria, a disease endemic to our part of the world.
This is a historic day for science and technology in India as well as for the pharmaceutical industry in the country. Today, India joins the elite and exclusive club of nations of the world that have demonstrated the capability of developing a new drug”. So Ranbaxy mixes a known active compound (piperaquine) with a new compound that someone else found to be active (arterolane) and claims that they developed a new drug?
In an interview in LiveMint, Sawhney says, “Ranbaxy spent around $30 million on Synriam and the contribution from DST [India’s Department of Science & Technology] was Rs.5 crore.
The drug went through several phases of development since the project began in 2003. We did not look at this as a commercial development. Instead, this is a CSR [Corporate Social Responsibility] venture for us.” That’s a give away because developing a new drug from scratch has to cost more than $30 million + Rs.50 million.
 now taken over by sun
now taken over by sun


SynriamTM
| Generic Name |
| Arterolane Maleate and Piperaquine Phosphate Tablets |
|
| Composition |
| Each film coated tablet contains: Arterolane maleate equivalent to Arterolane ……………………………150 mg Piperaquinephosphate……………750 mg |
|
| Dosage Form |
| Tablets |
|
|
|
|
|
| Inactive ingredients: |
| Microcrystalline cellulose, Crospovidone, Magnesium stearate, Hydroxypropyl methyl cellulose/Hypromellose, Titanium dioxide, Macrogol/ Polyethylene glycol, Talc, Ferric Oxide (Yellow), Ferric Oxide (Red) |
|
|
|
Description SynriamTM is a fixed dose combination of two antimalarial active ingredients arterolane maleate and piperaquine phosphate.
Arterolane maleate is a synthetic trioxolane compound. The chemical name of arterolane maleate is cis-adamantane-2-spiro-3’-8’-[[[(2’-amino-2’ methylpropyl) amino] carbonyl] methyl] 1’,2’,4’-trioxaspiro [4.5] decane hydrogen maleate. The molecular formula is C26H40N2O8 and molecular weight is 508.61. The structural formula is as follows:

| MALARIA |
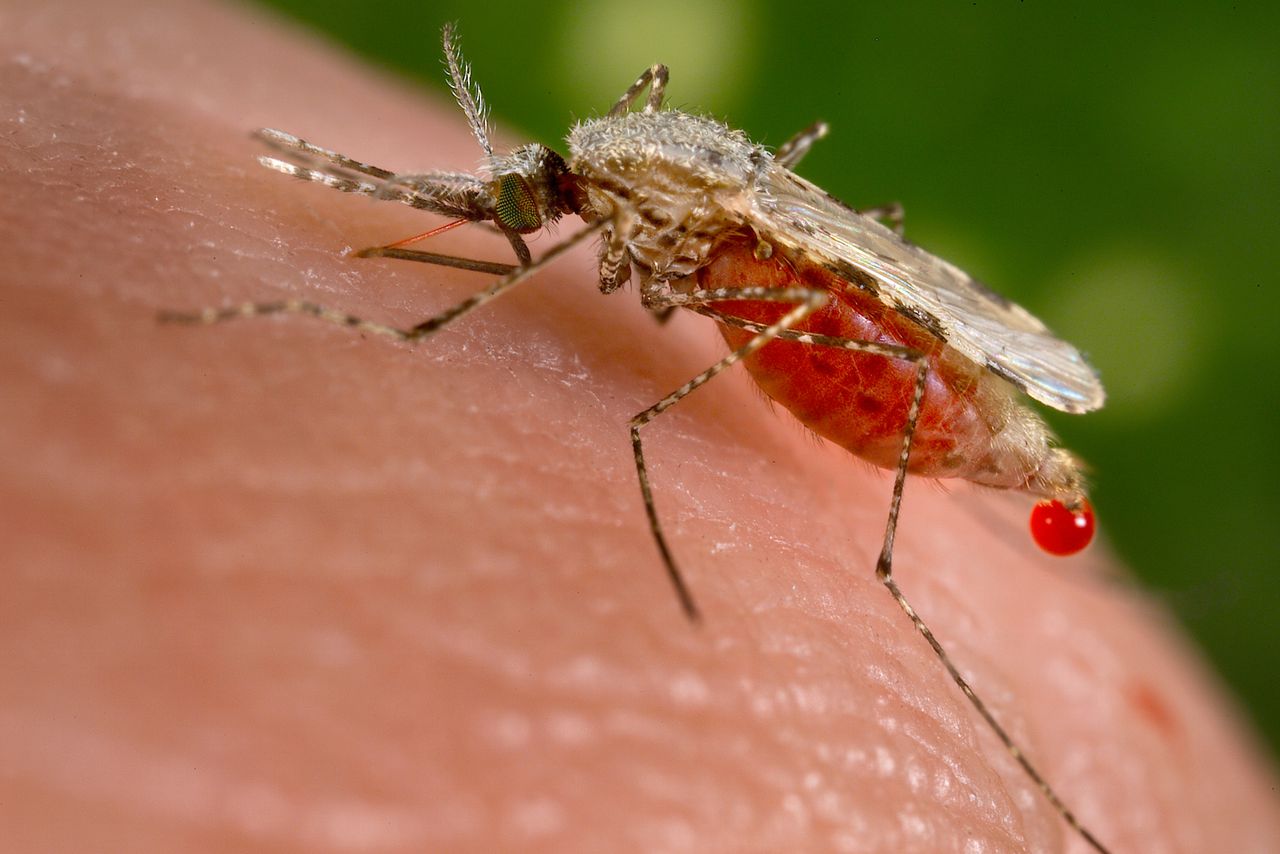
| Malaria is one of the most prevalent and deadly parasitic diseases in the world. Up to 289 million cases of malaria may have occurred in 2010, causing between 660,000 and 1.25 million deaths, mainly in Africa and mostly of children younger than 5 years. |
| (WHO: http://www.who.int/malaria/publications/world_malaria_report_2012/en/index.html; Fidock, D. A. Eliminating Malaria. Science 2013, 340, 1531-1533.) |
|

The most serious problem in malaria treatment is that the parasites causing the disease, particularly the deadly Plasmodium falciparum, have developed resistance to widely used drugs, particularly chloroquine (CQ). Currently, the most efficacious therapies are combinations of an artemisinin-type compound with a long-lasting partner drug like lumefantrine, amodiaquine or mefloquine.
|

Malaria, the most common parasitic disease of humans, remains a major health and economic burden in most tropical countries. Large areas of Central and South America, Hispaniola (Haiti and the Dominican Republic), Africa, the Middle East, the Indian subcontinent, Southeast Asia, and Oceania are considered as malaria-risk areas. It leads to a heavy toll of illness and death, especially amongst children and pregnant women.
According to the World Health Organization, it is estimated that the disease infects about 400 million people each year, and around two to three million people die from malaria every year. There are four kinds of malaria parasites that infect human: Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale and Plasmodium malariae.
Malaria spreads from one person to another by the bite of mosquito, Anopheles gambiae, which serves as vector. When a mosquito sucks the blood of human, sporozoites are transfused into the human body together with saliva of the mosquito. The sporozoites enter into the hepatocytes, reproduce asexually and finally enter into the blood stream. The parasites continue to multiply inside the red blood cells, until they burst and release large number of merozoites.
This process continues, destroying a significant number of blood cells and causing the characteristic paroxysm (“chills and fever”) associated with the disease. In the red blood cells, some of the merozoites become male or female gametocytes. These gametocytes are ingested by the mosquito when it feeds on blood. The gametocytes fuse in the vector’s gut; sporozoites are produced and are migrated to the vector’s salivary glands.
The clinical symptoms of malaria are generally associated with the bursting of red blood cells causing an intense fever associated with chills that can leave the infected individual exhausted and bedridden. More severe symptoms associated with repeat infections and/or infection by Plasmodium falciparum include anaemia, severe headaches, convulsions, delirium and, in some instances, death.
Quinine, an antimalarial compound that is extracted from the bark of cinchona tree, is one of the oldest and most effective drugs in existence. Chloroquine and mefloquine are the synthetic analogs of quinine developed in 1940’s, which due to their effectiveness, ease of manufacture, and general lack of side effects, became the drugs of choice. The downside to quinine and its derivatives is that they are short-acting and have bitter taste.
Further, they fail to prevent disease relapses and are also associated with side effects commonly known as “Chinchonism syndrome” characterized by nausea, vomiting, dizziness, vertigo and deafness. However, in recent years, with the emergence of drug- resistant strains of parasite and insecticide-resistant strains of vector, the treatment and/or control of malaria is becoming difficult with these conventional drugs.
Malarial treatment further progressed with the discovery of Artemisinin
(qinghaosu), a naturally occurring endoperoxide sesquiterpene lactone isolated from the plant Artemisia annua (Meshnick et al., Microbiol. Rev. 1996, 60, p. 301-315; Vroman et al., Curr. Pharm. Design, 1999, 5, p. 101-138; Dhingra et al., 2000, 66, p. 279-300), and a number of its precursors, metabolites and semi-synthetic derivatives which have shown to possess antimalarial properties. The antimalarial action of artemisinin is due to its reaction with iron in free heme molecules of the malaria parasite, with the generation of free radicals leading to cellular destruction. This initiated a substantial effort to elucidate its molecular mechanism of action (Jefford, dv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297) and to identify novel antimalarial peroxides (Dong and Vennerstrom, Expert Opin. Ther. Patents 2001, 1 1, p. 1753-1760).
Although the clinically useful artemisinin derivatives are rapid acting and potent antimalarial drugs, they have several disadvantages including recrudescence,
neurotoxicity, (Wesche et al., Antimicrob. Agents. Chemother. 1994, 38, p. 1813-1819) and metabolic instability (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43). A fair number of these compounds are quite active in vitro, but most suffer from low oral activity (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43 and van Agtmael et al., Trends Pharmacol. Sci., 1999, 20, p. 199-205). Further all these artemisinin derivatives are conventionally obtained from plant source and are therefore expensive.
As the cultivation of the plant material is dependent on many factors including the weather conditions, the supply source thus becomes finite and there are chances of varying yield and potency. This leads to quality inconsistencies and supply constraints. As malaria is more prevalent in developing countries, a switch to cheaper and effective medicine is highly desirable.
Thus there exists a need in the art to identify new peroxide antimalarial agents, especially those which are not dependent on plant source and can be easily synthesized, are devoid of neurotoxicity, and which possess improved solubility, stability and pharmacokinetic properties.
Following that, many synthetic antimalarial 1 ,2,4-trioxanes (Jefford, Adv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297), 1,2,4,5-tetraoxanes (Vennerstrom et al., J. Med. Chem., 2000, 43, p. 2753-2758), and other endoperoxides have been prepared. Various patents/applications disclose means and method for treating malaria using Spiro or dispiro 1,2,4-trioxolanes for example, U.S.
Patent Application No. 2004/0186168 and U.S. Patent Nos. 6,486, 199 and 6,825,230. The present invention relates to solid dosage forms of the various spiro or dispiro 1 ,2,4- trioxolanes antimalarial compounds disclosed in these patents/applications and are incorporated herein by reference.
Active compounds representing various Spiro and dispiro 1 ,2,4-trioxolane derivatives possess excellent potency, efficacy against Plasmodium parasites, and a lower degree of neurotoxicity, in addition to their structural simplicity and ease of synthesis. Furthermore, these compounds have half-lives which are believed to permit short-term treatment regimens comparing favorably to other artemisinin-like drugs. In general, the therapeutic dose of trioxolane derivative may range between about 0.1-1000 mg/kg/day, in particular between about 1-100 mg/kg/day. The foregoing dose may be administered as a single dose or may be divided into multiple doses. For malaria prevention, a typical dosing schedule could be, for example, 2.0-1000 mg/kg weekly beginning 1-2 weeks prior to malaria exposure, continued up to 1-2 weeks post-exposure.
Monotherapy with artemisinin (natural or synthetic) class of drugs might cure the patients within 3 days, however perceiving the potential threat of the malarial parasite developing resistance towards otherwise very potent artemisinin class of drugs, WHO had strictly called for an immediate halt to the provision of single-drug artemisinin malaria pills. Combination therapy in case of malaria retards the development of resistance, improve efficacy by lowering recrudescence rate, provides synergistic effect, and increase exposure of the parasite to the drugs.
Artemsinin based combinations are available in the market for a long time.
Artemether-lumafentrine (Co-artem®) was the first fixed dose antimalarial combination containing an artemisinin derivative and has been known since 1999. This combination has passed extensive safety and efficacy trials and has been approved by more than 70 regulatory agencies. Co-artem® is recommended by WHO as the first line treatment for uncomplicated malaria.
Other artemisinin based combinations include artesunate and amodiaquine (Coarsucam®), and dihydroartemisin and piperaquine (Eurartesim®). Unfortunately, all the available artemisinin based combinations have complicated dosage regimens making it difficult and inconvenient for a patient to comply completely with the total prescribed duration. For example, the dosage regimen of Co-artem®for an adult having body weight of more than 35 kg includes 6 doses over three days.
The first dose comprises four tablets initially, the second dose comprises four tablets after eight hours, the third to sixth doses comprise four tablets twice for another two days; making it a total of 24 tablets. The dosage regimen of Coarsucam® for an adult having body weight of more than 36 kg or age above 14 years includes three doses over three days; each dose comprises two tablets; making it a total of six tablets. The dosage regimen of Eurartesim® for an adult having body weight between 36 kg – 75 kg includes 3 doses over three days, each dose comprises of three tablets, making it a total of nine tablets.
It is evident that the available artemisinin-based combinations have a high pill burden on patients as they need to consume too many tablets. As noted above, this may increase the possibility of missing a few doses, and, consequently, could result in reduced efficacy due to non-compliance and may even lead to development of resistance for the drug. Therefore, there is an urgent and unmet need for anti-malarial combinations with a simplified daily dosing regimen that reduces the pill burden and would increase patient compliance.
Apart from simplifying the regimen, there are certain limitations for formulators developing formulations with trioxolones, the first being their susceptibility to degradation in presence of moisture that results in reduced shelf lives. Another is their bitter taste, which can result in poor compliance of the regimen or selection of another, possibly less effective, therapeutic agent.
……………………..
PATENT
http://www.google.st/patents/US6906205

……………………
PATENT
http://www.google.st/patents/WO2013008218A1?cl=en
structural Formula II.
Formula II
Active compound includes one or more of the various spiro and dispiro trioxolane derivatives disclosed in U.S. Application No. 2004/0186168 and U.S. Patent Nos.
6,486,199 and 6,825,230, which are incorporated herein by reference. These trioxolanes are relatively sterically hindered on at least one side of the trioxolane heterocycle which provides better in vivo activity, especially with respect to oral administration. Particularly, spiro and dispiro 1,2,4-trioxolanes derivatives possess excellent potency and efficacy against Plasmodium parasites, and a lower degree of neurotoxicity.
The term “Active compound I” herein means cis-adamantane-2-spiro-3′-8′-[[[(2′- amino-2′-methylpropyl)amino]carbonyl]-methyl]- 1 ‘,2′,4′-trioxaspiro[4.5]decane hydrogen maleate. The Active compound I may be present in an amount of from about 5% to about 25%, w/w based on the total dosage form.

………………
PATENT
http://www.google.st/patents/WO2007138435A2?cl=en
A synthetic procedure for preparing compounds of Formula I, salts of the free base c«-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]- 1 ‘, 2′, 4′-trioxaspiro [4.5] decane has been disclosed in U.S. 6,906,205.

The process for the preparation of compounds of Formula I wherein a compound of Formula II (wherein R is lower alkyl) is reacted with a compound of Formula III (wherein R is lower alkyl) to obtain compound of Formula IV;
Formula Formula IV
followed by hydrolysis of the compounds of Formula IV to give a compound of Formula V;
Formula V followed by the reaction of the compound of Formula V with an activating agent, for example, methyl chloroformate, ethyl chloroformate, propyl chloro formate, n-butyl chloro formate, isobutyl chloroformate or pivaloyl chloride leads to the formation of mixed anhydride, which is reacted in situ reaction with 1 ,2-diamino-2-methyl propane to give a compound of Formula VI; and
Formula Vl reacting the compound of Formula VI with an acid of Formula HX (wherein X can be the same as defined earlier) to give compounds of Formula I.
Example 1 : Preparation of O-methyl-2-adamantanone oxime
To a solution of 2-adamantanone (50 g, 0.3328 mol, 1 equiv.) in methanol (0.25 lit), sodium hydroxide solution (15 g, 0.3761mol, 1.13 equiv, in 50 mL water) was added followed by methoxylamine hydrochloride (37.5 g x 81.59% Purity= 30.596 g, 0.366 mol, 1.1 equiv) at room temperature under stirring. The reaction mixture was stirred at room temperature for 1 to 2 h. The reaction was monitored by HPLC. The reaction mixture was concentrated at 40- 45°C under vacuum to get a thick residue. Water (250 mL) was added at room temperature and the reaction mixture was stirred for half an hour. The white solid was filtered, washed with water (50 mL), and dried at 40 to 45°C under reduced pressure. O-methyl 2- adamantanone oxime (57 g, 95 % yield) was obtained as a white solid.
(M++l) 180, 1HNMR (400 MHz, CDCl3 ): δ 1.98 – 1.79 (m, 12H), 2.53 (s, IH), 3.46 ( s, IH), 3.81 (s, 3H).
Example 2: Preparation of 4-(methoxycarbonvmethvPcvclohexanone A high pressure autoclave was charged with a mixture of methyl (4- hydroxyphenyl)acetate (50 g, 0.30 mol), palladium ( 5g) (10 %) on carbon (50 % wet) and O- xylene (250 mL). The reaction mixture was stirred under 110 to 115 psi of hydrogen pressure for 7 to 8 h at 1400C. The reaction was monitored by HPLC. The reaction mixture was then cooled to room temperature, and the catalyst was filtered off. Filtrate was concentrated under reduced pressure to get 4-(methoxycarbonylmethyl)cyclohexanone as light yellow to colorless oily liquid (48.7 g, 97.4 %).
(M++!) 171, ‘ HNMR (400 MHz, CDCl 3): δ 1.48 – 1.51 ( m, 2H), 2.1 1-2.07 (m, 2H), 2.4- 2.23 (m, 7H), 3.7 (s, 3H).
Example 3: Preparation of methyl (Is, 4s)-dispiro [cyclohexane-l, 3′-f 1,2,4] trioxolane-5′, 2″-tricvclor3.3.1.13–71decan1-4-ylacetate
A solution of O-methyl-2-adamantanone oxime (example 1) (11.06 g, 61.7 mmol, 1.5 equiv.) and 4-(methoxycarbonymethyl)cyclohexanone (example 2) (7.0 g, 41.1 mmol, 1 equiv.) in cyclohexane ( 200ml) and dichloromethane (40 mL) was treated with ozone (ozone was produced with an OREC ozone generator [0.6 L/min. O2, 60 V] passed through an empty gas washing bottle that was cooled to -780C). The solvent was removed after the reaction was complete. After removal of solvents, the crude product was purified by crystallization from 80% aqueous ethanol (200 mL) to afford the title compound as a colorless solid. Yield: 10.83 g, 78%, mp: 96-980C; 1HNMR (500 Hz3CDCl3): δ 1.20-1.33 (m, 2H), 1.61-2.09 (m, 5 21H), 2.22 (d, J = 6.8Hz, 2H), 3.67(s,3H).
Example 4: Preparation of (Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″- tricvclo [3.3.1.13‘7] decanl-4-ylacetic acid
Sodium hydroxide (3.86 g, 96.57 mmol, 3 equiv.) in water (80 mL) was added to a solution of methyl (\s, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo
10 [3.3.1.I3‘7] decan]-4-ylacetate (example 3) (10.83 g, 32.19 mmol, 1 equiv.) in 95% ethanol (150 mL). The mixture was stirred at 500C for about 4 h, cooled to O0C, and treated with IM hydrochloric acid (129ml, 4 equiv). The precipitate was collected by filtration, washed with 50 % aqueous ethanol (150 mL) and dried in vacuum at 40 0C to give the title compound as colorless solid. Yield: 9.952 g, 96%, mp: 146-1480C ( 95% ethanol), 1HNMR (500 Hz,
15 CDCl3): δ 1.19-1.41 (m,2H), 1.60-2.05 (m,21H), 2.27 (d, J=6.8 Hz,2H).
Example 5: Preparation of c?s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, T , 4′-trioxaspiro [4.5] decane
Method A:
(Is, 4s)-dispiro[cyclohexane- 1 ,3 ‘-[ 1 ,2,4]trioxolane-5 ‘,2 ‘ ‘-tricyclo[3.3.1.13‘7]decan]-4-
.0 ylacetic acid (example 4) (5 g ,15.5mmol, 1 equiv) was mixed with triethylamine (2.5 g , 24.8 mmol, 1.6 equiv) in 100ml of dichloromethane. The reaction mixture was cooled to – 1O0C to 00C. Ethyl chloro formate (1.68 g, 17 mmol, 1.0 equiv) in 15 mL dichloromethane was charged to the above reaction mixture at – 100C to 00C. The reaction mixture was stirred at the same temperature for 10 to 30 minutes. The resulting mixed anhydride reaction mixture
15 was added dropwise to a previously prepared solution of l,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv), in 100 mL dichloromethane at -100C to O0C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the same temperature till the reaction was complete. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete
>0 within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (50 mL) was charged, organic layer was separated and washed with 10% sodium bicarbonate solution (50 mL) and water (50 mL) at room temperature. The organic layer was dried over sodium sulphate and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (50ml) was added to obtain residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. The solid was dried under reduced pressure at room 5 temperature.
Yield: 5.2 g (85.4 %), (M++l) 393, 1HNMR (400 MHz, DMSO-J6 ): δ 0.929 ( s, 6H), 1.105 – 1.079 (m, 2H), 1.887-1.641 (m, 21H), 2.030-2.017 (d, 2H), 2.928 (d, 2H).
Method B:
(Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo [3.3.1.I3‘7]
10 decan]-4-ylacetic acid (example 4) (10 g, 31mmol, 1 equiv) was treated with isobutyl chloroformate (4.5 g, 33mmol, 1.1 equiv) in presence of organic base like triethyl amine (5 g, 49.6mmol, 1.6 equiv) at 00C to 7°C in 250ml of dichloromethane. The solution was stirred at O0C to 7°C for aboutlO to 30 minutes. To the above reaction mixture, previously prepared solution of l,2-diamino-2-methylpropane (3.27 g, 37 mmol, 1.2 equiv), in 50 mL of
15 dichloromethane was added at O0C to 7°C in one lot. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. Reaction was complete within 2 h. The reaction nitrogen atmosphere was maintained throughout the reaction. Water (250 mL) was charged, organic
20 layer was separated and washed with 10% sodium bicarbonate solution (200 mL) and water (100 mL) at room temperature and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (100ml) was added to the residue, under stirring, at room temperature. The mixture was filtered and washed with chilled hexane (10 mL). The resultant solid was dried under reduced pressure at room temperature. Yield: 10.63 g (87%), (M++l) 393, 1HNMR
>5 (400 MHz, DMSO-J6 ) :δ 0.928 ( s, 6H), 1.102 – 1.074 (m, 2H), 1.859-1.616 (m, 21H), 2.031- 2.013 (d, 2H), 2.94-2.925 (d, 2H). Method C:
(\s, 4s)-dispiro[cyclohexane-l,3′-[l,2,4]trioxolane-5′,2″-tricyclo[3.3.1.13>7]decan]-4- ylacetic acid (example 4) (5 g, 15.5mmol, 1 equiv) was treated with pivaloyl chloride (1.87 g, 15.5 mmol, 1 equiv) and triethylamine (2.5gm, 24.8mmol, 1.6 equiv) at -15°C to -100C in dichloromethane (125 mL). The solution was stirred at -150C to -100C for aboutlO to 30 minutes. It resulted in the formation of mixed anydride. To the above reaction mixture, previously prepared solution of 1 ,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv) in 25 mL dichloromethane was added at -15°C to -100C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (125 mL) was charged, organic layer was separated and washed with 50 mL of 10% sodium bicarbonate solution and 125 mL of water, respectively at room temperature. Finally solvent was removed at 25 to 4O0C under reduced pressure. 50 mL of 5% Ethyl acetate – hexane solvent mixture was added to the residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. Solid was dried under reduced pressure at room temperature. Yield: 5.03 g (83 %), (M++l) 393, 1JINMR (400 MHz, OMSO-d6 ):δ 0.93 ( s, 6H), 1.113 – 1.069 (m, 2H), 1.861-1.644 (m, 21H), 2.033-2.015 (d, 2H), 2.948-2.933 (d, 2H).
Example 6: Preparation of c/s-adamantane-2-spiro-3′ -8 ‘-πT(2′-amino-2′ -methyl propyl) amino! carbonyl] methyli-l ‘, 2\ 4′-U-JoXaSpJrQ [4.51 decane maleate To a solution of c/s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, 2′, 4′-trioxaspiro [4.5] decane (example 5) (60 g, 0.153 moles) in ethanol (150 mL) was added a solution of maleic acid (17.3 g, 0.15 moles, 0.98 equiv. in ethanol 90 mL) and the reaction mixture was stirred for about 1 h. To this clear solution, n- heptane (720 mL) was added at room temperature in 1 h and the reaction mixture was stirred for 3 h. It was then cooled to 0 to 100C and filtered. The cake was washed with n-heptane (60 mL) and dried under vacuum at 40-450C.
Yield: 67 g, 77.4%, mp: 1490C (decomp), (M++l) 393.5, 1HNMR (300 MHz, DMSO-^ ): δ 1.05-1.11 (2H,m), 1.18 (6H,s), 1.64-1.89 (21H,m), 2.07(2H,d), 3.21 (2H,d), 6.06 (2H,d), 7.797 (2H, bs), 8.07 (IH, t).



References
- Dong, Yuxiang; Wittlin, Sergio; Sriraghavan, Kamaraj; Chollet, Jacques; Charman, Susan A.; Charman, William N.; Scheurer, Christian; Urwyler, Heinrich et al. (2010). “The Structure−Activity Relationship of the Antimalarial Ozonide Arterolane (OZ277)”. Journal of Medicinal Chemistry 53 (1): 481–91. doi:10.1021/jm901473s. PMID 19924861.
- Blow to Ranbaxy drug research plans at LiveMint.com, Sep 21 2007
- Vennerstrom, Jonathan L.; Arbe-Barnes, Sarah; Brun, Reto; Charman, Susan A.; Chiu, Francis C. K.; Chollet, Jacques; Dong, Yuxiang; Dorn, Arnulf et al. (2004). “Identification of an antimalarial synthetic trioxolane drug development candidate”. Nature 430 (7002): 900–4.doi:10.1038/nature02779. PMID 15318224.
- In the Pipeline: “Ozonides As Drugs: What Will They Think Of Next?”, by Derek Lowe, November 23, 2009, at Corante.com
- Indian company starts Phase III trials of synthetic artemisinin, May 4 2009, at the WorldWide Antimalarial Resistance Network
- http://www.nature.com/nature/journal/v430/n7002/full/nature02779.html
|
|
5-27-2011
|
PROCESS FOR THE PREPARATION OF DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS (OZ277)
|
|
|
2-13-2009
|
STABLE DOSAGE FORMS OF SPIRO AND DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS
|
|
|
6-15-2005
|
Spiro and dispiro 1,2,4-trioxolane antimalarials
|
|
|
11-31-2004
|
Spiro and dispiro 1,2,4-trixolane antimalarials
|
ANTIMALARIALS




http://www.rsc.org/chemistryworld/2013/03/new-antimalarial-drug-class-resistance-elq-300-quinolone

http://www.nature.com/nrd/journal/v9/n11/full/nrd3301.html
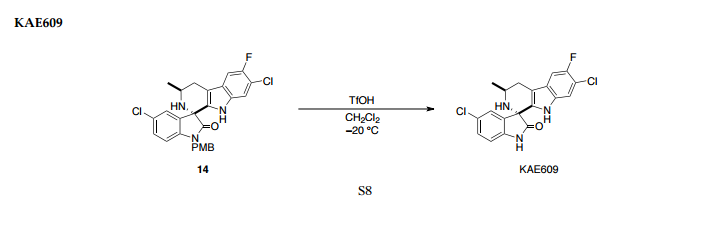
Structure of NITD609; the 1R,3Sconfiguration is fundamental for its antimalarial activity

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











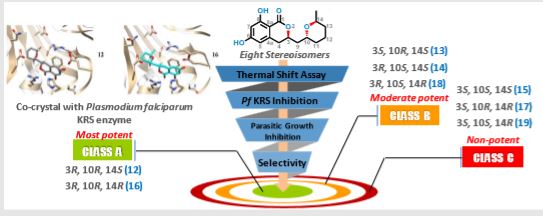


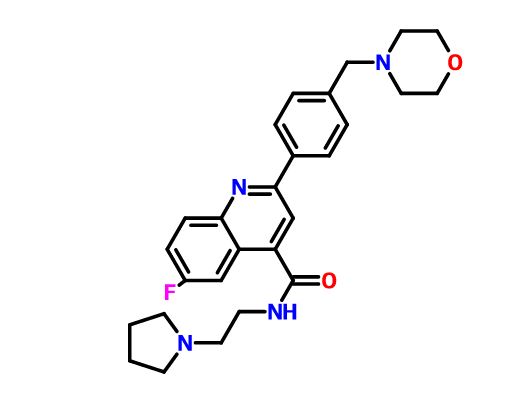



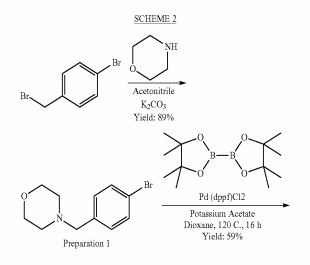
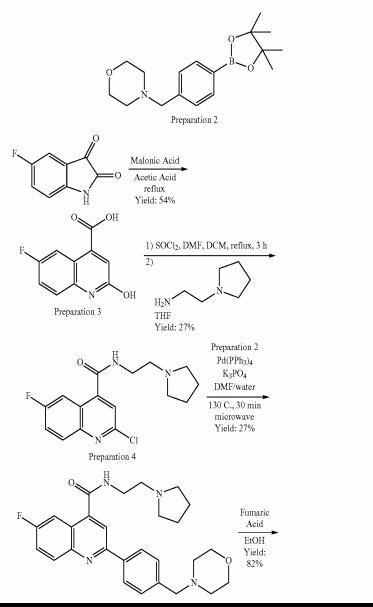
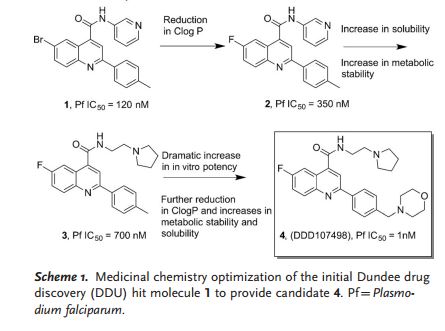
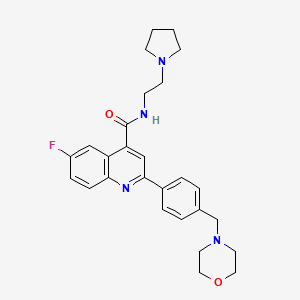 DDD498
DDD498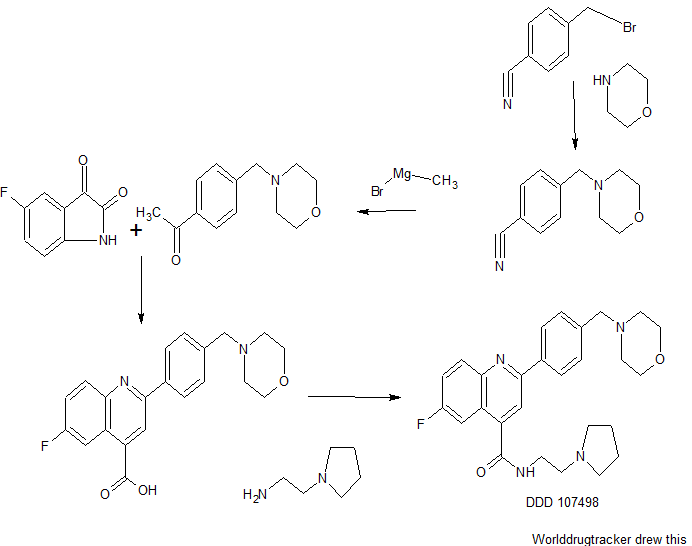































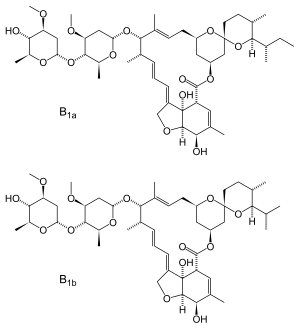
 C48H74O14, 875.09
C48H74O14, 875.09



 Professor Satoshi Ōmura
Professor Satoshi Ōmura
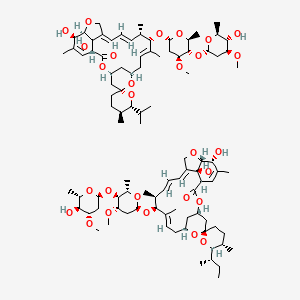
 IVERMECTIN
IVERMECTIN